

Background: In most bacteriophages, a long tail primarily built from tail tube proteins serves as a conduit for DNA delivery into the bacteria.
Results: The tail tube protein of phage SPP1 self-assembles into tubes exhibiting a phage tail-like helical architecture.
Conclusion: A three-dimensional model is proposed for the self-assembled tubes.
Significance: This work opens the way for the generation of artificial tubular structures.
Keywords: Bacteriophage, Fourier Transform IR (FTIR), Solid State NMR, Tertiary Structure, Virus Assembly, Electron Microscopy, Virion, Tail Tube
Abstract
The majority of known bacteriophages have long tails that serve for bacterial target recognition and viral DNA delivery into the host. These structures form a tube from the viral capsid to the bacterial cell. The tube is formed primarily by a helical array of tail tube protein (TTP) subunits. In phages with a contractile tail, the TTP tube is surrounded by a sheath structure. Here, we report the first evidence that a phage TTP, gp17.1 of siphophage SPP1, self-assembles into long tubes in the absence of other viral proteins. gp17.1 does not exhibit a stable globular structure when monomeric in solution, even if it was confidently predicted to adopt the β-sandwich fold of phage λ TTP. However, Fourier transform infrared and nuclear magnetic resonance spectroscopy analyses showed that its β-sheet content increases significantly during tube assembly, suggesting that gp17.1 acquires a stable β-sandwich fold only after self-assembly. EM analyses revealed that the tube is formed by hexameric rings stacked helicoidally with the same organization and helical parameters found for the tail of SPP1 virions. These parameters were used to build a pseudo-atomic model of the TTP tube. The large loop spanning residues 40–56 is located on the inner surface of the tube, at the interface between adjacent monomers and hexamers. In line with our structural predictions, deletion of this loop hinders gp17.1 tube assembly in vitro and interferes with SPP1 tail assembly during phage particle morphogenesis in bacteria.
Introduction
More than 96% of bacterial viruses (bacteriophages or phages) have a tail (1). This structure is designed for phage attachment to bacteria, penetration of the cell envelope, and delivery of the viral genome into the host. In long-tailed phages, the main elementary unit of the genome delivery tube is the phage tail tube protein (TTP).5 During assembly the tail adsorption apparatus is built first, thus providing a platform for helical polymerization of the TTP around a tape measure protein that defines the tail tube length (2, 3). The tube is then tapered by tail completion proteins that build the interface for binding the viral capsid connector (4, 5). When the tail adsorption apparatus recognizes its cognate receptor on the bacterial surface, a signal is transmitted through the tail to the head-to-tail connection, and viral DNA is ejected from the capsid to the targeted bacterium (6).
Structural analysis of long-tailed phages revealed that tails of Siphoviridae and Myoviridae phages share common features. In particular, they both exhibit a helical tube organization. The TTPs of Siphoviridae most frequently form stacked rotated hexamers with 6-fold rotational symmetry. The distance between hexamers is ∼40 Å, and the rotation angle between them is ∼20° (6–10). In addition, Siphoviridae tails are often decorated with protein domains encoded within the TTP gene (6, 10–12). Structures of the Siphoviridae phage SPP1 tail before and after DNA ejection showed that structural rearrangements occurred in the tail upon incubation with its cellular receptor (6).
The tail of Bacillus subtilis phage SPP1 includes two TTPs, gp17.1 (19.1 kDa) and gp17.1* (28.3 kDa), that share a common N-terminal sequence (11). A programmed translational frameshift of gene 17.1 leads to the production of gp17.1*, which contains an additional C-terminal domain as compared with gp17.1. In the wild-type phage, approximately one gp17.1* for every 3 eq of gp17.1 is incorporated into the tail structure. However, a virion coding for only gp17.1 is viable and infectious, demonstrating that the additional domain present in gp17.1* is dispensable for virion assembly and function. Here, we observe by electron microscopy (EM) that recombinant gp17.1 alone forms tubes constituted by stacked rings of TTP subunits with a helical organization similar to the one found in tails of SPP1 virions but with variable lengths. NMR and FTIR analyses show that gp17.1 adopts a partially folded structure in its monomeric form and undergoes further structural changes to build a β-sheet-rich helical tube. From EM and bioinformatics results, we propose a first model for the phage noncontractile tail TTP tube.
EXPERIMENTAL PROCEDURES
Cloning Procedures
A PCR fragment spanning from gene 16.1 to the end of gene 17.1 without a stop codon (coordinates 9641–11,011 of the SPP1 genome sequence, accession code X97918.2) was obtained by amplification of SPP1 wild-type DNA using oligonucleotides 5:pIA6For and 6:pIA6Rev 17.1 (Table 1). The sequence of oligonucleotide 6 replaces the rare proline codons CCC at the end of gene 17.1 by more abundant CCG or CCT codons, and it also codes for a noncleavable C-terminal hexahistidine tag. The purified PCR fragment product was cleaved with EcoRI and HindIII and inserted into the pPT25 shuttle vector (11) digested by the same restriction enzymes generating pIA6. This vector was used for the first production trials of gp17.1 with a noncleavable C-terminal hexahistidine tag, further named gp17.1SPP1.
TABLE 1.
Oligonucleotides used in this work
The sequence complementary to the 3′-terminus region of B. subtilis 16S RNA (37) is in lowercase underlined. Translation signals are in bold. Sequences homologous to SPP1 are in uppercase and underlined while mutated bases are only in uppercase. Other sequences are in lowercase. Relevant restriction sites are in italics. Sequences coding for histidine residues are boxed.

To increase the protein production yield, we further used a synthetic gene optimized for expression in Escherichia coli inserted into a pETM-13 vector (GeneScript, Piscataway, NJ). This gene also coded for gp17.1 with a noncleavable C-terminal hexahistidine tag. The only amino acid sequence difference between gp17.1SPP1 and gp17.1 is an extra glycine residue in position 2 in gp17.1. gp17.1Δ (gp17.1 deleted from loop 40–56) was similarly overproduced from a synthetic gene inserted in a pETM-13 vector. Plasmids were transformed into BL21Star (DE3) cells (Stratagene) and selected on kanamycin media at all stages.
To study gp17.1 assembly into the virion within the bacteria, the shuttle vector pIA14 (11, 13, 14), which replicates both in E. coli and in B. subtilis cells, was engineered to produce a gp17.1Δ form of the TTP. PIA14 produces only the short form of the SPP1 TTP, gp17.1, because the translational frameshift leading to the long TTP form, gp17.1*, has been disrupted by mutagenesis (11). Deletion of 51 bp of the17.1 gene coding the TTP residues 40–56 was done by the PCR-driven overlap extension method (15) with primers 1–4 generating pIA65 (Table 1).
Protein Labeling, Production, and Purification
E. coli BL21(DE 3) freshly transformed with plasmid pIA6 was grown at 37 °C in Luria-Bertani (LB) broth supplemented with erythromycin (30 μg ml−1) and chloramphenicol (10 μg ml−1) until reaching an absorbance at 600 nm of 1. The culture was then diluted 10-fold into a minimal medium containing (15NH4)2SO4 as the sole nitrogen source supplemented with the same antibiotics. Recombinant hexahistidine-tagged gp17.1 proteins were constitutively produced overnight at 37 °C. Pelleted bacteria were resuspended in lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Triton, 10 mm MgSO4) and disrupted by sonication. Crude extracts were centrifuged at 4 °C twice for 20 min at 27,000 × g, and gp17.1 proteins were purified by metal-chelating chromatography. The supernatant was loaded onto a 5-ml Hi-Trap Ni-NTA column (GE Healthcare) pre-equilibrated with buffer A (20 mm NaH2PO4, pH 7.4, 500 mm NaCl). The column was washed with increasing concentrations of buffer B (buffer A with 1 m imidazole) and the protein eluted at 250 mm imidazole. The eluate was dialyzed against a 50 mm sodium phosphate, pH 6.0, 150 mm NaCl buffer and concentrated by ultrafiltration (Vivaspin 20, 10-kDa molecular mass cutoff). At this step, the protein, further named gp17.1SPP1, was more than 95% pure as assessed by Coomassie Blue staining of SDS-PAGE. Protein concentration was determined by measurement of the UV absorbance at 280 nm, with an extinction coefficient of 18,910 m−1 cm−1. The purified protein was then characterized using solution-state NMR by recording 1H-15N HSQC and 1H-15N NOE experiments at 25 °C and 700 MHz (Fig. 1, A and B) as described below.
FIGURE 1.

Structural analysis of gp17.1SPP1 in 50 mm sodium phosphate, pH 6.0, and 150 mm NaCl. A and B, solution-state NMR study of gp17.1 SPP1 at 700 MHz and 25 °C. A, superimposition of the 1H-15N HSQC spectrum (blue) and the saturated 1H → 15N NOE spectrum (red and green). Only 15 NH belong to completely unstructured residues (green peaks in the saturated experiment). B, zoom views of 1H-15N HSQC spectra recorded 1 h after dissolution of the sample in 10% (black) or 100% (red) D2O. Only 12 NH are still protonated in 100% D2O. C, fluorescence analysis of the thermal denaturation of gp17.1SPP1 in the presence of SYPRO Orange. Evolution of the SYPRO Orange fluorescence signal is followed as a function of the temperature. The opposite of the first derivative of the fluorescence signal is plotted to clearly read the denaturation temperature as the curve minimum that is highlighted by the dashed green line.
gp17.1SPP1 was also analyzed using fluorescent-based thermal shift assay with a SYPRO Orange dye as a fluorescent probe (Invitrogen) (Fig. 1C). Reactions were carried out in duplicate in a 96-well fast PCR plate with 5 μg of protein in a final volume of 20 μl. The temperature gradient was performed in the range of 25-95 °C at a 3 °C min−1 rate on an Applied Biosystems 7900HT Fast real time PCR system. Fluorescence was recorded as a function of temperature in real time (excitation wavelength, 488 nm; emission spectrum, 500-660 nm), and the melting temperature was calculated with the ABI SDS2.4 software as the maximum of the derivative of the resulting denaturation curves.
E. coli BL21(DE3) strains transformed with the pETM-13 vectors coding for gp17.1 and gp17.1Δ were grown at 37 °C to an absorbance of 1.5, induced with 0.25 mm isopropyl 1-thio-β-d-galactopyranoside, and incubated at the same temperature overnight. Uniformly (>98% for 15N and 13C and >70% for 2H) 15N-labeled, 15N/13C-labeled, and 15N/13C/2H-labeled proteins were produced in minimal media containing 0.5 g/liter 15NH4Cl and 2 g/liter [12C6]glucose or [13C6]glucose as the sole nitrogen and carbon sources. For triple labeling, cells were first grown in LB at 37 °C overnight, then diluted 25-fold in a 99.9% 2H2O-based M9 minimal medium, and incubated at 37 °C overnight, and finally diluted 50-fold in the same deuterated minimal medium, induced with 0.25 mm isopropyl 1-thio-β-d-galactopyranoside and grown overnight at 37 °C. Cells were harvested by centrifugation, resuspended in 20 mm phosphate buffer, pH 7.5, 150 mm NaCl and 8 m urea to solubilize inclusion bodies, and lysed by sonication. After addition of benzonase and 10 mm MgCl2, cells were centrifuged at 20,000 rpm for 1 h on a rotor JLA25.50 (∼48,000 × g). Hexahistidine-tagged proteins were purified on about 7 ml of Ni-NTA resin (GE Healthcare) in denaturing conditions with 8 m urea, 20 mm phosphate buffer, pH 7.5 (buffer A). The resin was washed with buffer A containing 500 mm NaCl and 50 mm imidazole. Proteins were then refolded using a urea gradient from 8 to 0 m and eluted in buffer A containing 500 mm NaCl and 500 mm imidazole. gp17.1 exhibited a denaturation temperature similar to that of gp17.1SPP1 (55 °C against 55.5 °C) in the fluorescent-based thermal shift assay (data not shown).
For liquid-state NMR spectroscopy, the gp17.1 and gp17.1Δ proteins were dialyzed against 20 mm sodium phosphate, pH 7.4, NaCl from concentrations 100 to 500 mm depending on the sample, 1 mm EDTA, and 1 mm PMSF overnight. They were concentrated using ultracentrifugation in Vivaspin 5000 (Vivascience). The final samples consisted of the 15N-labeled, 15N/13C-labeled, or 15N/13C/2H-labeled proteins at a concentration of 8–10 mg/ml (0.4–0.5 mm) in 20 mm sodium phosphate, pH 7.4, NaCl from 150 to 500 mm depending on the sample, 1 mm EDTA, and 90% H2O, 10% D2O. For solid-state NMR (ssNMR) spectroscopy, the 15N,13C-labeled gp17.1 protein was dialyzed against 20 mm sodium phosphate, pH 7.4, 500 mm NaCl, 1 mm EDTA, and 1 mm PMSF overnight. Tube formation was obtained through incubation in this buffer at 37 °C for 1 week. Before NMR experiments, the tubes were dialyzed against 20 mm sodium phosphate, pH 7.4, 100 mm NaCl, 1 mm EDTA, and 10% D2O was added to the sample.
To follow gp17.1 assembly into tubes, gp17.1 and mixtures of gp17.1 with gp17.1Δ were loaded in denaturing conditions (8 m urea, 20 mm phosphate buffer, pH 7.5) on a Ni-NTA column, eluted by adding 500 mm imidazole to the buffer, and refolded by overnight dialysis against 20 mm sodium phosphate, pH 7.4, 50 mm NaCl, 1 mm EDTA. Proteins were concentrated by ultracentrifugation in Vivaspin 5000 (Vivascience) to concentrations of 0.5 to 8 mg/ml (0.025–0.4 mm). They were then run through size exclusion chromatography (SEC) in a Superdex 200 16/60 HR column (GE Healthcare) equilibrated in 20 mm phosphate buffer, pH 7.5, and 50 mm NaCl. Fractions corresponding to monomeric proteins were pooled and used for FTIR and EM assays.
Size Exclusion Chromatography
Analytical SEC and multiangle static light scattering (SEC-MALS) experiments were performed with a 2 mg/ml (0.1 mm) gp17.1 sample injected on a Shodex KW-804 column initially calibrated with BSA. The column was pre-equilibrated with 20 mm sodium phosphate buffer, pH 7.5, and 50 mm NaCl.
SEC experiments were also performed with gp17.1, gp17.1Δ, and gp17.1 mixed with gp17.1Δ. Mixtures corresponded to 1:3, 1:6, and 1:12 mass ratios of gp17.1Δ/gp17.1 monomers. All mixed samples were prepared from a defined amount of purified monomeric gp17.1Δ added to the supernatant of bacteria producing gp17.1 for which the yield of gp17.1 was well determined. The mixture was purified in denaturing conditions (8 m urea) on a Ni-NTA column as described above, dialyzed against a refolding buffer (20 mm sodium phosphate, pH 7.5, 50 mm NaCl) overnight, and injected on a Superdex 200 16/60 HR column calibrated before use.
Liquid-state NMR Spectroscopy
The two- and three-dimensional 1H/15N/13C/2H NMR experiments were performed at 25 °C on Bruker DRX600 and DRX700 spectrometers equipped with triple resonance TCI cryoprobes. All data were processed with Topspin 1.3 (Bruker Biospin, Germany) and analyzed using Sparky3 (T. D. Goddard and D. G. Kneller, University of California, San Francisco). First two-dimensional 15N-1H HSQC spectra were recorded on gp17.1SPP1 (Fig. 1, A and B). To obtain higher production yields, we then used an optimized gene for overproduction of gp17.1 in E. coli and purified it in the same buffer used for gp17.1SPP1 studies. At a concentration higher than 6 mg/ml (0.3 mm), the NMR signal corresponding to the monomeric protein disappeared within a week. Screening different buffer conditions showed that increasing salt concentrations slowed down this process. Therefore, the following NMR analyses of gp17.1 were carried out in 20 mm phosphate buffer, pH 7.5, 500 mm NaCl. Comparison of the two-dimensional 1H-15N HSQC spectra recorded on gp17.1SPP1 in the presence of 150 mm NaCl and on gp17.1 in the presence of 500 mm NaCl showed overlapping NMR spectra, confirming that the two proteins adopt the same three-dimensional conformation in this range of salt concentrations. All reported experiments were then carried out using the refolded protein gp17.1 in 500 mm NaCl. Three-dimensional HNCO, HNCA, HNCACO, HNCOCA, HNCACB, CBCA(CO)NH, HBHA(CO)NH, and 15N- and 13C-edited 1H-1H NOESY experiments were recorded on 15N/13C-labeled samples. Three-dimensional HNCO, HNCA, HNCACO, HNCOCA, HNCACB, HNCOCACB, NNHN, and NCONHN experiments were recorded on 15N/13C/2H-labeled samples. These experiments were analyzed to assign the backbone resonances of gp17.1. The assigned chemical shifts were submitted to Predator (16) to identify the gp17.1 secondary structure elements.
ssNMR Spectroscopy
gp17.1 tubes were obtained by incubation at 37 °C of the refolded protein in 20 mm sodium phosphate, pH 7.4, 500 mm NaCl, 1 mm EDTA, and 1 mm PMSF overnight. Tubes were spun down at 20,000 × g by centrifugation, and the pellet was transferred into a 3.2-mm zirconia rotor. All proton-driven spin diffusion (PDSD) experiments were conducted using a 3.2-mm triple resonance (1H,13C, and 15N) probe head at a static magnetic field of 18.8 tesla corresponding to 800 MHz 1H resonance frequency (Bruker Biospin). All experiments were performed at a magic angle spinning rate of 10 kHz at −20 °C with SPINAL 64 (17) 1H decoupling during evolution and detection periods. The liquid state NMR chemical shift-based cross-peaks were generated using the program FANDAS (18) and were analyzed with Sparky3. The average chemical shifts of the amino acid residues in a β-structure were obtained from the work of Wang and Jardetsky (19). Tentative assignments were obtained using a combination of two-dimensional correlation experiments that included two-dimensional 13C,13C PDSD correlations obtained using under weak coupling conditions (20) with spin diffusion times of 30 and 150 ms to encode intra-residue and sequential effects, respectively. The 15N,13C experiments were recorded using a SPECIFIC-CP (21) time of 3 ms. Two-dimensional 15N,13Cα-Cx and two-dimensional 15N,13C′-Cx were recorded with 13C-13C mixing times of 50 ms, respectively.
Fourier Transform Infrared (FTIR) Spectroscopy
Protein solution spectra were measured at 4 cm−1 resolution with a Bruker IFS 66 spectrophotometer equipped with a 45° N ZnSe attenuated total reflection attachment. After elution from the gel filtration column in 20 mm phosphate buffer, pH 7.5, and 50 mm NaCl, gp17.1 was lyophilized. For the FTIR experiments, gp17.1 samples were suspended in D2O and concentrated to about 3 mg/ml (0.15 mm). The concentrator flow-through was used as the reference buffer sample for measuring buffer signal. FTIR spectra of the protein sample and its corresponding buffer solution were initially recorded just after dissolution in D2O. Thereafter, spectra of both protein and buffer samples were recorded every 24 h to follow tube formation from 0 to 16 days. The protein sample was maintained at 37 °C between spectra acquisitions. Spectra of both protein and buffer samples were recorded with an average of 30 scans. The buffer spectra were subsequently subtracted from the protein spectra. Each spectrum was normalized to the area between 1530 and 1750 cm−1.
Electron Microscopy (EM)
To follow gp17.1 tube assembly, EM images of gp17.1 were obtained by transmission electron microscopy of negative-staining samples. About 5 μl of the sample solution was applied on a carbon-coated Formvar copper grid (Agar Scientific). After 2 min, the sample droplet was blotted with filter paper, and 5 μl of 0.5% phosphotungstic acid stain was applied onto the grid. After 2 min, the excess of staining solution was blotted with filter paper and then the grid was rinsed with deionized water and dried. Samples were imaged in a transmission electron microscope JEOL MET 1400 (120 kV) equipped with a CCD camera SC100 Orius at ×10,000 to 30,000 magnification.
To assess helical parameters of the gp17.1 tubes, images of samples negatively stained with 2% uranyl acetate were taken on a Tecnai F20, and data were recorded using a Gatan Ultrascan 4000 4 × 4 camera at a magnification of ×80,000. Determination of the contrast transfer function was done for each CCD image using CTFFIND3 (22). Segments of tubes were selected manually using BOXER (EMAN (23)). Images were corrected for the CTF effects using Spider on the cut-out segments. Selected particles (segments) were normalized to an average density of zero and the same standard deviation of 1 for all images. All segments were aligned to the vertical rectangle and subjected to multivariate statistical analysis and classification (24). Class averages contained ∼10 images/class, and their diffraction patterns were analyzed. The classes that produced the most symmetrical diffraction patterns were analyzed further. The maxima intensities on a layer line are defined by a distance R from the meridian and radius r of the virus that was described by a Bessel function J of order n: Jn(2πRr) (25). The values of Bessel function orders were determined, and the parameters of the tube helix were defined using the selection rule for the helical symmetry as described in Stewart (25). The distance between gp17.1 rings was determined from the positions of layer lines in the diffraction patterns, and the angle of rotation around the tube axis between the rings was derived from the helical parameters of the tube.
Bioinformatics and Molecular Modeling
Search for the gp17.1 fold was done using the HHpred server (26), which identified four structural homologs. N-terminal gp17.1 residues 1–138 were predicted to adopt the fold of the N-terminal domain of the TTP gpV of phage λ (PDB code 2K4Q) with a probability of 99%. They were also aligned with residues 44–152 of the base plate protein ORF46 from Lactococcus phage TP901-1 (PDB code 4DIV) with a probability of 64%, residues 35–137 of the base plate protein gp19.1 of SPP1 (PDB code 2X8K) with a probability of 53%, and residues 30–136 of the prophage protein XKDM encoded by B. subtilis (PDB code 2GUJ) with a probability of 46%. All these protein fragments share a similar fold, organized around a β-sandwich decorated by a α-helix. To model the whole gp17.1 protein fragment 1–177, we used the I-TASSER server (27). It produced five models of full-length gp17.1, in which fragment 1–138 was structurally similar to the phage λ gpV structure, as predicted by HHpred. The convergence between the five models is high because the average root mean square deviation on their secondary structural elements of the β-sandwich is lower than 2.0 ± 0.2 Å. All these models present a large unfolded loop between residues 40 and 56. I-TASSER calculated a TM score of 0.42 for the first model, indicating a high probability to predict a correct gp17.1 topology from this model.
A three-dimensional model for hexameric gp17.1 was computed from the HHpred and I-TASSER results. We used a multitemplate MODELLER procedure (28) in which we combined gpV (PDB code 2K4Q), gp19.1 (PDB code 2X8K), and I-TASSER structures to build the β-sandwich, the large β-sheet formed by loop β2β3 (residues 40–56) and the C-terminal region of gp17.1, respectively. The SPP1 tail cap gp19.1 structure was solved in a hexameric form, and its loop (gp19.1 residues 37–53) corresponding to gp17.1 loop β2β3 is an essential element of the intermolecular interfaces (PDB code 2X8K (29)). We modeled the gp17.1 loop structure from the corresponding loop structure observed in gp19.1. Then the gp17.1 hexamer was assembled by superimposing the monomeric gp17.1 model onto each of the gp19.1 subunits. Loop β2β3 forms a β-sheet located at the intermolecular interfaces in our hexameric gp17.1 model. Finally, from this model and the helical parameters derived from EM analysis of the tubes, we calculated a model of three stacked hexamers of gp17.1 that is representative of the tube structure.
Tail Assembly in B. subtilis
Phage SPP1sus45 (11, 30) was amplified by infection of the B. subtilis HA101B permissive strain as described previously (31). Phage titers and reversion rates in the lysates were determined by titration in strains HA101B and YB886, respectively. Culture infection and phage purification through a discontinuous CsCl density gradient with preformed layers of 1.7, 1.5, and 1.45 g cm−3 CsCl in TBT buffer (100 mm NaCl, 10 mm MgCl2, 100 mm Tris-Cl, pH 7.5) were carried out as described previously (11, 32).
Phage SPP1sus45 was titrated in B. subtilis YB886 bearing pIA14 or pIA65 plasmids coding for full-length gp17.1 or for gp17.1Δ, respectively. The titers were expressed as a percentage of the titer obtained by infection of the permissive B. subtilis strain HA101B to determine the level of complementation (33). Extracts of B. subtilis were prepared and analyzed by Western blot as described previously (34).
RESULTS
Monomeric SPP1 TTP Is Only Partially Folded in Solution
In long-tailed bacteriophages, the tail tube is formed by a large number of TTP subunits. NMR analyses demonstrated that in phage λ, the TTP gpV N-terminal domain adopts a β-sandwich fold in its monomeric state (35). Several studies suggested that this fold is conserved in most TTPs of long-tailed phages (35, 36) and might be a recurrent fold found in other components that extend the inner tube channel at the long tail extremities (5, 36). Analysis of the sequence of the SPP1 TTP protein using HHpred also suggested that fragment 1–138 of gp17.1 adopts the fold of phage λ gpV N-terminal domain, even if these protein regions only share 14% of sequence identity.
To characterize the three-dimensional structure of gp17.1, 15N-labeled gp17.1SPP1 was produced and purified by affinity chromatography. The protein is mainly monomeric at 4 °C and 100 μm as assessed by SEC (data not shown). Its NMR 1H-15N HSQC spectrum displays well dispersed cross-peaks, revealing that the protein is at least partially folded (Fig. 1A). NMR 1H → 15N NOE measurements indicated that only 15 residues are completely unstructured in gp17.1SPP1 (Fig. 1A). Thermal stability monitored by a fluorescent-based thermal shift assay showed that gp17.1SPP1 unfolds cooperatively with a single transition (Tm of 55.5 °C), demonstrating that it exhibits a folded hydrophobic core (Fig. 1C). However, the large heterogeneity in the 1H-15N HSQC peak line widths observed at 25 °C (Fig. 1, A and B) marks the presence of conformational exchange. This line width heterogeneity is similarly found on a large temperature range, from 10 to 40 °C (data not shown). Moreover, following H-D exchange by NMR at 25 °C showed that only few amide protons are not exchanged with deuterium after 1 h at 30 °C (Fig. 1B). This demonstrates that most amide protons form marginally stable hydrogen bonds within gp17.1SPP1, which confirms that the protein structure is dynamic. Similar observations were made for gp17.1, which possesses only one amino acid difference relative to gp17.1SPP1, but it was produced and purified using a different protocol (simultaneous elution and refolding from an affinity chromatography column), yielding larger protein yields.
Triple 15N, 13C, and 2H labeling was necessary to achieve 63% of the gp17.1 backbone frequency assignment (supplemental Table 1). About 110 peaks remained unassigned on the 1H-15N HSQC spectrum, corresponding to 67 unassigned non-proline residues and about 40 residues in minor conformations. Further assignment was prevented by the lack of signal corresponding to these peaks on the three-dimensional 1H, 15N, and 13C NMR experiments. Analysis of the assigned gp17.1 NMR chemical shifts using Predator showed that very few secondary structure elements could be unambiguously identified on the basis of NMR data; only eight assigned residues showed backbone chemical shifts characteristic of β-strands, and no assigned residues corresponded to α-helices. Further NMR chemical shift assignment of residues belonging to predicted secondary structure elements (Fig. 2A) was not possible, most probably because of the presence of conformational exchange processes. This hindered determination of the three-dimensional solution structure of gp17.1.
FIGURE 2.

Molecular modeling of the gp17.1 monomer. A, sequence alignment between residues 1 and 138 of SPP1 gp17.1 (177 amino acids) and residues 6 and 152 of the N-terminal domain of phage λ gpV (156 amino acids), as proposed by HHpred. gp17.1_SS and gpV_SS correspond to secondary structure predictions using PsiPred and GpV_NMR to secondary structure elements determined using DSSP from the NMR structure of gpV (PDB code 2K4Q). B and C, gp17.1 model calculated by I-TASSER, based on the structural analogy between gp17.1 and gpV. B, ribbon is colored based on the gp17.1 NMR chemical shift analysis as follows: unassigned residues in gray; assigned residues in secondary structure elements in red (as deduced from chemical shift analysis using Predator); and assigned residues not involved in secondary structures in yellow and green (for loop 40–56). The eight residues in red (Thr-66, Tyr-67, Tyr-68, Asp-120, Gly-121, Val-126, Glu-127, and Ile-128) are labeled. C, ribbon is colored based on the gp17.1 1H → 15N NOE analysis as follows: unassigned residues are rendered in dark gray; assigned residues with reduced local mobility in cyan (NOE >0.6); assigned residues in flexible regions in orange (0 < NOE < 0.6); and assigned residues in unstructured regions in red (NOE <0).
A model of the gp17.1 fold was calculated using I-TASSER. This model is consistent with the gp17.1 secondary structure predictions (Fig. 2A). It is based on the sequence alignment between the fragment 1–138 of gp17.1 and the N-terminal region of gpV (Fig. 2A). It also exhibits an additional α-helix in the region 139–177 of gp17.1 (Fig. 2, B and C). To evaluate whether the NMR analysis of gp17.1 supports the gp17.1 model proposed by HHpred and I-TASSER, we projected the secondary structure elements identified using Predator onto this atomic model. Fig. 2B shows that the NMR observable β-strands correspond to β3, β7, and β6, all belonging to the same β-sheet. In contrast, no residues belonging to β-strands β1, β4, and β5 could be assigned. Analysis of the 1H-15N HSQC spectrum of gp17.1 recorded after 1 h of incubation in D2O consistently revealed that three amide protons, corresponding to Tyr-68(β3), Ile-128, and Ser-129(β7), are involved in stable hydrogen bonds, together with nine other amide protons belonging to unassigned residues (Fig. 1B). Finally, we projected the 59 1H → 15N NOE values that could be assigned to a gp17.1 residue (supplemental Table 1) onto the gp17.1 model (Fig. 2C). Most values higher than 0.6, corresponding to well structured residues, are found in the β-sheet formed by β3, β7, and β6 (Fig. 2C, cyan ribbon). On the opposite, most of the negative values, corresponding to unstructured residues, are located in the C-terminal fragment 166–173, which adopts no secondary structure in the gp17.1 model (Fig. 2C, red ribbon). Altogether, the NMR and I-TASSER results suggest that the β-sheet formed by β3, β7, and β6 is well structured, and the β-sheet formed by β1, β4, and β5 exhibits large conformation exchange processes. The NMR signals corresponding to the two predicted α-helices are partially assigned, but their chemical shifts indicate that they are not structured as α-helices within monomeric gp17.1.
Purified gp17.1 Forms Tubes Rich in β-Structure
NMR analysis of gp17.1 at a concentration of 8 mg/ml (0.4 mm) and a temperature of 25 °C revealed that the NMR signal of this protein reproducibly disappears with time. To follow its association state as a function of time, we purified unfolded gp17.1 in urea by affinity chromatography and then dialyzed the protein in a refolding buffer and injected the protein at 2 mg/ml (0.1 mm) on a SEC-MALS system (Fig. 3A). In these conditions, gp17.1 is eluted in two peaks. Measurement of the differential refractive index and the light scattering intensity corresponding to each peak provided an estimate of their mass. The second peak corresponds to a mass of 28.8 ± 2.4 kDa, slightly higher than the mass of a monomer (20.2 kDa), whereas the first peak corresponds to a heterogeneous population of multimers with a molecular mass superior to 1000 kDa. These multimers (50 μm) incubated for 3 days at 37 °C form long tubes of different lengths as observed by EM (Fig. 3B).
FIGURE 3.
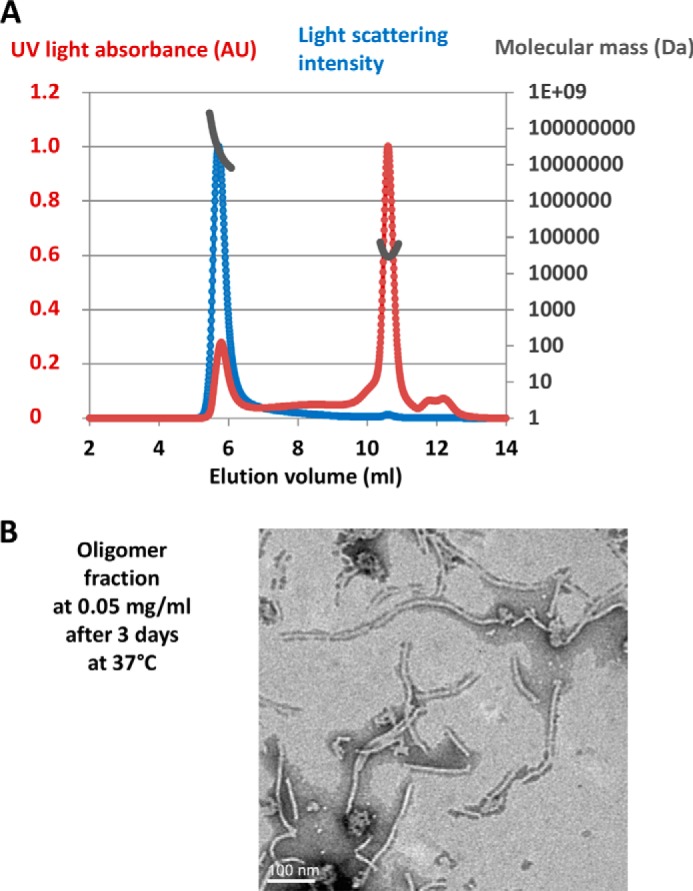
Association of gp17.1 with time. A, SEC-MALS experiment performed on a 2 mg/ml gp17.1 sample injected on a Shodex KW-804 column pre-equilibrated with 20 mm sodium phosphate buffer, pH 7.5, and 50 mm NaCl. The UV light absorbance (UV), differential refractive index, and light scattering intensity were measured as a function of the elution volume. Molecular masses calculated by the MALS-UV-refractive index method (gray) are plotted against elution volume with UV light absorbance (red) and light scattering intensity (blue) traces overlaid. The second peak corresponds to a mass of 28.8 ± 2.4 kDa, slightly higher than the mass of a monomer (20.2 kDa), whereas the first peak corresponds to largely heterogeneous oligomers with a molecular mass superior to 1000 kDa. B, micrograph of material from the oligomer peak at 0.05 mg/ml, 3 days after elution from the chromatography column. The sample was negatively stained with 0.5% phosphotungstic acid.
To follow the formation of tubes from gp17.1 monomers, we recorded FTIR spectra and EM images of a refolded and SEC-purified sample of monomeric gp17.1 incubated at 37 °C (Fig. 4). We observed tubes 48 h after collection from the SEC column at 3 mg/ml (0.15 mm; Fig. 4A). The tubes increased in length over time. FTIR spectra showed a shift of a band near 1645 cm−1 assigned to random coil toward a band at 1620 cm−1 corresponding to β-sheet in the amide I band spectrum over the reaction time course (Fig. 4B). Changes in gp17.1 secondary structure were highlighted by calculating the difference absorbance spectra corresponding to the amide I band (bottom panel in Fig. 4B). They were obtained by subtracting the initial protein spectrum from spectra obtained at different incubation times at 37 °C. The increase in absorbance around 1620 cm−1 with time is indicative of β-sheet formation in gp17.1. This increase of β-sheet content correlates with formation of gp17.1 tubes as observed by EM.
FIGURE 4.

Time-dependent gp17.1 tube formation followed using TEM (A) and FTIR (B) on a protein sample at 3 mg/ml in 20 mm phosphate buffer, pH 7.5, and 50 mm NaCl. A, TEM images were obtained after incubation of gp17.1 monomers at 37 °C during 24, 48, 72, and 240 h. The samples were negatively stained with 0.5% phosphotungstic acid. In parallel, FTIR measurements were performed for the same samples (B). FTIR buffer-subtracted spectra of the 1530–1730 cm−1 region are displayed in the upper part of B as a function of the gp17.1 incubation time at 37 °C as follows: black bold lines at time 0; black dotted lines and after 48 h (· · ·) 72 h (-●-), 96 h (●●●●), 168 h (— —), 192 h (-●●-); gray lines after 216 h (solid) and 348 h (dotted). Difference absorbance spectra calculated by subtracting the buffer-subtracted spectrum corresponding to time 0 to the buffer-subtracted spectra recorded at different times are shown in the lower part of B.
gp17.1 Tubes Structure Studied by ssNMR
Self-assembly of gp17.1 into long tubes prompted their structural analysis by ssNMR. Tubes were obtained from uniformly 13C,15N-labeled gp17.1 proteins incubated at 37 °C for 1 week and pelleted by centrifugation. 13C-13C PDSD experiments were recorded on the resulting sample at 253 K. The favorable 13C line widths observed at 800 MHz in 13C-13C PDSD experiments below 1 ppm suggested that the gp17.1 polypeptide chain is well folded in the tubes (Fig. 5A). A 13C-13C PDSD spectrum was calculated in silico on the basis of the solution NMR chemical shifts; this enabled comparison between the solid-state (black peaks) and solution-state (green symbols) NMR fingerprints of gp17.1 (Fig. 5A). In several regions of the 13C-13C PDSD spectrum, including regions 3 and 4, the experimental and calculated peaks poorly superimpose. If, as strongly predicted, gp17.1 adopts a β-sandwich-like fold (Fig. 5A, blue symbols), we observe a significant improvement in the correlation between the experimental and calculated NMR cross-peaks (Fig. 5A). In the soluble monomer, most threonine and serine Cα-Cβ correlations exhibit secondary chemical shifts expected for random coil folds. In contrast, the same region in the ssNMR spectrum is dominated by cross-peak correlations typically found in β-strand conformations (Fig. 5, A and B, zoom 4) (19). Thus, additional β-strands rich in serine and threonine residues are observed in tubes. Furthermore, in the region corresponding to proline Cα-Cβ, most solid-state peaks are found at β-structure-specific chemical shifts (Fig. 5, A and B, zoom 3). Finally, in the region corresponding to alanine and leucine Cα-Cβ correlations, several solid-state peaks are found at either α-helix or β-strand-specific chemical shifts (Fig. 5B, zooms 1 and 2). Altogether, we conclude that the gp17.1 protein fold is stabilized in the self-assembled tubes with Ser, Thr, Pro, Ala, and Leu found in β-strand regions and α-helix conformations. This secondary structure amino acid content is consistent with the β-strands and α-helix predicted for gp17.1 (Figs. 2B and 5B), supporting the presence of the corresponding β-sandwich organization in the tubes.
FIGURE 5.

13C-13C ssNMR spectrum of a uniformly 13C,15N-labeled gp17.1 tube sample recorded at 800 MHz and 253 K (peaks are displayed in black). A, green symbols reflect correlations predicted on the basis of solution NMR assignments, and blue symbols correspond to chemical shifts calculated for a 100% β-structure. B, zooms of the spectrum that display 13C-13C correlations corresponding to alanines (1), leucines (2), prolines (3), and threonines and serines (4). Spectral regions corresponding to random coil 13C chemical shifts are colored in gray, and those corresponding to β-strand and α-helix chemical shifts are colored in blue and red, respectively (42). The amino acid sequences of the secondary structure elements identified by PSIPRED in gp17.1 are displayed next to the spectra. In these sequences, alanines, leucines, prolines, threonines, and serines are colored in blue (β-strands) and red (α-helices). Predicted β-strands belonging to intermolecular β-sheets in the pseudo-atomic model of gp17.1 tubes (Figs. 7 and 8) are marked as “im” (for intermolecular).
gp17.1 Assembles into Helical Tubes of Stacked 6-Fold Symmetric Rings
Comparison of gp17.1 tubes with tails from SPP1 virions indicated that they have similar diameters of 11 nm and share a common morphology (Fig. 6, A and B). Stain penetration revealed by a dark line inside the tube shows that the tubes are hollow as observed for empty tail tubes of virions that ejected their DNA following incubation with the SPP1 bacterial receptor (Fig. 6A) (38). Class averages (∼10 images/class) of aligned segments of gp17.1 tubes were obtained, and their diffraction patterns were calculated (Fig. 6C). Analysis of these diffraction patterns showed that tubes have 6-fold symmetry and therefore are formed by stacked hexameric rings. The rings are 4.0 ± 0.2 nm in height and are rotated 21.4 ± 0.2° to form the helical tube (Fig. 6D). SPP1 virion tails are composed of ∼40 stacked rings of ∼40 Å in height, and each tail ring is rotated by ∼21° relative to the previous one. Thus, the recombinant gp17.1 tubes assembled in absence of any other phage SPP1 protein show the same organization found for gp17.1 in the virion tail.
FIGURE 6.

TEM analysis of self-assembled gp17.1 tubes. A, gp17.1 tubes mixed with SPP1 virions challenged with the receptor YueB780 to visualize the empty tail tube (38). B, images of self-assembled gp17.1 tubes. Tilting of the tube ends toward the viewer, probably due to gp17.1 hydrophobic regions sticking to the carbon film, provides very clear images of the tail ring. A and B, samples were negatively stained with 2% uranyl acetate. C, four classes of 10 images (top) from side views of three tube rings and their corresponding diffraction patterns. D, order of the Bessel functions (reflections) and parameters of the helical packing (see “Experimental Procedures”). The lattice corresponding to the helix is superimposed on the diffraction pattern. Layer lines are indicated.
Modeling the gp17.1 Tube Structure Suggests a Critical Role for Loop β2β3 in Tube Assembly
Although the gp17.1 monomer does not adopt a stable three-dimensional structure in solution, characterization of self-assembled gp17.1 by ssNMR showed that the gp17.1 protein acquires a stable fold in the tube (Fig. 5). The secondary structure content and ssNMR signature (Figs. 4B and 5) of gp17.1 in the tubes are consistent with a tertiary structure similar to the β-sandwich structure of phage λ TTP (Fig. 2, B and C). The same β-sandwich fold is found in the N-terminal part of SPP1 gp19.1, a phage protein of the Dit family (2, 3). Dit proteins form a hexamer at the tail tube end that extends the tail channel to the tail adsorption apparatus (29). The x-ray structure of the gp19.1 hexamer (PDB code 2X8K) was used as a template to model the gp17.1 hexamer (Fig. 7). gp17.1 hexamers were then positioned using the helical parameters determined by EM to simulate a gp17.1 tube (Fig. 8A). In that model, the interface between two subunits consists of β-sheets connecting adjacent β-sandwiches (Fig. 8B). The large loop β2β3, which is unstructured in the monomer (Fig. 2), is located at the interface between subunits and hexamers (Fig. 8B). Its structure was modeled from the corresponding segment in gp19.1 that forms a β-sheet at the gp19.1 hexamer inter-subunit interface. Loop β2β3 from one gp17.1 subunit forms a two-stranded β-sheet that interacts with a β-strand of the β-sandwich of the neighboring gp17.1 subunit and may thus stabilize the three-dimensional structure of this subunit. This model suggests that loop β2β3 is essential for tube assembly by contributing to the formation of a large interface between adjacent subunits of the tail hexamers (Figs. 7B and 8B). Moreover, in this model, loop β2β3 participates in the contact surface between stacked hexamers (Fig. 8B).
FIGURE 7.

Modeling of the gp17.1 hexamer. A, sequence alignment of SPP1 gp17.1 with residues 12–152 of phage λ gpV N-terminal domain and residues 26–138 of SPP1 gp19.1 N-terminal domain. Sequences corresponding to loop β2β3 of gp17.1 and homologous loops in gpV and gp19.1 are boxed in red. B, top views of the gp17.1 hexamer model, either superimposed in green to the gp19.1 x-ray structure rendered in blue (left, loop β2β3 of gp17.1 is shown in red and the corresponding large loop of gp19.1 is in orange) or displayed using a specific color for each subunit (right).
FIGURE 8.
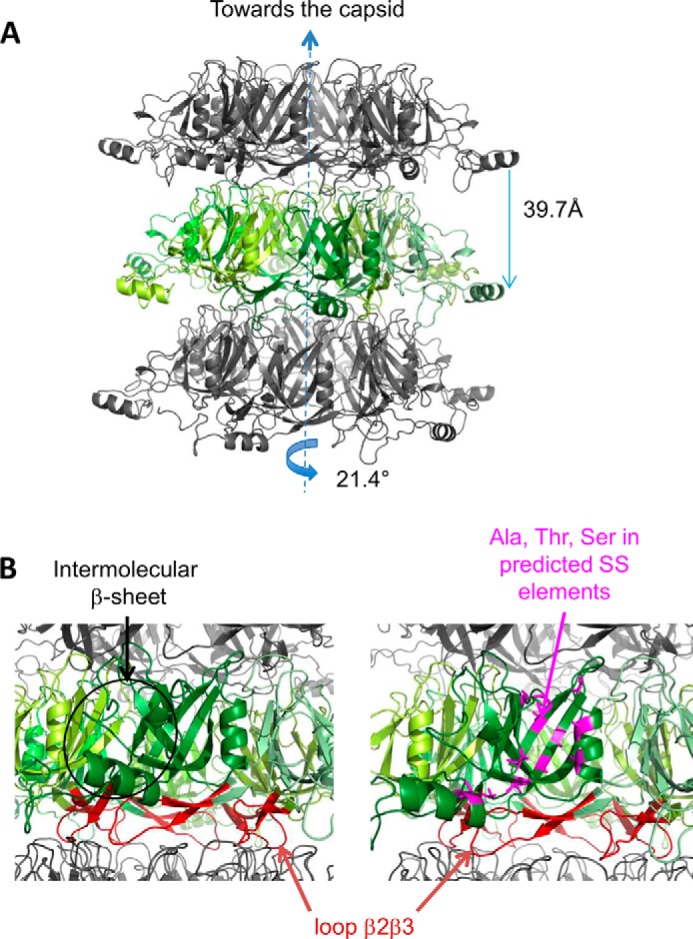
Modeling of the atomic structure of self-assembled gp17.1 tubes. A, view of three stacked gp17.1 hexamers. The central hexamer is colored in green, with each monomer being characterized by a specific tone of green. B, zooms of A with loops β2β3 colored in red. On the right view, alanine, threonine, and serine residues belonging to predicted secondary structure elements (Fig. 5A) are represented by magenta sticks.
Loop β2β3 Is Necessary for gp17.1 Tube Self-assembly
A gp17.1 protein deleted for the segment 40–56 (gp17.1Δ) was stably produced. Comparison of the NMR 1H-15N HSQC spectra of gp17.1SPP1 and gp17.1Δ monomers revealed that they have a similar fold (Fig. 9). Attempts to assign the gp17.1Δ 1H-15N HSQC spectrum faced the same difficulties encountered when assigning the spectrum of gp17.1. The gp17.1Δ HSQC signals that could be assigned corresponded to residues also assigned in gp17.1, and about 40% of the residues could not be assigned in gp17.1Δ. The heterogeneity of the gp17.1Δ 1H-15N HSQC line widths showed that this protein is also subjected to conformational exchange processes. 1H-15N HSQC peaks corresponding to minor conformations are still observed in gp17Δ. However, SEC revealed that gp17.1Δ does not form oligomers upon incubation for 3 days at 37 °C in contrast to gp17.1 (Fig. 10A). Similarly EM did not show detectable formation of tubes when gp17.1Δ at 3 mg/ml (0.15 mm) was incubated for 3 days at 37 °C (data not shown).
FIGURE 9.
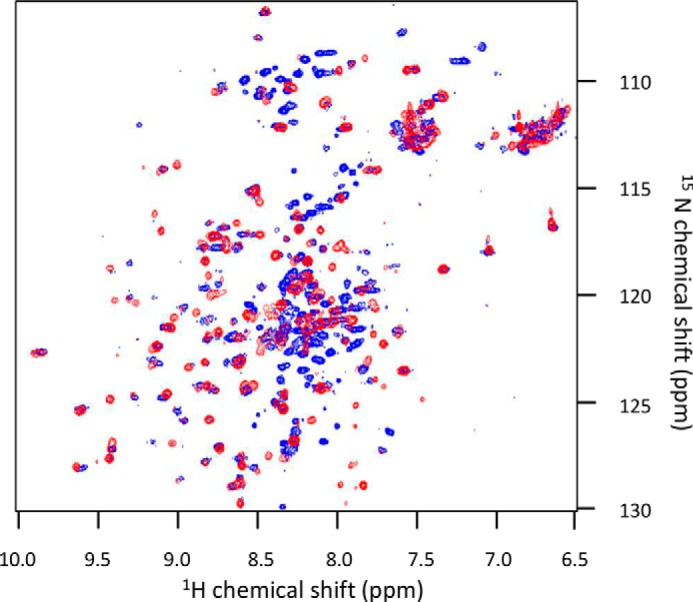
Superimposition of the 1H-15N HSQC spectra of gp17.1SPP1 and gp17.1Δ acquired in low salt conditions (100–150 mm NaCl) at 25 °C. The blue spectrum was acquired on gp17.1SPP1 and is the same as in Fig. 1A. The red spectrum was obtained on gp17.1Δ in 20 mm sodium phosphate, pH 7.0, 100 mm NaCl.
FIGURE 10.

SEC behavior of gp17.1Δ and its interference with gp17.1 oligomerization. A, SEC of gp17.1 (black) and gp17.1Δ (gray) after 3 days at 37 °C (thick lines) or 1 week at 4 °C (thin lines). All protein samples were injected at ∼2 mg/ml on a Superdex 200 16/60 HR column equilibrated in 20 mm sodium phosphate, pH 7.5, 50 mm NaCl. Calibration of the column showed that P1 and P2 would correspond to molecular masses higher than 500 kDa and equal to 21 kDa in the case of globular proteins, respectively. B, SEC of gp17.1 (—), gp17.1 with 33% of gp17.1Δ (—··), 17% of gp17.1Δ (—·), and 9% of gp17.1Δ (--) after 3 weeks at 4 °C. Concentrations of all the injected samples were between 1.5 and 2 mg/ml. Percentages corresponded to mass ratios. C, SDS-PAGE of fractions eluted in B. One fraction of peak P1 (top gel) and several fractions covering peak P2 (middle and bottom gels) of the different samples run in B were analyzed. A gp17.1Δ sample was added as a reference in the top gel. Only gp17.1 was detected in peak P1, and both gp17.1 and gp17.1Δ were eluted in peak P2.
After 3 weeks at 4 °C, gp17.1 still exists in the monomeric (peak P2) and oligomeric (peak P1) forms as revealed by SEC (Fig. 10B). Its mixing with gp17.1Δ reduces the amount of gp17.1 oligomers. Analysis on SDS-PAGE of each gel filtration peak confirms that the peaks containing the oligomers are only composed of gp17.1, whereas both gp17.1 and gp17.1Δ are found in the monomer-containing peaks (Fig. 10C). Altogether, these chromatography results suggest that gp17.1Δ is capable of interacting with gp17.1 and reduces gp17.1 propensity to assemble into oligomers.
gp17.1Δ Is Incompetent for Building SPP1 Phage Tails and Interferes with Wild-type gp17.1 Tail Assembly
The biological activity of gp17.1 deleted from residues 40 to 56 (gp17.1Δ) was assessed by complementation assays. The TTP (gp17.1/gp17.1*)-deficient mutant SPP1sus45 did not multiply (<3%) in the nonpermissive B. subtilis strain YB886 (pIA65) that codes for gp17.1Δ, whereas strain YB886 bearing plasmid pIA14 that codes for wild-type gp17.1 led to full complementation (>95%). Thus, gp17.1Δ did not assemble biologically active tails.
We then tested whether gp17.1Δ could interfere with assembly of wild-type TTPs to build the tail structure during wild-type infection. The mutant protein was produced as a soluble protein in B. subtilis YB886 (pIA65) in an amount comparable with the amount of wild-type TTP synthesized during SPP1 infection (Fig. 11A, lane 6). The efficiency of plating of SPP1 wild-type in cells producing gp17.1Δ was only slightly reduced (∼80%, Fig. 11B), but the phage plaque size was considerably smaller than in normal SPP1 infections. The presence of gp17.1Δ thus had a negative impact on phage multiplication in the cell (Fig. 11B). Analysis of the viral particles produced in the presence and absence of the gp17.1Δ separated through a discontinuous CsCl density gradient showed that a large amount of DNA-filled capsids accumulate when gp17.1Δ is present during infection in contrast to normal wild-type infections in which complete phage particles predominate (Fig. 11C). This observation strongly suggests that the amount of tail particles competent for virus morphogenesis becomes limiting when assembly occurs in the presence of gp17.1Δ.
FIGURE 11.

Impact of gp17.1Δ on SPP1 tail assembly. A, TTP production before (lanes 1, 3, and 5) or after (lanes 2, 4, and 6) infection with SPP1 wild type of YB886 strains bearing the plasmids indicated within parentheses. The gp17.1, gp17.1*, and gp17.1Δ proteins were detected in cell lysates using a polyclonal antiserum raised against the N-terminal region of gp17.1 (12). The different TTPs are identified by black arrows on the right and the position of migration of molecular mass markers is shown on the left. B, plaque assays. Morphology of SPP1 wild-type phage plaques in strains YB886, YB886 (pIA14), and YB886 (pIA65) at 30 °C. The efficiency of plating (EOP) is indicated below each titration tested, and standard deviations were calculated from five independent experiments. C, isopycnic centrifugation of phages produced during SPP1 wild-type infection of YB886 bearing plasmid pIA65 or without plasmid (control). The picture shows centrifuge tubes after centrifugation of the virions through a discontinuous density gradient with preformed layers of 1.7, 1.5, and 1.45 g cm−3 CsCl in TBT buffer. The upper band corresponds to entire phage particles and the lower band to tailless capsids filled with DNA (11).
DISCUSSION
Here, we show that the TTP of phage SPP1, gp17.1, self-assembles into tubes. The organization of gp17.1 in the tubes is remarkably similar to the one found in virus tails. They are both formed by stacked hexamers, rotated by ∼21°, and shifted along the tail axis by ∼4 nm (Fig. 6) (6). A similar helical tail tube organization has been recently observed in the Siphoviridae Araucaria (10) and in the giant Myoviridae ΦRSL1 (9).
gp17.1 monomers, which do not adopt a stable fold in solution (Fig. 2), undergo a conformational change characterized by stabilization or formation of β-structures to assemble tubes (Figs. 4 and 5). Bioinformatics, FTIR, and ssNMR results suggest that this structure is a β-sandwich similar to the structures of phage λ TTP protein (35) and Dit protein N-terminal domains, including the domain of Dit gp19.1 of phage SPP1 (29). The pseudo-atomic model of the gp17.1 tube built by homology modeling and using the tube helical parameters indicates that, within gp17.1 hexamers, intermolecular interfaces might result from interaction between gp17.1 β-sandwich structures through pairing of accessible β-strands. Recent docking of a TTP model into the cryo-EM density of the ΦRSL1 virion also suggested that large intermolecular β-sheets were responsible for hexamer assembly within the tail tube (9). These interactions should stabilize the β-sandwich fold in the tube structure.
In our experiments, gp17.1 loop β2β3 spanning residues 40–56 is essential for gp17.1 tube formation in vitro (Fig. 10) and assembly of phage tails in vivo (Fig. 11). Positioning of this loop in the model of the helical tube suggests that it participates both in hexamer intersubunit contacts and in inter-hexamer interactions. gp17.1Δ, in which the loop was deleted, interferes with formation of wild-type gp17.1 tubes showing that the two gp17.1 forms interact and gp17.1Δ disrupts the TTP assembly reaction. A disordered loop 50–78 was also observed between strands β2 and β3 of gpV (35), which corresponds to SPP1 loop 40–56, as proposed by secondary and tertiary structure predictions (Fig. 2B). As the gpV loop β2β3 is strongly and negatively charged, its functional role was assessed by mutating Asp-61 and Asp-62 into alanines (35). It was shown that the mutant D61A/D62A is completely devoid of biological activity and that it displays a dominant negative phenotype in that it inhibits the growth of wild-type λ phage. The authors suggested that mutant gpVD61A/D62A incorporates into tail assembly intermediates but that its presence inhibits further tail tube growth. On this basis, it was postulated that loop β2β3 mediates hexamer-hexamer interactions essential for phage λ tail polymerization. The pseudo-atomic model of gp17.1 tube structures consistently suggests a role of SPP1 loop β2β3 in intermolecular interactions (Figs. 7B and 8B). In this model, loop β2β3 stabilizes the gp17.1 fold both by forming a β-sheet with the β-sandwich of the adjacent subunit within the hexameric ring and by contributing to interactions between rings of the gp17.1 helical tube.
The phage TTP fold is also found in bacterial Hcp-like proteins, which form a channel in type VI secretion systems (35). Interestingly, all Hcp-like proteins purified so far are hexamers in solution with the exception of EpvC from Edwardsiella tarda, which was found in both dimeric and hexameric states in solution (39). Furthermore, no in vitro self-association of these hexamers was reported to date. Surface plasmon resonance experiments showed that Hcp hexamers transiently interact with each other but are incapable of forming tubes alone at micromolar concentrations (40). After intermolecular disulfide bond engineering, Hcp1 from Pseudomonas aeruginosa self-assembles into nonhelical nanotubes that are observable by EM (41). These tubes can be readily disassembled by addition of a reducing agent. Thus, our study supports the view that the evolutionarily related phage and T6SS bacterial tubes are built from a common structural element that can be observed in a hexameric state. However, assembly of this element into tubes follows different pathways and might lead to different tubular architectures.
In conclusion, we report here the first structural analysis of a phage TTP self-assembled tube. Bacteriophage SPP1 TTP exists in a monomeric form that is partially folded. This form is probably favored in infected bacteria to prevent the formation of TTP tubes competing with assembly of biologically active tail structures. However, in vitro, increasing the gp17.1 concentration at physiological temperature leads to the formation of TTP tubes. We propose that tube assembly involves formation of β-sheets between adjacent β-sandwich structures and contacts involving loop β2β3. Our results strongly suggest that these intermolecular interactions are present both in self-assembled tubes and in phage tails. However, it remains to be explored how they are modified after bacterial recognition and during DNA ejection. gp17.1 structural plasticity could be important for the observed SPP1 tail reorganization triggered during infection (6). Finally, our study demonstrates that the SPP1 TTP has the inherent property to fold and form tubes in the absence of any other phage protein. This remarkable feature opens the way for generation of TTP-based tubular structures grafted with molecules exhibiting specific functionalities. Such engineered nanotubes could be encoded by a single gene resulting from fusion of the TTP gene with the gene coding for the grafted protein (as observed in TTPs comprising a tube assembly domain and an additional accessory domain (11, 12)), or it could be formed by chemically linking the TTP to the molecule to be grafted.
Acknowledgments
We thank Rudi Lurz (Max-Planck Institute for Molecular Genetics, Berlin, Germany) for the EM images of tubes and virions on the same grid; Eric Jacquet (Imagif, Gif-sur-Yvette, France) for fluorescence-based thermal shift assays; Christophe Velours (Imagif, Gif-sur-Yvette, France) for the SEC-MALS analysis; and Frédéric Gobeaux and Pierre Chervy (CEA Saclay, France) for helping with the use of the EM microscope to follow tube assembly. We also acknowledge the IMAGIF TEM facility for access to their microscopes.
This work was supported in part by French Agence Nationale de la Recherche Program Grant ANR-09-BLAN-0149-01 (to P. T. and S. Z. J.), French National Infrastructure FRISBI Program, and European Community's Seventh Framework Program, BioNMR Project Contract 211800.

This article contains supplemental Table 1.
- TTP
- tail tube protein
- Ni-NTA
- nickel-nitrilotriacetic acid
- ssNMR
- solid-state NMR
- SEC
- size exclusion chromatography
- SEC-MALS
- SEC and multiangle static light scattering
- PDSD
- proton-driven spin diffusion
- PDB
- Protein Data Bank
- HSQC
- heteronuclear single quantum coherence
- TEM
- transmission electron microscopy.
REFERENCES
- 1. Ackermann H. W. (2007) 5500 phages examined in the electron microscope. Arch. Virol. 152, 227–243 [DOI] [PubMed] [Google Scholar]
- 2. Katsura I. (1990) Mechanism of length determination in bacteriophage λ tails. Adv. Biophys. 26, 1–18 [DOI] [PubMed] [Google Scholar]
- 3. Veesler D., Cambillau C. (2011) A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev. 75, 423–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. King J. (1971) Bacteriophage T4 tail assembly: four steps in core formation. J. Mol. Biol. 58, 693–709 [DOI] [PubMed] [Google Scholar]
- 5. Davidson A. R., Cardarelli L., Pell L. G., Radford D. R., Maxwell K. L. (2012) Long noncontractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 726, 115–142 [DOI] [PubMed] [Google Scholar]
- 6. Plisson C., White H. E., Auzat I., Zafarani A., São-José C., Lhuillier S., Tavares P., Orlova E. V. (2007) Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 26, 3720–3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kostyuchenko V. A., Chipman P. R., Leiman P. G., Arisaka F., Mesyanzhinov V. V., Rossmann M. G. (2005) The tail structure of bacteriophage T4 and its mechanism of contraction. Nat. Struct. Mol. Biol. 12, 810–813 [DOI] [PubMed] [Google Scholar]
- 8. Fokine A., Zhang Z., Kanamaru S., Bowman V. D., Aksyuk A. A., Arisaka F., Rao V. B., Rossmann M. G. (2013) The molecular architecture of the bacteriophage T4 neck. J. Mol. Biol. 425, 1731–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Effantin G., Hamasaki R., Kawasaki T., Bacia M., Moriscot C., Weissenhorn W., Yamada T., Schoehn G. (2013) Cryo-electron microscopy three-dimensional structure of the jumbo phage PhiRSL1 infecting the phytopathogen Ralstonia solanacearum. Structure 21, 298–305 [DOI] [PubMed] [Google Scholar]
- 10. Sassi M., Bebeacua C., Drancourt M., Cambillau C. (2013) The first structure of a mycobacteriophage, the Mycobacterium abscessus subsp. bolletii phage Araucaria. J. Virol. 87, 8099–8109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Auzat I., Dröge A., Weise F., Lurz R., Tavares P. (2008) Origin and function of the two major tail proteins of bacteriophage SPP1. Mol. Microbiol. 70, 557–569 [DOI] [PubMed] [Google Scholar]
- 12. Fraser J. S., Maxwell K. L., Davidson A. R. (2007) Immunoglobulin-like domains on bacteriophage: weapons of modest damage? Curr. Opin. Microbiol. 10, 382–387 [DOI] [PubMed] [Google Scholar]
- 13. Haima P., Bron S., Venema G. (1987) The effect of restriction on shotgun cloning and plasmid stability in Bacillus subtilis Marburg. Mol. Gen. Genet. 209, 335–342 [DOI] [PubMed] [Google Scholar]
- 14. Le Grice S. F. (1990) Regulated promoter for high-level expression of heterologous genes in Bacillus subtilis. Methods Enzymol. 185, 201–214 [DOI] [PubMed] [Google Scholar]
- 15. Heckman K. L., Pease L. R. (2007) Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932 [DOI] [PubMed] [Google Scholar]
- 16. Berjanskii M. V., Neal S., Wishart D. S. (2006) PREDATOR: a web server for predicting protein torsion angle restraints. Nucleic Acids Res. 34, W63–W69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fung B. M., Khitrin A. K., Ermolaev K. (2000) An improved broadband decoupling sequence for liquid crystals and solids. J Magn. Reson. 142, 97–101 [DOI] [PubMed] [Google Scholar]
- 18. Gradmann S., Ader C., Heinrich I., Nand D., Dittmann M., Cukkemane A., van Dijk M., Bonvin A. M., Engelhard M., Baldus M. (2012) Rapid prediction of multi-dimensional NMR data sets. J. Biomol. NMR 54, 377–387 [DOI] [PubMed] [Google Scholar]
- 19. Wang Y., Jardetzky O. (2002) Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 11, 852–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seidel K., Lange A., Becker S., Hughes C. E., Heise H., Baldus M. (2004) Protein solid-state NMR resonance assignments from (13C,13C) correlation spectroscopy. Physical Chemistry Chemical Physics 6, 5090–5093 [Google Scholar]
- 21. Baldus M., Petkova A. T., Herzfeld J., Griffin R. G. (1998) Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol. Physics 95, 1197–1207 [Google Scholar]
- 22. Mindell J. A., Grigorieff N. (2003) Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 [DOI] [PubMed] [Google Scholar]
- 23. Ludtke S. J., Baldwin P. R., Chiu W. (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 [DOI] [PubMed] [Google Scholar]
- 24. van Heel M. (2001) Do single (ribosome) molecules phase themselves? Cold Spring Harb. Symp. Quant. Biol. 66, 77–86 [DOI] [PubMed] [Google Scholar]
- 25. Stewart M. (1988) Computer image processing of electron micrographs of biological structures with helical symmetry. J. Electron Microsc. Tech. 9, 325–358 [DOI] [PubMed] [Google Scholar]
- 26. Söding J., Biegert A., Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics 2006. Chapter 5, Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Veesler D., Robin G., Lichière J., Auzat I., Tavares P., Bron P., Campanacci V., Cambillau C. (2010) Crystal structure of bacteriophage SPP1 distal tail protein (gp19.1): a baseplate hub paradigm in Gram-positive infecting phages. J. Biol. Chem. 285, 36666–36673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Behrens B., Lüder G., Behncke M., Trautner T. A., Ganesan A. T. (1979) The genome of B. subtilis phage SPP1: physical arrangement in phage genes. Mol. Gen. Genet. 175, 351–357 [DOI] [PubMed] [Google Scholar]
- 31. Chai S., Bravo A., Lüder G., Nedlin A., Trautner T. A., Alonso J. C. (1992) Molecular analysis of the Bacillus subtilis bacteriophage SPP1 region encompassing genes 1 to 6. The products of gene 1 and gene 2 are required for pac cleavage. J. Mol. Biol. 224, 87–102 [DOI] [PubMed] [Google Scholar]
- 32. Sambrook J., Fritsh E. F., Maniatis T. (1989). Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 33. Dröge A., Tavares P. (2000) In vitro packaging of DNA of the Bacillus subtilis bacteriophage SPP1. J. Mol. Biol. 296, 103–115 [DOI] [PubMed] [Google Scholar]
- 34. Isidro A., Santos M. A., Henriques A. O., Tavares P. (2004) The high-resolution functional map of bacteriophage SPP1 portal protein. Mol. Microbiol. 51, 949–962 [DOI] [PubMed] [Google Scholar]
- 35. Pell L. G., Kanelis V., Donaldson L. W., Howell P. L., Davidson A. R. (2009) The phage λ major tail protein structure reveals a common evolution for long-tailed phages and the type VI bacterial secretion system. Proc. Natl. Acad. Sci. U.S.A. 106, 4160–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lopes A., Tavares P., Petit M. A., Guérois R., Zinn-Justin S. (2014) Automated identification of tailed bacteriophages and classification according to their neck organization. BMC Genomics 15, 1027–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moran C. P., Jr., Lang N., LeGrice S. F., Lee G., Stephens M., Sonenshein A. L., Pero J., Losick R. (1982) Nucleotide sequences that signal the initiation of transcription and translation in Bacillus subtilis. Mol. Gen. Genet. 186, 339–346 [DOI] [PubMed] [Google Scholar]
- 38. São-José C., Lhuillier S., Lurz R., Melki R., Lepault J., Santos M. A., Tavares P. (2006) The ectodomain of the viral receptor YueB forms a fiber that triggers ejection of bacteriophage SPP1 DNA. J. Biol. Chem. 281, 11464–11470 [DOI] [PubMed] [Google Scholar]
- 39. Jobichen C., Chakraborty S., Li M., Zheng J., Joseph L., Mok Y. K., Leung K. Y., Sivaraman J. (2010) Structural basis for the secretion of EvpC: a key type VI secretion system protein from Edwardsiella tarda. PLoS one 5, e12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Douzi B., Spinelli S., Blangy S., Roussel A., Durand E., Brunet Y. R., Cascales E., Cambillau C. (2014) Crystal structure and self-interaction of the type VI secretion tail-tube protein from enteroaggregative Escherichia coli. PLoS One 9, e86918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ballister E. R., Lai A. H., Zuckermann R. N., Cheng Y., Mougous J. D. (2008) In vitro self-assembly of tailorable nanotubes from a simple protein building block. Proc. Natl. Acad. Sci. U.S.A. 105, 3733–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wishart D. S., Sykes B. D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]