

Background: VEGF plays a causal role in diabetic retinopathy.
Results: Hyperglycemia induced VEGF and REDD1 expression in the retina of wild-type, but not REDD1 knock-out mice.
Conclusion: REDD1 is necessary for hyperglycemia mediated effects on VEGF expression in the retina of diabetic mice.
Significance: Molecular therapies targeting REDD1 could prove useful in preventing pathological effects of hyperglycemia.
Keywords: Diabetes, Eukaryotic Translation Initiation Factor 4E-binding Protein 1 (EIF4EBP1), mTOR Complex (mTORC), Retina, Vascular Endothelial Growth Factor (VEGF), DDIT4, REDD1, RTP801
Abstract
Vascular endothelial growth factor (VEGF) is considered a major role player in the pathogenesis of diabetic retinopathy, yet the mechanisms regulating its expression are not fully understood. Our laboratory previously demonstrated that diabetes-induced VEGF expression in the retina was dependent on the repressor of mRNA translation 4E-BP1. Interaction of 4E-BP1 with the cap-binding protein eIF4E regulates protein expression by controlling the selection of mRNAs for translation. The process is regulated by the master kinase mTOR in complex 1 (mTORC1), which phosphorylates 4E-BP1, thus promoting its disassociation from eIF4E. In the present study, we investigated the role of the Akt/mTORC1 repressor REDD1 (regulated in development and DNA damage) in diabetes-induced VEGF expression. REDD1 expression was induced by hyperglycemia in the retina of diabetic rodents and by hyperglycemic conditions in Müller cells concomitant with increased VEGF expression. In Müller cells, hyperglycemic conditions attenuated global rates of protein synthesis and cap-dependent mRNA translation concomitant with up-regulated cap-independent VEGF mRNA translation, as assessed by a bicistronic luciferase reporter assay. Hyperglycemic conditions also attenuated mTORC1 signaling and enhanced 4E-BP1 binding to eIF4E. Furthermore, ectopic expression of REDD1 in Müller cells was sufficient to promote both increased 4E-BP1 binding to eIF4E and VEGF expression. Whereas the retina of wild-type mice exhibited increased expression of VEGF and tumor necrosis factor alpha (TNF-α) 4 weeks after streptozotocin administration, the retina of REDD1 knock-out mice failed to do so. Overall, the results demonstrate that REDD1 contributes to the pathogenesis of diabetes in the retina by mediating the pathogenic effects of hyperglycemia.
Introduction
Diabetic retinopathy (DR)2 is the leading cause of blindness in working age Americans, affecting more than one-third of the nearly 21 million individuals with diabetes. The primary cause of diabetes-related vision loss is neurovascular complications that result from hyperglycemia (1). The Diabetes Control and Complications Trial demonstrated that intensive glycemic control is associated with reduced incidence and progression of DR (2), yet the molecular mechanisms whereby hyperglycemia mediates neurovascular dysfunction remain incompletely understood. As a consequence, current clinical therapies largely address symptoms rather than the molecular mechanisms of the disease.
Levels of the pro-angiogenic cytokine vascular endothelial growth factor (VEGF) are elevated in the vitreous fluid from eyes of patients with DR (3), and the cytokine is considered a major molecular role player in the neurovascular complications of DR. While VEGF expression is classically thought of as being regulated transcriptionally, more recent studies have indicated that it is also regulated at the level of mRNA translation (4–6). Ribosome recruitment to the mRNA 5′-end serves as the rate-controlling step in translation and in mammalian cells is classically thought of as occurring through mechanisms that are either dependent on or independent of a m7GTP cap. The VEGF mRNA transcript has an exceptionally long (1038 bases) G-C base pair rich 5′-untranslated region (UTR) that contains alternative start sites and stop codons in frame with the classical start site. Two independent internal ribosome entry sites (IRES) within the UTR control start codon selection (7) and allow the VEGF mRNA to be translated independent of the 5′-cap under conditions in which cap dependent translation is inhibited (4).
The Akt/mTORC1 (master kinase mammalian target of rapamycin complex 1) signaling pathway regulates mRNA translation by governing assembly of the mRNA cap-binding complex, eIF4F, which is composed of eIF4E, eIF4G, and eIF4A (8). In their hypophosphorylated state, eIF4E-binding proteins (4E-BPs) sequester the cap-binding protein eIF4E away from eIF4G and thus inhibit cap-dependent mRNA translation. However upon phosphorylation by mTORC1, 4E-BPs are released from eIF4E, allowing assembly of the eIF4F-complex and thus a concomitant increase in cap-dependent and a decrease in cap-independent mRNA translation. We previously demonstrated that hyperglycemic conditions enhanced relative cap-independent translation in the liver of diabetic mice and in cells in culture and that 4E-BP1 is necessary for the effect (9). Furthermore, we also showed that 4E-BP1, but not 4E-BP2, is necessary for increased VEGF expression in both the retina during diabetes and in cells maintained under hyperglycemic conditions (9, 10). These previous studies from our laboratory (18) demonstrate that the effect of diabetes-induced hyperglycemia on 4E-BP1 is mediated at least in part through the covalent addition of O-linked N-acetylglucosamine (O-GlcNAcylation). Regulation of mRNA translation and specifically sequestration of eIF4E by 4E-BP1 potentially represent a key mechanism in the pathological VEGF expression associated with DR. However, the relative contribution of reduced phosphorylation and increased O-GlcNAcylation of 4E-BP1 in diabetes-induced VEGF expression is unexplored. Thus, we set out to evaluate the signaling mechanisms whereby hyperglycemia alters mRNA translation in the retina.
The protein Regulated in Development and DNA Damage 1 (REDD1; also known as DNA-damage-inducible transcript 4 (DDIT4), dexamethasone-induced gene 2 (Dig2), and RTP801) governs phosphorylation of 4E-BP1 through inhibition of Akt/mTORC1 signaling (11). Our laboratory previously reported increased REDD1 protein and mRNA expression in the muscle of diabetic mice (12), and more recent reports have shown enhanced REDD1 mRNA expression in the retinal pigment epithelium/choroid of diabetic rats (13). Thus, in the present study we set out to test the hypothesis that diabetes-induced REDD1 protein expression represents a translational control mechanism that contributes to enhanced VEGF expression in the retina. The results show that hyperglycemia, rather than reduced insulin availability, plays a dominant role in inducing both VEGF mRNA translation and REDD1 expression. Moreover, diabetes failed to promote expression of either VEGF or the proinflammatory marker TNF-α in the retina of REDD1 knock-out mice, demonstrating a critical role for REDD1 in diabetes-induced pathology of the retina.
EXPERIMENTAL PROCEDURES
Materials
Protease inhibitor mixture was purchased from Sigma, and ECL Western blot detection reagent from Pierce. Horseradish peroxidase-conjugated goat anti-rabbit and mouse IgG antibodies, as well as anti-phospho-S6K1 (Thr389) were purchased from Bethyl Laboratories. Anti-GAPDH (sc-32233), VEGF (sc-7269), TNF-α (sc-12744), and horseradish peroxidase-conjugated goat anti-hamster IgG antibodies were purchased from Santa Cruz Biotechnology. Anti-REDD1 antibody was purchased from Proteintech. Anti-actin was purchased from Sigma. Preparation of the 4E-BP1 and eIF4E antibodies has been previously described (14, 15). All other antibodies were purchased from Cell Signaling Technology.
Animals
Age-matched male Sprague-Dawley rats (Charles River, MA) received a single intraperitoneal injection containing 65 mg/kg streptozotocin (STZ) dissolved in sodium citrate buffer (pH 4.5) to induce diabetes. Male wild-type and RTP801 (REDD1) knock-out C57Bl/6 × 129SvEv mice (16) were treated with 50 mg/kg STZ for 5 consecutive days. Control rodents were injected with equivalent volumes of sodium citrate buffer. Diabetic phenotype was confirmed by blood glucose concentration >250 mg/dl in freely fed animals. Glycemic control was achieved in diabetic rats by twice-daily subcutaneous phlorizin (200 mg/kg) injections during the last 3 full days of the experiment, as well 3 h prior to tissue harvest. Retinas were harvested following 4 weeks of diabetes, flash-frozen in liquid nitrogen, and later sonicated in 250 μl of extraction buffer as previously described (10). The homogenate was centrifuged at 10,000 × g for 5 min at 4 °C, and the supernatant was collected for analysis.
Cell Culture
TR-MUL retinal Müller cells were provided by K. Hosoya (Toyama Medical and Pharmaceutical University), Moorfields/Institute of Ophthalmology-Müller 1 (MIO-M1) cells were provided by G. A. Limb (University College of London), and REDD1+/+ and REDD1−/− MEFs were provided by L. Ellison (Massachusetts General Hospital Cancer Center). Cells were maintained in DMEM containing 5 mm glucose and supplemented with 10% heat-inactivated FBS (Atlas Biologicals) and 1% penicillin/streptomycin (Invitrogen). Cells were maintained at either 37 °C (MIO-M1 and MEFs) or 33 °C (TR-MUL) and 5% CO2 atmosphere. For studies on the effects of hyperglycemic conditions, cell culture medium was replaced with medium containing 10% FBS and either 30 mm glucose or 5 mm glucose with 25 mm mannitol as an osmotic control added 16 h prior to harvest or as indicated. Global rates of protein synthesis were calculated by the incorporation of 35S-labeled methionine into TCA-precipitable protein as previously described (17). Transfections were performed using Xtremegene HP (Roche). The following previously described plasmids (11) were used to evaluate the effect of REDD1 expression: empty pCMV vector, pCMV-HA-REDD1, empty pTREtight vector, and pTREtight-HA-REDD1. pTREtight plasmids were co-transfected with pRevTet-On (Clontech), which expresses the reverse tetracycline-controlled transactivator. Cell culture medium was supplemented with doxycycline (1 μg/ml; Clontech) for 6 h to induce HA-REDD1 protein expression from the pTREtight-HA-REDD1 plasmid. Renilla and firefly luciferase activity was measured in TR-MUL cell lysates using a bicistronic luciferase reporter plasmid containing the VEGF IRES (9) either 4 h after of exposure to medium containing a high glucose concentration or 6 h after doxycycline administration by Dual-Glo Luciferase Assay (Promega). Anti-eIF4E or anti-4E-BP1 antibodies were used as previously described (18) to immunoprecipitate eIF4E or 4E-BP1.
Protein Analysis
A fraction of the supernatant was added to 1× SDS sample buffer, boiled for 5 min, and analyzed by Western blotting as previously described (10). Another fraction of the supernatant was evaluated with a Mouse VEGF Quantikine ELISA (R&D systems).
RNA Isolation and Quantitative Real-time PCR Analysis
RNA was extracted from cells with TRIzol reagent according to the manufacturer's instructions (Invitrogen). RNA (1 μg) was reverse transcribed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and subjected to quantitative real-time PCR using QuantiTect SYBR Green master mix (Qiagen). QuantiTect SYBR Green primers were used for rat Vegfa and Actb (Qiagen). Primers for mouse Vegfa were as follows: forward 5′-gag aga ggc cga agt cct tt-3′, reverse 5′-ttg gaa ccg gca tct tta tc-3′. Primers for mouse Gapdh were as previously described (19).
Statistical Analysis
The data are expressed as mean ± S.E. Analysis of variance was used to identify differences between group means. When identified, Student's t test was used post hoc to compare differences among groups. p < 0.05 was considered statistically significant.
RESULTS
Hyperglycemia Produces a Coordinate Increase in REDD1 and VEGF Expression in the Retina of Diabetic Rats
REDD1 protein expression was assessed in the retina of diabetic rats by Western blot analysis. Following 4 weeks of STZ-induced diabetes, REDD1 expression was increased in the retina of diabetic rats as compared with non-diabetic controls (Fig. 1A). We have previously reported decreased phosphorylation of 4E-BP1 on Thr37/46 concomitant with elevated VEGF abundance in the retina of diabetic rats (10). When analyzed by SDS-PAGE, 4E-BP1 can be resolved into multiple phospho-isoforms based upon a reduced migratory capacity that is associated with increased phosphorylation. In the present study, relative 4E-BP1 phosphorylation was lower in the retina of diabetic rats compared with control rats as assessed by the proportion of the protein present in the hyperphosphorylated γ isoform relative to total 4E-BP1 (i.e. the sum of the α, β, and γ isoforms, Fig. 1B). As expected, the abundance of VEGF was also increased as compared with non-diabetic cohorts (Fig. 1C). To specifically evaluate the contribution of diabetes-induced hyperglycemia to these changes, normalization of blood glucose levels by phlorizin was used to achieve glycemic control. As previously observed (20), twice-daily administration of phlorizin reduced serum glucose levels in diabetic rats close to normal levels independently of insulin (145.0 ± 23.0 mg.dL−1 in control versus 547.3 ± 21.5 mg.dL−1 in diabetic and 214.9 ± 21.1 mg.dL−1 in diabetic treated with phlorizin). Normalization of serum glucose concentrations also reduced the abundance of REDD1 (Fig. 1A), enhanced phosphorylation of 4E-BP1 (Fig. 1B), and reduced VEGF abundance (Fig. 1C) in the retina of diabetic rats, such that levels were no longer different than those observed in non-diabetic controls. These findings demonstrate that hyperglycemia contributes to the effect of diabetes on these key signaling proteins in the retina.
FIGURE 1.
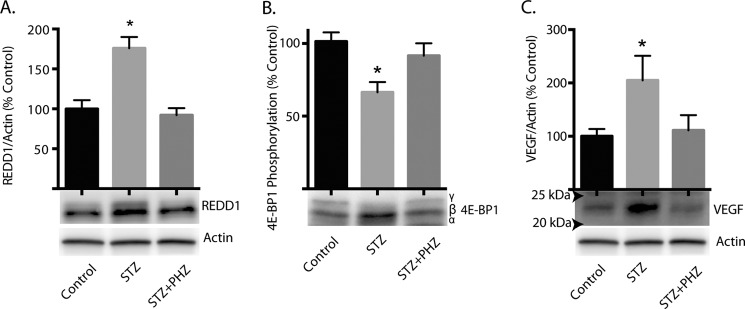
Effect of diabetes-induced hyperglycemia on expression of REDD1 and VEGF and phosphorylation of 4E-BP1 in the retina. Diabetes was induced in rats by STZ injection. Four weeks after the induction of diabetes, STZ-treated rats were subcutaneously injected with phlorizin (PHZ) to lower blood glucose concentrations. Retina supernatant fractions were prepared and the abundance of REDD1 (A), phosphorylation of 4E-BP1 (B), and the abundance of VEGF (C) were assessed by Western blot. Molecular weight markers are indicated with arrows in C. SDS-PAGE was performed using 15% Tris-HCl polyacrylamide gels. Values are means + S.E. relative to actin (n = 8 in A & C, 5 in B). Statistical significance is denoted *, p < 0.05.
Hyperglycemic Conditions Promote REDD1 and VEGF Expression in Müller Cells in Culture
To further evaluate the effect of hyperglycemia on REDD1 and VEGF abundance, we employed Müller cells in culture exposed to medium containing either control (5 mm) or high (30 mm) concentrations of glucose (i.e. a hyperglycemic condition). Müller cells are the main glial cells of the retina and a likely target of hyperglycemia (21, 22). Furthermore, Müller cells are activated early in the progression of DR (21) and are among the cells responsible for the synthesis and expression of a number of growth factors including VEGF that play a causative role in retinal inflammation, vascular lesions, and increased permeability that characterize the disease (23, 24). In both human MI0-M1 (Fig. 2A) and rat TR-MUL (Fig. 2B) Müller cells in culture, exposure to medium containing 30 mm glucose increased the abundance of REDD1 and VEGF as compared with control medium containing 5 mm glucose with 25 mm mannitol added as an osmotic control. Furthermore, increased REDD1 and VEGF abundance was detected in TR-MUL cells as early as 4 h after exposure to high glucose and was maintained through 24 h (Fig. 2C).
FIGURE 2.

Effect of hyperglycemic conditions on REDD1 and VEGF expression in Müller cells in culture. Müller cell cultures were maintained in DMEM containing 5 mm glucose and supplemented with 10% heat-inactivated FBS. Human MI0-M1 (A) or rat TR-MUL Müller (B) cells were exposed to medium containing either 30 mm glucose or 5 mm glucose with 25 mm mannitol as an osmotic control. C, TR-MUL cells were exposed to medium containing 30 m glucose as indicated. The abundance of VEGF, REDD1, and GAPDH was assessed by Western blot. Representative blots are shown. D, TR-MUL cells were treated as described in B, and relative VEGF mRNA abundance was assessed by RT-qPCR. E, rate of protein synthesis was measured by incorporation of 35S-labeled methionine into protein. dpm, disintegrations per minute. F, diagram of bicistronic luciferase reporter where translation of Renilla luciferase (LucR) occurs in a cap-dependent manner and translation of Firefly luciferase (LucF) is regulated by the VEGF IRES in a cap-independent manner. G and H, TR-MUL cells were exposed to medium containing 30 mm glucose as indicated and cap-dependent (G) and cap-independent (H) translation were assessed in lysates using the bicistronic reporter described in F. RLU, relative light units. I, ratio of LucF to LucR activity in TR-MUL cell lysate supernatant fractions represented as the relative activity from each reporter. Values are means + S.E. (n = 6). Statistical significance is denoted *, p < 0.05.
Hyperglycemic conditions did not alter VEGF mRNA expression as compared with an osmotic control (Fig. 2D). Although transcriptional changes are strongly correlated with those seen in VEGF protein following prolonged exposure of retinal cells to hyperglycemic conditions (i.e. >72 h), elevated VEGF protein expression precedes the change in VEGF mRNA (25). Thus, a post-transcriptional control mechanism allows retinal cells to acutely responded to hyperglycemia. To evaluate the effect of hyperglycemic conditions on global rates of protein synthesis, the incorporation of 35S-labeled methionine into cellular protein was assessed. Global protein synthesis was reduced in TR-MUL cells following exposure to 30 mm glucose (Fig. 2E). Thus, an increase in the overall rate of protein synthesis was not responsible for the induction of VEGF protein expression following exposure to hyperglycemic conditions. To specifically evaluate VEGF mRNA translation in TR-MUL cells, we ectopically expressed a bicistronic plasmid containing two open reading frames (ORF) encoding two distinct luciferase enzymes, LucR and LucF (9). Translation of the first ORF (LucR) occurs in a cap-dependent manner, whereas translation of the second ORF (LucF) is driven by the VEGF IRES that is located between the two cistrons (Fig. 2F). Thus, LucF activity functions as a reporter of cap-independent translation. Whereas hyperglycemia attenuated cap-dependent translation (Fig. 2G), cap-independent translation was enhanced (Fig. 2H). Overall, the relative LucF/LucR activity ratio in TR-MUL lysates was increased by 54% upon exposure to hyperglycemic conditions (Fig. 2I).
Hyperglycemic Conditions Attenuate Akt/mTORC1 Signaling in Retinal Müller Cells in Culture
The best characterized mechanism for controlling the shift from cap-dependent to cap-independent mRNA translation involves assembly of the eIF4F complex (27), a process that is regulated by the mTORC1 signaling pathway (8). Since the effects of hyperglycemia are potentially mediated by repression of mTORC1, we initially evaluated Akt/mTORC1 signaling in TR-MUL cells following exposure to medium containing high glucose concentrations. We recently demonstrated that REDD1 acts to repress mTORC1 by promoting the association of protein phosphatase 2A with Akt, leading to the site-specific dephosphorylation of the kinase on Thr308, a subsequent reduction in Akt-mediated phosphorylation of TSC2, and a fall in the proportion of Rheb in the active GTP-bound state (11). Direct interaction of Rheb-GTP, but not Rheb-GDP, with mTORC1 results in its activation and thus promotes hyperphosphorylation of 4E-BP1 and p70S6K1 (28). Following exposure to medium containing high glucose concentrations, Akt phosphorylation was attenuated at the REDD1-sensitive Thr308 site, but not at Ser473 following (Fig. 3A). Furthermore, as compared with osmotic controls, hyperglycemic conditions attenuated mTORC1 signaling in both MI0-M1 (Fig. 3B) and TR-MUL Müller (Fig. 3C) cells in culture as assessed by p70S6K1 phosphorylation on Thr389. Importantly, the high glucose-induced phosphorylation changes on both Akt and p70S6K1 were inversely proportional to the magnitude of increase in REDD1 abundance (Figs. 2–3).
FIGURE 3.

Effect of hyperglycemic conditions on Akt/mTORC1 signaling and eIF4E sequestration in Müller cells. TR-MUL cell cultures were maintained in DMEM containing 5 mm glucose and supplemented with 10% heat-inactivated FBS. Cells were then exposed to medium containing 30 mm glucose. A–D, phosphorylation of Akt at Thr308 and Ser473, p70S6K1 phosphorylation on Thr389, and 4E-BP1 at Ser65 as well as abundance of Akt, 4E-BP1, p70S6K1, and GAPDH was assessed in cell lysates by Western blot. D, binding of 4E-BP1 to eIF4E was assessed in cell lysates by co-immunoprecipitation. SDS-PAGE was performed using either 15% (A) or 4–15% Tris-HCl polyacrylamide gels (B–D). Representative blots are shown. Values are means + S.E. (n = 8). Statistical significance is denoted *, p < 0.05.
To evaluate the effects of high glucose on eIF4F complex assembly, we assessed phosphorylation of 4E-BP1 in TR-MUL cells. In support of the observation in the retina, hyperglycemic conditions increased the abundance of the faster migrating 4E-BP1 isoforms, indicating a reduction in the phosphorylation state of the protein (Fig. 3A). Moreover, site-specific phosphorylation of 4E-BP1 on Ser65, which is exclusively detected in the 4E-BP1 γ-isoform, was also attenuated following exposure to medium containing high glucose concentrations (Fig. 3A). Although insufficient to block eIF4E binding alone, phosphorylation of 4E-BP1 on Ser65 occurs after phosphorylation of both Thr70 and Thr37/46 (29). To specifically assess binding of 4E-BP1 to eIF4E, 4E-BP1 was immunoprecipitated from TR-MUL cells. Hyperglycemic conditions enhanced the co-immunoprecipitation of eIF4E with 4E-BP1 as compared with the osmotic control (Fig. 3D). Overall the results are consistent with reduced mTORC1 signaling and impaired assembly of the eIF4F complex in TR-MUL cells following exposure to hyperglycemic conditions.
REDD1 Regulates VEGF Expression
To investigate the necessity of REDD1 in the effects of hyperglycemic conditions on mTORC1 signaling and VEGF expression, REDD1+/+ and REDD1−/− MEFs were exposed to medium containing either control or high glucose concentrations. Consistent with our observations in Müller cells, hyperglycemic conditions attenuated phosphorylation of p70S6K1 and 4E-BP1 and enhanced REDD1 abundance in REDD1+/+ MEFs (Fig. 4A). However, in REDD1−/− MEFs, phosphorylation of p70S6K1 and 4E-BP1 was unchanged following exposure to hyperglycemic conditions as compared with REDD1+/+ MEFs. Furthermore, exposure to hyperglycemic conditions increased VEGF protein abundance in REDD1+/+ but not REDD1−/− MEFs (Fig. 4A) as assessed by ELISA. The relative abundance of VEGF mRNA was not altered by hyperglycemic conditions in either REDD1+/+ or REDD1−/− MEFs (Fig. 4B). Overall, these data demonstrate that REDD1 is necessary for the hyperglycemia-mediated effects on mTORC1 signaling and VEGF expression.
FIGURE 4.

REDD1 is necessary for the effect of hyperglycemic conditions on VEGF expression. Wild-type and REDD1 knock-out mouse embryonic fibroblasts (MEF) were maintained in 5 mm glucose medium as described above. MEF were exposed to medium containing either 30 mm glucose or 5 mm glucose with 25 mm mannitol as an osmotic control. VEGF protein abundance was assessed by ELISA. The abundance of GAPDH, REDD1, and p70S6K1, and phosphorylation of p70S6K1 on Thr389 were assessed in cell lysates by Western blot. SDS-PAGE was performed using 15% Tris-HCl polyacrylamide gels. Representative blots are shown. B, relative VEGF mRNA abundance was assessed by RT-qPCR. Values are means + S.E. (n = 6). Statistical significance is denoted *, p < 0.05.
To further evaluate the role of REDD1 in the effect of hyperglycemic conditions on eIF4F complex assembly and VEGF expression, HA-tagged REDD1 was ectopically expressed in TR-MUL cells. Ectopically expressed REDD1 both increased the co-immunoprecipitation of 4E-BP1 with eIF4E (Fig. 5A) and reduced phosphorylation of 4E-BP1 on Ser65 (Fig. 5B) as compared with an empty vector control. Furthermore, ectopically expressed REDD1 induced VEGF protein abundance by 4-fold when compared with an empty vector (Fig. 5B). Ectopic REDD1 also induced a small, but significant increase in relative VEGF mRNA expression (Fig. 5C). To specifically evaluate the effect of ectopic REDD1 on VEGF mRNA translation, we used the previously described dual luciferase reporter (Fig. 2F). Following REDD1 induction, the relative LucF/LucR activity ratio in TR-MUL lysates was increased as compared with an empty vector control (Fig. 5D). Overall, REDD1 was sufficient to promote sequestration of eIF4E, enhance relative cap-independent mRNA translation driven by the VEGF 5′-UTR, and induce expression of VEGF protein in Müller cells.
FIGURE 5.

Effect of REDD1 overexpression on 4E-BP1 binding to eIF4E and VEGF expression. TR-MUL cell cultures were maintained in DMEM containing 5 mm glucose and supplemented with 10% heat-inactivated FBS. Cells were transiently transfected with either an empty vector (EV) control plasmid or a plasmid encoding HA-tagged REDD1 (REDD1). A, binding of 4E-BP1 with eIF4E was assessed by co-immunoprecipitation. B, abundance of VEGF, HA-tagged REDD1, eIF4E, 4E-BP1, and tubulin was assessed in cell lysates by Western blot. SDS-PAGE was performed using 4–15% Tris-HCl polyacrylamide gels. Representative blots are shown. C, relative VEGF mRNA abundance was assessed by RT-qPCR. D, REDD1 expression was induced in TR-MUL cells after which cap-dependent and cap-independent translation was assessed using the bicistronic luciferase reporter described in 2F. Results represent the ratio of the relative LucF to LucR activity in TR-MUL cell lysate. REDD1 expression was induced by 6 h of doxycycline administration. Values are means + S.E. (n = 6). Statistical significance is denoted *, p < 0.05.
Ablation of REDD1 Prevents Diabetes-induced VEGF Expression in Mouse Retina
To evaluate the role of REDD1 in the effect of diabetes on VEGF expression in the retina, mice with a germline disruption of REDD1 were administered STZ. Post-prandial blood glucose concentrations were not different between wild-type (454.1 ± 33.9 mg/dl) and REDD1 knock-out (502.3 ± 43.9 mg/dl) mice following 4-weeks of diabetes. Similar to STZ-treated rats, REDD1 abundance was increased in the retina of wild-type diabetic mice as compared with age-matched non-diabetic animals (Fig. 6A). As expected, REDD1 expression was undetectable in the retina of REDD1 knock-out mice (Fig. 6A). It was previously demonstrated that p70S6K1 activity is decreased in the retina of rats following 4 weeks of diabetes (30). In support of the previous data, phosphorylation of p70S6K1 on Thr389 was attenuated in the retina of wild-type mice following the induction of diabetes (Fig. 6B). However, no difference in phosphorylation of p70S6K1 on Thr389 was observed between non-diabetic and diabetic REDD1 knock-out mice (Fig. 6B). This observation demonstrates a regulatory role for REDD1 in the repression of mTORC1 signaling in the retina during diabetes.
FIGURE 6.

REDD1 is necessary for attenuated mTORC1 signaling and enhanced VEGF expression in the retina of diabetic mice. Diabetes was induced in mice by STZ injection. Four weeks after the induction of diabetes, mice were fasted 16 h and the abundance of REDD1 (A) and p70S6K1 phosphorylation on Thr389 (B) were assessed in retina homogenate supernatants by Western blot. C, VEGF abundance was assessed in the same fractions by ELISA. D, TNF-α was assessed in retina homogenate supernatants by Western blot. Values are means + S.E. (n = 8). Statistically significant difference are denoted * versus control injection or # versus wild-type mice, p < 0.05.
To assess VEGF expression, ELISA was performed on whole retina homogenates. VEGF protein expression was elevated in the retina of diabetic wild-type mice as compared with non-diabetic controls (Fig. 6C). Yet, retinal levels of VEGF were no different between non-diabetic and diabetic REDD1 knock-out mice. Importantly, VEGF expression was significantly lower in the retina of diabetic REDD1 knock-out mice as compared with their diabetic wild-type counterparts, such that levels were similar to those observed in the retina of non-diabetic wild-type mice (Fig. 6C).
Müller cell-derived VEGF plays a causative role in retinal inflammation (24), which has been implicated in the etiology of DR (31). Expression of the proinflammatory marker tumor necrosis factor alpha (TNF-α) is elevated in the vitreous of patients with DR (32, 33) and Müller cell specific disruption of VEGF inhibits diabetes-induced overexpression of TNF-α (24). Since diabetes failed to enhance VEGF in the retina of REDD1 knock-out mice, we also evaluated TNF-α expression. Whereas retinas from diabetic wild-type mice exhibited more than a 2-fold increase in TNF-α expression compared with non-diabetic cohorts, diabetes failed to promote TNF-α expression in the retina of REDD1 knock-out mice (Fig. 6D). This demonstrates that REDD1 is not only necessary for diabetes-induced overexpression of VEGF, but also contributes to the inflammatory signaling associated with DR.
DISCUSSION
The findings of the present study provide further insight into the mechanisms responsible for the effects of diabetes on VEGF expression in the retina. The abundance of any given protein in a cell (aka gene expression) is the sum of transcription, mRNA stability, mRNA translation, and protein degradation. Whereas the pathological consequences of altered transcription and degradation have received much attention (34, 35), the effect of diabetes on mRNA translation has been less well explored, despite the fact that next-generation sequencing and large-scale proteomics demonstrate a dominant contribution of mRNA translation to gene expression (36). We recently reported the existence of a diabetes-induced hyperglycemia-mediated shift in gene expression that was associated with down-regulation of cap-dependent and concomitant up-regulation of cap-independent mRNA translation in both STZ-treated mice and cells in culture (9). The results reported here support a model whereby hyperglycemia-induced REDD1 expression promotes VEGF production in the retina by contributing to the activation of a switch in mRNA translation (Fig. 7).
FIGURE 7.

Working model for the role of REDD1 in the effect of diabetes-induced hyperglycemia on VEGF expression. In both the retina of diabetic rodents and Müller cells in culture, hyperglycemic conditions increase the abundance of the protein REDD1. REDD1 acts to repress Akt/mTORC1 signaling in a manner that attenuates phosphorylation of 4E-BP1 and promotes sequestration of the mRNA cap-binding eIF4E. In turn, global protein synthesis is repressed. However, whereas cap-dependent translation is attenuated, cap-independent translation by the VEGF IRES is activated. Thus, hyperglycemia-induced VEGF abundance results as a consequence of REDD1 in the retina.
In the present study, we demonstrate that the effect of diabetes on VEGF expression is mediated by hyperglycemia per se, as phlorizin administration to normalize blood glucose levels prevented the effect. This observation is supported by previous studies with retinal cells, where hyperglycemic conditions increased VEGF abundance (10, 37, 38). Herein we extend the previous studies to demonstrate that the effect is also dependent on REDD1. In the retina of diabetic rodents, VEGF protein abundance was not only elevated in a manner that paralleled REDD1 abundance, but VEGF did not increase following STZ administration to REDD1 knock-out mice, despite the increase in blood glucose concentrations. Overall, the results demonstrate that REDD1 is necessary for diabetes-induced VEGF expression in the retina.
The abundance of VEGF protein is increased in the retina of both STZ-induced diabetic rats and mice and Ins2Akita mice without a change in either VEGF mRNA expression or the expression of HIF1-α (10). These observations suggest that important post-transcriptional mechanisms exist for the regulation of VEGF abundance. Indeed, the results presented herein confirm that hyperglycemic conditions promote an increase in translation of VEGF mRNA. Furthermore, the increased abundance of VEGF protein expression in response to diabetes and hyperglycemic conditions was observed in the absence of a change in VEGF mRNA expression, supporting the importance of posttranscriptional mechanisms. Under one condition employed in the present studies (i.e. the ectopic expression of REDD1) we did observed a modest increase in VEGF mRNA expression. However, this was accompanied by a much greater (5-fold) increase in VEGF protein expression, implying that mechanisms other than transcription were involved. Indeed, under those same conditions, a 54% increase in cap-independent VEGF mRNA translation was observed. Nonetheless, these observations do raise the possibility that conditions exist, such as long-term diabetes, in which transcriptional as well as translational control mechanisms contribute to the hyperglycemia-induced changes in VEGF expression.
We previously demonstrated that STZ-induced diabetes reduces global rates of protein synthesis in the retina of rats and that blood glucose normalization with phlorizin is sufficient to reverse the effect (39). Here, we found that protein synthesis was also attenuated in Müller cells following acute exposure to hyperglycemic conditions. This observation is supported by a previous report that insulin-stimulated protein synthesis is reduced in cells pretreated with medium containing high glucose concentrations (40). Hyperglycemic conditions also decreased cap-dependent mRNA translation and increased cap-independent translation mediated by the VEGF IRES. Similar to the effect of hyperglycemia, global protein synthesis is inhibited by exposure to hypoxia, which causes an overall loss of polysomes (41). Furthermore, hypoxia also controls a cap-dependent to cap-independent translation switch that is mediated by eIF4E availability and increases the selective translation of mRNAs containing IRESs including VEGF (42). Intriguingly, REDD1 was initially identified as a gene induced by hypoxia (43) and disruption of REDD1 blocks hypoxia-induced inhibition of mTORC1 (44). Thus, the molecular mechanisms through which hyperglycemia and hypoxia act to induce gene-specific changes in mRNA translation may be similar. Indeed, the concept of “hyperglycemic psuedohypoxia” in the retina has been long argued to result from an increase in the cytosolic NADH:NAD+ ratio as a consequence of hyperglycemia, despite normal oxygen partial pressure (45).
REDD1 was linked to oxygen-induced retinopathy shortly after its discovery (16). Brafman et al. (16) demonstrated that REDD1 is induced in the retina under ischemic conditions, and that ablation of REDD1 conferred significant reduction in both retinal pathologic neovascularization and apoptosis within the inner nuclear layer (16). Notably, in oxygen-induced retinopathy, VEGF mRNA expression is similar in the retina of wild-type and REDD1 knock-out mice (16). Thus, while it is clear that the effects of oxygen tension on VEGF gene expression are mediated at least in part by hypoxia inducible transcription factors (46), the protective effects of REDD1 ablation on retinopathy are not likely to function at this level.
Overexpression of REDD1 was not only sufficient to induce VEGF in Müller cells in culture, but ablation of REDD1 prevented increased VEGF expression in the retina of diabetic mice. Regulation of VEGF gene expression is particularly important, because VEGF plays a causal role in the development and progression of complications associated with DR (24). Elevated VEGF expression precedes retinal neovascularization in patients with diabetes and is largely localized to glial cells of the inner retina (47). While it is likely that VEGF is initially increased in preclinical DR as a mechanism to maintain retinal vascular bed integrity, continued elevation of VEGF concentrations in ischemic areas leads to the increased vascular permeability and neovascularization that characterize the later stages of the disease (48). Intravitreal injections of recombinant VEGF are sufficient to produce vascular abnormalities in primates (49). Similarly, overexpression of VEGF in the rodent retina induces neovascularization (50). In contrast, emerging treatments that attempt to neutralize VEGF such as anti-VEGF antibodies show great promise, particularly with regards to management of the later proliferative stages of DR (51). However, anti-VEGF therapies are not specific to vascular cells, as VEGF regulates proliferation, differentiation, and survival of both glial and neuronal cells in the adult retina (52). Systemic VEGF neutralization produces significant neural cell death and decline of retinal function as measured by electroretinogram (ERG) (26). Thus, the therapeutic blockade of VEGF is accompanied by important caveats.
In the present study, ablation of REDD1 not only prevented the diabetes-induced elevation in VEGF abundance in the retina, but in both diabetic and non-diabetic REDD1 knock-out mice VEGF was maintained at levels similar to those observed in non-diabetic wild-type cohorts. This observation demonstrates that molecular therapies designed to target REDD1, such as PF-04523655 (13), could prove useful in preventing the pathological effect of hyperglycemia on VEGF abundance without abolishing all VEGF function. In fact, Timothy et al. (Timothy et al. IOVS 2005; 46:ARVO E-Abstract 427) determined that diabetes fails to induce retinal vascular permeability or ERG abnormalities in REDD1 knock-out mice. The results of the present study suggest that the reportedly protective effects of REDD1 ablation are potentially mediated through modulation of VEGF expression.
Overall, the results support a model where diabetes-induced hyperglycemia increases REDD1 expression, attenuates Akt/mTORC1 signaling, and promotes cap-independent translation from the VEGF IRES (Fig. 7). In both the retina of diabetic rodents and Müller cells in culture, hyperglycemic conditions increased the abundance REDD1, leading to hypophosphorylation of the translational repressor 4E-BP1. Hyperglycemia repressed both global protein synthesis and cap-dependent mRNA translation. However, hyperglycemic conditions or ectopic REDD1 expression was sufficient to promote cap-independent translation by the VEGF IRES and expression of the cytokine. Importantly, increased REDD1 expression was critical for the effect of hyperglycemia on VEGF expression in both cells in culture and the retina. Moreover, REDD1 is not required for basal VEGF expression, but rather an increase in its expression represents a stress response to hyperglycemic conditions. Thus, REDD1 is necessary for hyperglycemia-induced VEGF expression in the retina.
Acknowledgments
We thank Dr. E. Feinstein (Quark Pharmaceuticals) for permission to use the REDD1 knock-out mice and Dr. D. Williamson (SUNY Buffalo) for providing breeding pairs. We thank Dr. K. Hosoya (Toyama Medical and Pharmaceutical University) for kindly providing TR-MUL cells, Dr. G. A. Limb (University College of London) for kindly providing Moorfields Institute of Ophthalmology-Müller 1 (MIO-M1) cells, and Dr. L. Ellisen (Massachusetts General Hospital Cancer Center) for kindly providing REDD1 wild-type and knock-out MEFs. We also thank Chen Yang and Tony Martin for technical assistance in performance of the studies described herein.
This work was supported by the American Diabetes Association Pathway to Stop Diabetes Grant 1-14-INI-04 (to M. D. D.), and National Institutes of Health Grants EY023612 (to M. D. D.), DK15658 and DK13499 (to L. S. J.), and EY020895 (to P. E. F.).
- DR
- diabetic retinopathy
- REDD
- regulated in development and DNA damage
- VEGF
- vascular endothelial growth factor
- mTORC
- mTOR complex
- UTR
- untranslated region
- IRES
- internal ribosome entry site
- STZ
- streptozotocin.
REFERENCES
- 1. Sheetz M. J., King G. L. (2002) Molecular understanding of hyperglycemia's adverse effects for diabetic complications, JAMA 288, 2579–2588 [DOI] [PubMed] [Google Scholar]
- 2. The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus, N. Engl. J. Med. 329, 977–986 [DOI] [PubMed] [Google Scholar]
- 3. Miller J. W., Adamis A. P., Shima D. T., D'Amore P. A., Moulton R. S., O'Reilly M. S., Folkman J., Dvorak H. F., Brown L. F., Berse B. (1994) Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model, Am. J. Pathol. 145, 574–584 [PMC free article] [PubMed] [Google Scholar]
- 4. Bornes S., Prado-Lourenco L., Bastide A., Zanibellato C., Iacovoni J. S., Lacazette E., Prats A. C., Touriol C., Prats H. (2007) Translational induction of VEGF internal ribosome entry site elements during the early response to ischemic stress, Circ. Res. 100, 305–308 [DOI] [PubMed] [Google Scholar]
- 5. Bastide A., Karaa Z., Bornes S., Hieblot C., Lacazette E., Prats H., Touriol C. (2008) An upstream open reading frame within an IRES controls expression of a specific VEGF-A isoform, Nucleic Acids Res. 36, 2434–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morris M. J., Negishi Y., Pazsint C., Schonhoft J. D., Basu S. (2010) An RNA G-quadruplex is essential for cap-independent translation initiation in human VEGF IRES, J. Am. Chem. Soc. 132, 17831–17839 [DOI] [PubMed] [Google Scholar]
- 7. Huez I., Créancier L., Audigier S., Gensac M. C., Prats A. C., Prats H. (1998) Two independent internal ribosome entry sites are involved in translation initiation of vascular endothelial growth factor mRNA, Mol. Cell. Biol. 18, 6178–6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thoreen C. C. (2013) Many roads from mTOR to eIF4F, Biochem. Soc. Trans. 41, 913–916 [DOI] [PubMed] [Google Scholar]
- 9. Dennis M. D., Shenberger J. S., Stanley B. A., Kimball S. R., Jefferson L. S. (2013) Hyperglycemia mediates a shift from cap-dependent to cap-independent translation via a 4E-BP1-dependent mechanism, Diabetes 62, 2204–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schrufer T. L., Antonetti D. A., Sonenberg N., Kimball S. R., Gardner T. W., Jefferson L. S. (2010) Ablation of 4E-BP1/2 prevents hyperglycemia-mediated induction of VEGF expression in the rodent retina and in Muller cells in culture, Diabetes 59, 2107–2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dennis M. D., Coleman C. S., Berg A., Jefferson L. S., Kimball S. R. (2014) REDD1 enhances protein phosphatase 2A-mediated dephosphorylation of Akt to repress mTORC1 signaling, Sci. Signal 7, ra68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McGhee N. K., Jefferson L. S., Kimball S. R. (2009) Elevated corticosterone associated with food deprivation upregulates expression in rat skeletal muscle of the mTORC1 repressor, REDD1, J. Nutr. 139, 828–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rittenhouse K. D., Johnson T. R., Vicini P., Hirakawa B., Kalabat D., Yang A. H., Huang W., Basile A. S. (2014) RTP801 gene expression is differentially upregulated in retinopathy and is silenced by PF-04523655, a 19-Mer siRNA directed against RTP801, Invest Ophthalmol. Vis. Sci. 55, 1232–1240 [DOI] [PubMed] [Google Scholar]
- 14. Kimball S. R., Horetsky R. L., Jefferson L. S. (1998) Implication of eIF2B rather than eIF4E in the regulation of global protein synthesis by amino acids in L6 myoblasts, J. Biol. Chem. 273, 30945–30953 [DOI] [PubMed] [Google Scholar]
- 15. Kimball S. R., Jurasinski C. V., Lawrence J. C., Jr., Jefferson L. S. (1997) Insulin stimulates protein synthesis in skeletal muscle by enhancing the association of eIF-4E and eIF-4G, Am. J. Physiol. 272, C754–759 [DOI] [PubMed] [Google Scholar]
- 16. Brafman A., Mett I., Shafir M., Gottlieb H., Damari G., Gozlan-Kelner S., Vishnevskia-Dai V., Skaliter R., Einat P., Faerman A., Feinstein E., Shoshani T. (2004) Inhibition of oxygen-induced retinopathy in RTP801-deficient mice, Invest. Ophthalmol. Vis. Sci. 45, 3796–3805 [DOI] [PubMed] [Google Scholar]
- 17. Dennis M. D., Jefferson L. S., Kimball S. R. (2012) Role of p70S6K1-mediated phosphorylation of eIF4B and PDCD4 in the regulation of protein synthesis, J. Biol. Chem. 287, 42890–42899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dennis M. D., Schrufer T. L., Bronson S. K., Kimball S. R., Jefferson L. S. (2011) Hyperglycemia-Induced O-GlcNAcylation and Truncation of 4E-BP1 Protein in Liver of a Mouse Model of Type 1 Diabetes, J. Biol. Chem. 286, 34286–34297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gordon B. S., Steiner J. L., Lang C. H., Jefferson L. S., Kimball S. R. (2014) Reduced REDD1 expression contributes to activation of mTORC1 following electrically induced muscle contraction, Am. J. Physiol. Endocrinol. Metab. 307, E703–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fort P. E., Losiewicz M. K., Reiter C. E., Singh R. S., Nakamura M., Abcouwer S. F., Barber A. J., Gardner T. W. (2011) Differential roles of hyperglycemia and hypoinsulinemia in diabetes induced retinal cell death: evidence for retinal insulin resistance, PLoS One 6, e26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mizutani M., Gerhardinger C., Lorenzi M. (1998) Muller cell changes in human diabetic retinopathy, Diabetes 47, 445–449 [DOI] [PubMed] [Google Scholar]
- 22. Zhong Y., Li J., Chen Y., Wang J. J., Ratan R., Zhang S. X. (2012) Activation of endoplasmic reticulum stress by hyperglycemia is essential for Muller cell-derived inflammatory cytokine production in diabetes, Diabetes 61, 492–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shelton M. D., Distler A. M., Kern T. S., Mieyal J. J. (2009) Glutaredoxin regulates autocrine and paracrine proinflammatory responses in retinal glial (muller) cells, J. Biol. Chem. 284, 4760–4766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang J., Xu X., Elliott M. H., Zhu M., Le Y. Z. (2010) Muller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage, Diabetes 59, 2297–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Donovan K., Alekseev O., Qi X., Cho W., Azizkhan-Clifford J. (2014) O-GlcNAc Modification of Transcription Factor Sp1 Mediates Hyperglycemia-Induced VEGF-A Upregulation in Retinal Cells, Invest Ophthalmol Vis. Sci. 55, 7862–7873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saint-Geniez M., Maharaj A. S., Walshe T. E., Tucker B. A., Sekiyama E., Kurihara T., Darland D. C., Young M. J., D'Amore P. A. (2008) Endogenous VEGF is required for visual function: evidence for a survival role on muller cells and photoreceptors, PLoS One 3, e3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Svitkin Y. V., Herdy B., Costa-Mattioli M., Gingras A. C., Raught B., Sonenberg N. (2005) Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation, Mol. Cell. Biol. 25, 10556–10565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Long X., Lin Y., Ortiz-Vega S., Yonezawa K., Avruch J. (2005) Rheb binds and regulates the mTOR kinase, Curr. Biol. 15, 702–713 [DOI] [PubMed] [Google Scholar]
- 29. Gingras A. C., Raught B., Gygi S. P., Niedzwiecka A., Miron M., Burley S. K., Polakiewicz R. D., Wyslouch-Cieszynska A., Aebersold R., Sonenberg N. (2001) Hierarchical phosphorylation of the translation inhibitor 4E-BP1, Genes Dev. 15, 2852–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reiter C. E., Wu X., Sandirasegarane L., Nakamura M., Gilbert K. A., Singh R. S., Fort P. E., Antonetti D. A., Gardner T. W. (2006) Diabetes reduces basal retinal insulin receptor signaling: reversal with systemic and local insulin, Diabetes 55, 1148–1156 [DOI] [PubMed] [Google Scholar]
- 31. Gologorsky D., Thanos A., Vavvas D. (2012) Therapeutic interventions against inflammatory and angiogenic mediators in proliferative diabetic retinopathy, Mediators Inflam 2012, 629452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schoenberger S. D., Kim S. J., Sheng J., Rezaei K. A., Lalezary M., Cherney E. (2012) Increased prostaglandin E2 (PGE2) levels in proliferative diabetic retinopathy, and correlation with VEGF and inflammatory cytokines, Invest Ophthalmol. Vis. Sci. 53, 5906–5911 [DOI] [PubMed] [Google Scholar]
- 33. Zhou J., Wang S., Xia X. (2012) Role of intravitreal inflammatory cytokines and angiogenic factors in proliferative diabetic retinopathy, Curr. Eye Res. 37, 416–420 [DOI] [PubMed] [Google Scholar]
- 34. Aghdam S. Y., Gurel Z., Ghaffarieh A., Sorenson C. M., Sheibani N. (2013) High glucose and diabetes modulate cellular proteasome function: Implications in the pathogenesis of diabetes complications, Biochem. Biophys. Res. Commun. 432, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim M. Y., Bae J. S., Kim T. H., Park J. M., Ahn Y. H.( 2012) Role of transcription factor modifications in the pathogenesis of insulin resistance, Exp. Diabetes Res. 2012, 716425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control, Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 37. Hu J., Wu Q., Li T., Chen Y., Wang S. (2013) Inhibition of high glucose-induced VEGF release in retinal ganglion cells by RNA interference targeting G protein-coupled receptor 91, Exp. Eye Res. 109, 31–39 [DOI] [PubMed] [Google Scholar]
- 38. Li J., Zhao S. Z., Wang P. P., Yu S. P., Zheng Z., Xu X. (2012) Calcium mediates high glucose-induced HIF-1alpha and VEGF expression in cultured rat retinal Muller cells through CaMKII-CREB pathway, Acta Pharmacol. Sin 33, 1030–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fort P. E., Losiewicz M. K., Pennathur S., Jefferson L. S., Kimball S. R., Abcouwer S. F., Gardner T. W. (2014) mTORC1-independent reduction of retinal protein synthesis in type 1 diabetes, Diabetes 63, 3077–3090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aas V., Hessvik N. P., Wettergreen M., Hvammen A. W., Hallén S., Thoresen G. H., Rustan A. C. (2011) Chronic hyperglycemia reduces substrate oxidation and impairs metabolic switching of human myotubes, Biochim Biophys Acta 1812, 94–105 [DOI] [PubMed] [Google Scholar]
- 41. Koritzinsky M., Seigneuric R., Magagnin M. G., van den Beucken T., Lambin P., Wouters B. G. (2005) The hypoxic proteome is influenced by gene-specific changes in mRNA translation, Radiother. Oncol. 76, 177–186 [DOI] [PubMed] [Google Scholar]
- 42. Braunstein S., Karpisheva K., Pola C., Goldberg J., Hochman T., Yee H., Cangiarella J., Arju R., Formenti S. C., Schneider R. J. (2007) A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer, Mol. Cell 28, 501–512 [DOI] [PubMed] [Google Scholar]
- 43. Shoshani T., Faerman A., Mett I., Zelin E., Tenne T., Gorodin S., Moshel Y., Elbaz S., Budanov A., Chajut A., Kalinski H., Kamer I., Rozen A., Mor O., Keshet E., Leshkowitz D., Einat P., Skaliter R., Feinstein E. (2002) Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis, Mol. Cell. Biol. 22, 2283–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brugarolas J., Lei K., Hurley R. L., Manning B. D., Reiling J. H., Hafen E., Witters L. A., Ellisen L. W., Kaelin W. G., Jr. (2004) Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex, Genes Dev. 18, 2893–2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Williamson J. R., Chang K., Frangos M., Hasan K. S., Ido Y., Kawamura T., Nyengaard J. R., van den Enden M., Kilo C., Tilton R. G. (1993) Hyperglycemic pseudohypoxia and diabetic complications, Diabetes 42, 801–813 [DOI] [PubMed] [Google Scholar]
- 46. Levy A. P., Levy N. S., Wegner S., Goldberg M. A. (1995) Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia, J. Biol. Chem. 270, 13333–13340 [DOI] [PubMed] [Google Scholar]
- 47. Amin R. H., Frank R. N., Kennedy A., Eliott D., Puklin J. E., Abrams G. W. (1997) Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy, Invest Ophthalmol. Vis. Sci. 38, 36–47 [PubMed] [Google Scholar]
- 48. Witmer A. N., Vrensen G. F., Van Noorden C. J., Schlingemann R. O. (2003) Vascular endothelial growth factors and angiogenesis in eye disease, Prog. Retin Eye Res. 22, 1–29 [DOI] [PubMed] [Google Scholar]
- 49. Tolentino M. J., Miller J. W., Gragoudas E. S., Chatzistefanou K., Ferrara N., Adamis A. P. (1996) Vascular endothelial growth factor is sufficient to produce iris neovascularization and neovascular glaucoma in a nonhuman primate, Arch. Ophthalmol. 114, 964–970 [DOI] [PubMed] [Google Scholar]
- 50. Okamoto N., Tobe T., Hackett S. F., Ozaki H., Vinores M. A., LaRochelle W., Zack D. J., Campochiaro P. A. (1997) Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization, Am. J. Pathol. 151, 281–291 [PMC free article] [PubMed] [Google Scholar]
- 51. Salam A., Mathew R., Sivaprasad S. (2011) Treatment of proliferative diabetic retinopathy with anti-VEGF agents, Acta Ophthalmol. 89, 405–411 [DOI] [PubMed] [Google Scholar]
- 52. D'Amore P. A. (2007) Vascular endothelial cell growth factor-a: not just for endothelial cells anymore, Am. J. Pathol. 171, 14–18 [DOI] [PMC free article] [PubMed] [Google Scholar]