

Summary
Celastrol binds CIP2A and enhances CIP2A–CHIP interaction, leading to ubiquitination/degradation of CIP2A and inhibition of lung cancer cells in vitro and in vivo. Celastrol potentiates cisplatin’s efficacy by suppressing the CIP2A–Akt pathway, and therefore CIP2A inhibitors may represent novel therapeutics for cancer.
Abstract
Cancerous inhibitor of protein phosphatase 2A (CIP2A) is an oncoprotein overexpressed and inversely associated with prognosis in lung and many other human cancers. It modulates phospho-Akt and stabilizes c-Myc, and is required for cell proliferation and malignant transformation, indicating that CIP2A may play an important role in carcinogenesis. We reported here that a small compound celastrol could induce a rapid degradation of CIP2A, through the ubiquitin–proteasome pathway with the carboxyl terminus of Hsp70-interacting protein (CHIP) as the E3 ligase. Celastrol directly bound CIP2A protein and promoted CIP2A–CHIP interaction, leading to subsequent degradation of CIP2A in non-small-cell lung cancer cells. Furthermore, celastrol effectively inhibited cell proliferation and induced apoptosis in non-small-cell lung cancer cells, whereas CIP2A silencing enhanced these effects. Celastrol also suppressed tumor growth in xenograft murine models. In addition, celastrol potentiated the inhibitory effect of cytotoxic agent cisplatin on lung cancer cells in vitro and in vivo via inhibition of CIP2A–Akt pathway. These data indicate that celastrol is a CIP2A-targeting agent that may have therapeutic potentials in lung cancer.
Introduction
Lung cancer, the leading cause of cancer deaths worldwide, is a tumor of lung epithelial cells or neural crest with genetic (1) and epigenetic abnormalities (2). Identification of genetic/epigenetic alterations not only sheds new insights into the elusive molecular carcinogenesis, defines distinct cancer subsets, but also provides therapeutic targets for development of novel therapies exemplified by epidermal growth factor receptor-targeting agents (3). Though the 5-year overall survival of lung cancer of all stages combined is only 15% (4), these efforts inspire the development of more effective therapeutic approaches to combat this deadly disease.
Cancerous inhibitor of protein phosphatase 2A (CIP2A), an autoantigen previously known as KIAA1524 (5), has been shown to be an oncoprotein capable of modulating phospho-Akt (pAkt) (6,7) and stabilizing c-Myc (8). CIP2A is overexpressed in most human cancers, including lung, breast, colon, gastric, prostate cancer and neck and head carcinomas (5,7–10), and is inversely correlated with disease outcome in non-small-cell lung cancer (11), gastric cancer (10), ovarian cancer (12) and chronic myeloid leukemia (13). E2F1 can promote the expression of CIP2A, which in turn, by inhibiting protein phosphatase 2A activity, increases stabilizing serine 364 phosphorylation of E2F1. Increased activity of E2F1–CIP2A feedback renders breast cancer cells resistant to senescence induction (14). CIP2A is required for cell proliferation and transformation (7,8), and is associated with doxorubicin resistance (15). Mammary tumorigenesis is impaired in a CIP2A-deficient mouse model, and CIP2A-deficient tumors display markers of senescence induction (14). These results suggest that CIP2A may have an important role in carcinogenesis, and inactivation of CIP2A may have therapeutic potential.
Studies show that ETS1 may probably mediate high CIP2A expression in human cancer with increased EGFR-MEK1/2-ERK pathway activity (16) and c-Jun N-terminal kinase-2 regulates CIP2A transcription via ATF2 (17). However, the precise mechanisms underlying posttranslational modification and degradation of CIP2A are largely unknown, and strategy to inactivate CIP2A protein for cancer therapy is still lacking. In this study, we aimed to identify small compounds that are capable of inducing CIP2A degradation and investigate the mechanisms underlie from the natural compounds stored in our lab (7,18–23). Fortunately, we reported that a natural compound celastrol (also known as tripterine), which was isolated from a traditional Chinese medicinal herb thunder god vine or Tripterygium wilfordii Hook. F. (24), induced a rapid and proteasome-mediated degradation of CIP2A. Celastrol showed potent antilung cancer activity and enhanced the effects of cisplatin on lung cancer cells in vitro and in vivo, providing proof of principle for the development of CIP2A inhibitor to treat malignant neoplasms.
Materials and methods
Cell culture
The lung adenocarcinoma cell lines A549, NCI-H1975 and HCC827, large cell lung cancer line NCI-H460 and human embryonic kidney HEK293T cells were obtained from the American Tissue Culture Collection. Human lung squamous cell carcinoma cell line L78, lung adenocarcinoma cell lines SPC-A-1 and Glc-82, highly metastatic large cell lung cancer line 95D, hepatocellular carcinoma cell line HepG2, breast cancer cell line MCF-7 and gastric cancer cell line SGC7901 were obtained from the Cell bank of Chinese Academy of Sciences (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium or RPMI 1640 (Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (Gibco BRL), 100U/ml penicillin and 100mg/ml streptomycin as described (7).
Agents
Celastrol, MDL-28170 and MG-132 were purchased from Calbiochem (San Diego, CA). Cel-Biotin was synthesized by Bestsyn Synthetic Technologies (Shenzhen, China). Wilforol A was synthesized by WuXi PharmaTech (Shanghai, China). PS-341 was obtained from Millennium Pharmaceuticals (Cambridge, MA), Z-Ile-Glu(OtBu)-Ala-Leu-CHO (PSI) and epoxomycin were purchased from Peptide Institute (Osaka, Japan). Lysosomal pathway inhibitors (NH4Cl and chloroquine), anti-FLAG M2 Affinity gel and cisplatin were purchased from Sigma–Aldrich (St Louis, MO). Cycloheximide (CHX), LY94002 and nocodazole were obtained from Beyotime Institute of Biotechnology (Jiangsu, China). Z-VAD-FMK was obtained from Promega. 3-(4,5)-dimethylthiahiazo (-z-y1)-3, 5-di- phenytetrazoliumromide (MTT) was purchased from Amresco (Solon, OH). High Capacity Streptavidin Agarose Resins was obtained from Thermo Scientific Pierce Protein Research Products.
Antibodies
The antibodies used in this study were as follows: anti-β-actin (Sigma); anti-Casp-9, Casp-8, PARP, pS6K, DYKDDDDK(Flag) Tag antibody (Cell Signaling Technology); anti-CIP2A, AKT1/2/3, CHIP, pAKT1/2/3 (Ser 473)-R, normal mouse IgG (Santa Cruz Biotechnology); monoclonal antibody against mono- and polyubiquitinylated conjugates (FK2) (Enzo Life Sciences); HRP-labeled Streptavidin, Myc Tag antibody (Beyotime Institute of Biotechnology); Fluorescein-Conjugated AffiniPure Goat Anti-Mouse IgG (H+L) (Zhong Shan Golden Bridge Biological Technology CO.,LTD).
Cell proliferation, apoptosis and clonogenic assays
Cell proliferation was evaluated using MTT assay as described (19). The potential synergistic, additive or antagonistic effect between celastrol and cisplatin was assessed using the Calcusyn Software (Biosoft, Cambridge, UK) as described (25). Cell apoptosis was measured using Annexin V Apoptosis Detection Kit (BD Biosciences, San Jose, CA) according to the manufacturer’s instruction. Briefly, the cells were treated with or without celastrol for 24h, harvested, and after washing twice with cold phosphate-buffered saline (PBS), the cells were resuspended in binding buffer and stained with annexin V-PE and 7-aminoactinomycin D at room temperature for 15min in dark, and then detected by flow cytometry (BD FACSCallbur) within 1h. For clonogenic assay, A549 cells transfected with negative control (NC) or CIP2A-specific small interfering RNA (siRNA) for 72h were seeded in triplicate onto 35mm plates (300 cells per plate) and treated with 0.5–2.5 μΜ celastrol. After 9 days of culturing, the colonies were fixed in methanol and stained with 0.005% crystal violet (Sigma). The colony-forming units with >50 cells were counted using a light microscope (7).
siRNA assays and cell transfection
Cells were transfected with indicated double-stranded siRNA oligonucleotides as described (7). The siRNA sequences were 5′-GAAGAAGCG CTGGAACAGC-3′ [carboxyl terminus of Hsp70-interacting protein (CHIP) siRNA] (26), 5′-CUGUGGUUGUGUUUGCACUTT-3′ (CIP2A siRNA) (7) and 5′-UUCUCCGAACGUGUCACGUTT-3′ (NC siRNA). For transfection, HEK293T cells were transfected with target plasmids using Lipofectamine 2000 (Invitrogen) according to the recommended protocol by the manufacturer.
Lentiviral constructs and stable cell lines
Lentivirus was packaged by cotransfection of pLKO.1 short hairpin RNA plasmid targeting CIP2A messenger RNA (mRNA), psPAX2 packaging plasmid and pMD2.G envelope plasmid into 293FT cells by lipofectamine 2000 (Invitrogen). The sequences were as follow: NC short hairpin RNA, 5′-CAACAAGATGAAGAGCACCAA-3′; shCIP2A 1#, 5′-TGCGGCACTTGG AGGTAATTT-3′ and shCIP2A 2#, 5′-GACAGAAACTCACACGACTAT-3′. Media containing lentiviral particles was harvested and pooled from cells at 24 and 48h after transfection, and spinned to remove any 293FT cells. A549 cells were infected by lentiviral particle solution in the presence of 8 μg/ml polybrene. Puromycin at 1 μg/ml was used to screen for stable CIP2A or non-target (NC) knockdown cell lines.
Proteasome activity assay
A549 cells (5000) were seeded in 96-well plates for 24h, treated with PS-341 or celastrol at indicated concentrations and timepoints, followed by additional 2h incubation with Suc-LLVY-AMC (chymotrypsin-like activity substrate, at 40 μM) or Z-LLE-AMC (peptidylglutamyl peptide-hydrolyzing activity substrate, at 40 μM). The free hydrolyzed 7-amino-4-methylcoumarin was measured using Synergy 4 hybrid microplate reader (BioTek) at 380/460nm.
Immunoblotting and immunoprecipitation
Cells were treated with indicated protocols and then lysed in radioimmunoprecipitation assay lysis buffer [50mM Tris-HCl pH 7.4, 150mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% deoxycholate, 1% Triton X-100, 1mM ethylenediaminetetraacetic acid, 1 mM phenylmethanesulfonyl fluoride, 5mM NaF, 1mM sodium vanadate and protease inhibitors cocktail (Sigma)] or SDS loading buffer. Equal amounts of proteins were subjected to SDS–polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane and immunoblotted with indicated antibodies. For preparation of Triton X-100 soluble and insoluble fractions, cells were pretreated with PS-341 or MG132 followed by the treatment of celastrol, lysed in radioimmunoprecipitation assay lysis buffer. After centrifugation, the lysates were collected as Triton X-100 soluble fraction, and the pellets were boiled in SDS loading buffer at 99°C for 15 min to dissolve the Triton X-100 insoluble proteins. For immunoprecipitation, cells were harvested in lysis buffer (Beyotime, P0013). Cell lysate (1ml) containing 100–500 μg total protein was incubated with 1–2 μg primary antibody for 1 h at 4°C and added 20 μl protein A/G-conjugated beads (Santa Cruz Biotechnology) at 4°C overnight. After washed with lysis buffer or PBS, the pellet was resuspended in SDS sample buffer. The cells transfected with Flag-tagged expression constructs were lysed in lysis buffer, incubated with anti-FLAG M2 affinity gel (20–30 μl/ml cell lysates) at 4°C on a shaker overnight. The precipitated proteins were eluted with SDS loading buffer and analyzed by western blot.
Immunofluorescence microscopy
Cells were grown on cover slides and fixed with 4% formaldehyde, then permeabilized with 0.3% Triton X-100/PBS at room temperature. Cells were blocked in 5% bovine serum albumin/PBS for 30min at room temperature and then incubated with primary antibody overnight at 4°C, followed by incubation with fluorescein isothiocyanate-conjugated secondary antibodies for 1.5h at room temperature. 4′,6-diamidino-2-phenylindole was used to stain nuclei. The coverslips were mounted with antifade medium and observed under a confocal microscope (Zeiss LSM 510 Meta).
Protein purification and in vitro ubiquitination assays
For protein purification, Flag-tagged proteins were transfected into HEK293T cells and purified using an anti-FLAG M2 Affinity Gel (Sigma) according to the manufacturer’s protocol, then the precipitated proteins were eluted by competition with Flag peptide. For in vitro ubiquitination assays, 20 μl M2 beads of Flag-CIP2A were incubated in 50 μl reaction mixture [50mM Tris-HCl, pH 7.4, 5mM MgCl2, 2mM adenosine triphosphate, 0.4mM dithiothreitol, 0.5 μg E1 (Boston Biochem), 2.5 μg Ubc4 (Boston Biochem), 2.5 μg ubiquitin (Boston Biochem), 1 μg purified Flag-CHIP protein] at 30°C for 2h. After incubation, the reaction was terminated with SDS loading buffer, and the ubiquitinated CIP2A was detected using western blot analysis. For celastrol binding, the CIP2A-M2 beads were incubated with celastrol (0.5mM) and then washed to remove free celastrol before in vitro ubiquitination assay.
Streptavidin agarose affinity assay
Cells upon Cel-Biotin were lysed with lysis buffer (Beyotime). Cell lysates were incubated with streptavidin agarose overnight at 4°C. After washing with lysis buffer, streptavidin agarose beads were resuspended in SDS loading buffer. For celastrol competition, A549 cell lysates were pretreated with celastrol (100, 200 μM) for 1h, followed by 50 μM Cel-Biotin treatment for 2h at 4°C, and streptavidin agarose affinity assays were performed.
Murine models
All animal studies were conducted according to protocols approved by the Animal Ethics Committee of the Institute of Zoology, Chinese Academy of Sciences. BALB/c nude mice (5–6 weeks old) used in this study were bred and maintained in a specific pathogen-free environment. A549 (6×106) or H1975 (3×106) cells were injected subcutaneously into the right flank of nude mice. To test the in vivo antilung cancer efficacy of celastrol, animals were randomized into four groups (n = 10 for each group) and treated intraperitoneally with vehicle [10% dimethyl sulfoxide, 70% cremophor/ethanol (3:1) and 20% PBS] or celastrol (1 or 3mg/kg daily or 5mg/kg twice a week for 3 weeks). To test the combined effect of celastrol and cisplatin, animals were randomized into four groups (n = 8 for each group) and treated intraperitoneally with vehicle, celastrol (1mg/kg, every other day), cisplatin (1mg/kg, every other day) or combination of the two. Tumor size was measured every alternate day with electronic caliper and calculated by the formula: volume (mm3) = ½ (width)2 × length. After treatment of indicated days, the mice were euthanized by cervical dislocation. Tumor tissues were excised, homogenated and lysed for western blot analysis.
Statistical analysis
Differences between data groups were evaluated for significance using two-sided Student’s t-test. The tumor volume was analyzed with one-way analysis of variance and independent sample t-test using the software SPSS 12.0 for Windows (Chicago, IL). P values <0.05 were considered statistically significant. All experiments were repeated at least three times and the data are presented as the mean ± SD unless noted otherwise.
Results
Celastrol triggers CIP2A protein degradation
In our previous work, we reported that (7) a natural compound rabdocoetsin B is able to downregulate CIP2A at mRNA levels at relatively high concentrations (5–20 μM). In this work, we found that, treatment with celastrol (Figure 1A) at 1–5 μM for 24h could downregulate CIP2A expression in A549 cells (Figure 1B). Treatment with celastrol at 5 μM for 6–24h also downregulated CIP2A in lung cancer cells HCC827, H1975 and H460, hepatocellular carcinoma HepG2, breast cancer MCF-7 and gastric cancer SGC7901 cells (Figure 1C), indicating that celastrol-triggered CIP2A downregulation is not a cell type-specific event. We further showed that celastrol (5 μM) induced CIP2A downregulation in a time-dependent fashion in that CIP2A was markedly reduced in 1–2h and became undetectable in 4h (Figure 1D). Celastrol also decreased CIP2A expression in HEK293T cells transfected with Flag-CIP2A (Figure 1E). We therefore carefully dissected the mechanisms underlying celastrol-triggered CIP2A downregulation in this study.
Fig. 1.
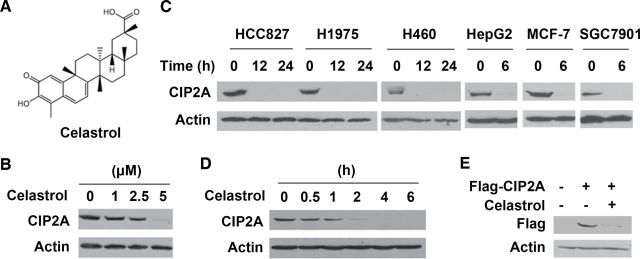
Celastrol induces degradation of CIP2A. (A) Chemical structures of celastrol. (B) A549 cells were treated with celastrol at indicated concentrations for 24h, lysed, and western blotting was performed using anti-CIP2A and anti-Actin antibodies. (C) Western blot analyses of CIP2A expression in lung cancer cells HCC827, H1975 and H460, hepatocellular carcinoma HepG2, breast cancer MCF-7 and gastric cancer SGC7901 cells treated with 5 μM of celastrol for indicated timepoints. (D) A549 cells were treated with celastrol at 5 μM for indicated timepoints. Cell lysates were prepared and analyzed by immunoblotting with antibodies to CIP2A and Actin. (E) HEK293T cells transfected with Flag-CIP2A plasmid for 48h were treated with celastrol (5 μM) for 6h, lysed, and analyzed by western blot assay with antibodies to Flag and Actin.
Celastrol-induced CIP2A degradation is mediated by the ubiquitin–proteasome pathway
Understanding of the mechanism by which CIP2A undergoes proteolytic breakdown is important to develop effective strategies for elimination of CIP2A for lung cancer therapy. To analyze the mechanisms of CIP2A decrease, we first tested the effect of celastrol on CIP2A at mRNA level in A549, L78 and H1975 cells, and reported that this compound did not markedly affect CIP2A expression (Figure 2A). In A549 cells upon protein synthesis inhibitor cycloheximide, CIP2A was decreased only very slightly at 10h (Figure 2B, left panel). However, in cells coincubated with cycloheximide and celastrol, CIP2A was downregulated at 2–6h and became undetectable in 10h (Figure 2B, right panel), indicating that celastrol can cause its proteolytic degradation.
Fig. 2.

Celastrol-triggered CIP2A turnover is mediated by ubiquitin–proteasome pathway. (A) Reverse transcription–polymerase chain reaction assays of cells upon celastrol at 5 μM for 6h. (B) Effects of 50 μg/ml cycloheximide (CHX) alone or in combination with 5 μM celastrol on CIP2A expression evaluated by western blotting in A549 cells. (C and D) A549 cells were preincubated with z-VAD-fmk (20 μM) (C) or MDL-28170 (MDL, 50 μM) (D) for 2h, followed by celastrol (5 μM) treatment for 6h. Cell lysates were subjected to immunoblotting analysis with indicated antibodies. (E) A549 cells were pretreated with chloroquine (Chl; 80 μM) or NH4Cl (20mM) for 2h, followed by addition of celastrol (5 μM) for 6h. Cells were lysed and analyzed by western blot using antibodies against CIP2A and Actin. (F) Western blot analysis of CIP2A expression in A549 cells incubated with PS-341 or epoxomycin (EPO) at indicated concentrations for 6h. (G) Western blot analysis of CIP2A expression in A549 cells preincubated with PS-341 (100nM), MG-132 (10 μM), EPO (100nM), PSI (10 μM) for 2h followed by treatment of celastrol (5 μM) for additional 6h. (H) A549 cells were pretreated with or without PS-341(100nM) for 2h, followed by celastrol incubation for 1h. Cell lysates were subjected to immunoprecipitation with CIP2A antibody followed by immunoblot with antibodies against CIP2A and ubiquitin. *Ubiquitinated CIP2A.
Caspase and calpain are families of cysteine proteases, which have important roles in cell death and cleavage of substrate proteins (27). To determine the role for caspases to play in CIP2A proteolysis, A549 cells were pretreated with pan-caspase inhibitor z-VAD-fmk (z-VAD) followed by celastrol treatment; however, z-VAD did not inhibit celastrol-induced CIP2A degradation (Figure 2C). Similarly, calpain inhibitor III MDL-28170 (MDL) could not prevent CIP2A catabolism (Figure 2D). Lysosome is an organelle containing a broad array of secluded proteases that can mediate intracellular proteolysis (28). The lysosomal protease inhibitors, NH4Cl and chloroquine, were employed to suppress the organelle before A549 cells were incubated with celastrol. As shown in Figure 2E; however, these inhibitors failed to prevent CIP2A degradation.
The ubiquitin–proteasome pathway provides the main pathway for degradation of intracellular proteins in eukaryotic cells (28). We then tried to use classic proteasome inhibitors in vitro to examine the effect of these inhibitors on the degradation of CIP2A induced by celastrol. To our surprise, although proteasome inhibitors PS-341, MG-132, PSI and epoxomycin (29) did not influence CIP2A stability within 6 h (Figure 2F and G), pretreatment of A549 cells with these compounds dramatically blocked celastrol-triggered CIP2A degradation (Figure 2G). We further showed that in A549 cells, celastrol increased polyubiquitinated CIP2A, whereas PS-341 enhanced this effect (Figure 2H). These data indicate that celastrol-triggered CIP2A degradation is mediated by the ubiquitin–proteasome pathway.
Unveiling CHIP as the E3 ubiquitin ligase for celastrol-induced CIP2A degradation
During the course of study on the CIP2A degradation mechanisms, an interesting experimental phenomenon was discovered to show that, in cells pretreated with PS-341 followed by celastrol treatment, CIP2A was shifted from the supernatant of cell lysates to the Triton X-100 insoluble pellet (Figure 3A, left panel). Similar result was also observed in A549 cells incubated with celastrol and MG-132 (Figure 3A, right panel). We then investigated the localization of CIP2A protein upon celastrol alone or in combination with proteasome inhibitor. By immunofluorescence analysis, we showed that CIP2A exhibited a diffuse cytoplasmic localization pattern (Figure 3B), in consistent with previous reports (5,8). Interestingly, in >90% of A549 cells exposed to celastrol in combination with PS-341, CIP2A staining shifted to a large juxtanuclear inclusion body (Figure 3B), suggestive of aggresome formation (30). It was reported that disruption of microtubules could block, whereas inhibition of proteasome could promote aggresome formation (30). We found that microtubular depolymerizing agent nocodazole inhibited CIP2A juxtanuclear inclusion body formation in cells treated with celastrol and PS-341, turning the juxtanuclear structure into small aggregates (Figure 3B), further indicating that the juxtanuclear inclusion body is aggresome. Studies showed that the E3 ligase CHIP plays a critical role in the aggresome pathway (31). We therefore tested whether CHIP participated in CIP2A aggresome formation by siRNA-mediated silencing (Figure 3C). Interestingly, our results showed that CHIP silencing resulted in inhibition of CIP2A aggresome formation in cells treated with both PS-341 and celastrol and reversal of CIP2A downregulation in cells treated with celastrol (Figure 3D), suggesting that CHIP promotes sequestration of CIP2A to form aggresome and involves in CIP2A stability.
Fig. 3.

CHIP is the ubiquitin E3 ligase mediating celastrol-triggered CIP2A degradation. (A) A549 cells were pretreated with 100nM PS-341 or 10 μM MG-132 for 2h, followed by celastrol treatment for additional 6h, lysed, and western blot was conducted using supernatant of cell lysates or Triton X-100-insoluble pellet, as described in Materials and methods. (B) Celastrol causes formation of CIP2A aggresome at the presence of PS-341. A549 cells pretreated with PS-341 (100nM) and/or nocodazole (10 μg/ml) for 2h, followed by celastrol (5 μM) incubation for 6h. The cells were then analyzed by immunofluorescence assay using an anti-CIP2A, a fluorescein isothiocyanate-conjugated secondary antibody and 4′,6-diamidino-2-phenylindole (DAPI). (C and D) A549 cells were transfected with negative control (NC) or CHIP-specific siRNA (100nM) for 72h and CHIP expression was evaluated by western blot (C). The cells were then pretreated with PS-341 (100nM) for 2h, followed by celastrol (5 μM) treatment for 6h, and assessed by immunofluorescence assay using an anti-CIP2A antibody and DAPI (D). (E) A549 cells were transfected with NC or CHIP-specific siRNA (100nM) for 72h, followed by treatment with 5 μM celastrol for indicated timepoints. The cells were lysed, and western blot was performed to detect the expression of CIP2A and CHIP. (F) A549 cells were treated with 5 μM celastrol (Cel) for 1h. Whole cell lysates were immunoprecipitated with anti-CIP2A antibody. Immunoprecipitates were then subjected to western blot using indicated antibodies. (G and H) HEK293T cells were cotransfected with Flag-CIP2A and Myc-CHIP constructs for 72h, followed by celastrol (5 μM) treatment for 1h, lysed, and the lysates were immunoprecipitated with anti-Flag M2 beads (G) or anti-Myc antibody (H). Immunoprecipitates were subjected to western blots with anti-Flag or anti-Myc antibody. (I) Flag-CIP2A or Flag-CHIP were transfected into HEK293T cells, proteins were purified by Flag-M2 affinity gel and in vitro ubiquitination assay was performed as described in Materials and methods. *Ubiquitinated CIP2A.
CHIP is a chaperone-binding ubiquitin ligase (32). Our above data pointed to the possibility that CHIP might be an E3 ligase of CIP2A and play a role in celastrol-triggered CIP2A degradation. To test this hypothesis, A549 cells were transfected with CHIP-specific siRNA followed by celastrol treatment. We reported that in cells transfected with NC siRNA, celastrol caused CIP2A degradation within 1–4h (Figure 3E, left panel), whereas in cells transfected with CHIP-specific siRNA, CIP2A degradation was markedly inhibited (Figure 3E, right panel). Furthermore, coimmunoprecipitation and western blot analysis revealed that in A549 cells the binding affinity between CIP2A and CHIP was weak, whereas celastrol could dramatically enhance CIP2A–CHIP interaction (Figure 3F). We also confirmed this interaction in HEK293T cells cotransfected with Flag-CIP2A and Myc-CHIP, and reciprocal coimmunoprecipitation experiments demonstrated that when celastrol was presented, CIP2A–CHIP interaction was dramatically enhanced (Figure 3G and H). An in vitro ubiquitination assay also showed that CHIP was able to directly induce ubiquitination of CIP2A (Figure 3I). These results indicate that CHIP is the ubiquitin E3 ligase involved in celastrol-induced CIP2A degradation.
Celastrol interacts directly with CIP2A protein and promotes its ubiquitination by CHIP
Since celastrol promotes the interaction of CIP2A with its E3 ligase CHIP and led to subsequent degradation of CIP2A, we next explore the detailed mechanisms involved. Previous studies reported that the C2 on A-ring and C6 on B-ring of celastrol had a high susceptibility toward a nucleophilic attack, suggesting that one or both of these carbons could interact with the nucleophilic amino acids residues (e.g. cysteine) of target proteins to form covalent adducts (33,34). We showed that the C6 group might be required for celastrol-triggered CIP2A catabolism because wilforol A (Figure 4A, left panel), which bears an altered quinone methide (35), failed to induce CIP2A turnover (Figure 4B). Furthermore, a biotinylated celastrol (Cel-Biotin, Figure 4A, right panel), which retained the biological activity and could be used as an affinity probe to identify the potential targets of celastrol (36), was synthesized and added into A549 cells to test whether celastrol had the ability to bind CIP2A protein. We found that CIP2A was pulled down by streptavidin agarose in A549 cells (Figure 4C). When the whole cell lysates of A549 cells were immunoprecipitated with an anti-CIP2A antibody, CIP2A-bound celastrol was detected by denaturing gel electrophoresis and western blot analysis using horseradish peroxidase-conjugated streptavidin (Figure 4D), indicating that celastrol could bind CIP2A covalently (37,38). CIP2A/Cel-Biotin binding could be attenuated by unlabeled celastrol competition (Figure 4E), and an in vitro binding experiment using Flag-CIP2A protein purified from HEK293T cells and Cel-Biotin coincubation further confirmed the CIP2A/Cel-Biotin covalent binding (Figure 4F). To directly demonstrate the role for celastrol binding to play in CIP2A degradation, another in vitro ubiquitination assay was conducted. The data showed that CHIP was able to directly induce ubiquitination of CIP2A, and more importantly, preincubation of CIP2A with celastrol (followed by washing out this agent and adding CIP2A to the system) markedly potentiated this effect (Figure 4G), suggesting that the covalent binding of celastrol to CIP2A promotes the turnover of this protein.
Fig. 4.
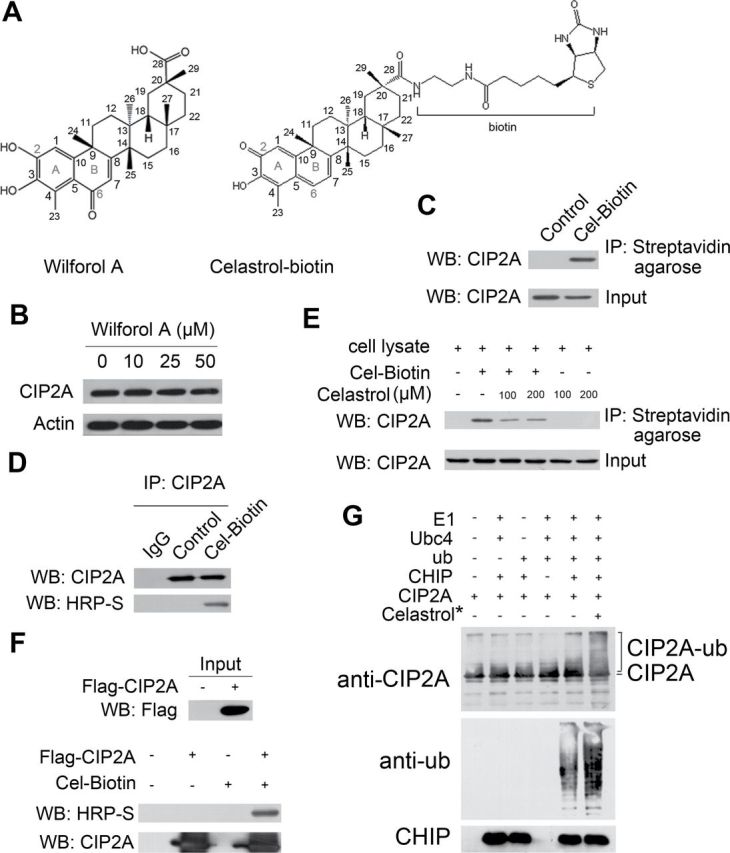
Celastrol directly binds CIP2A and promotes its ubiquitination. (A) Chemical structure of wilforol A and biotinylated celastrol (Cel-Biotin). (B) Western blot analysis of CIP2A expression from lysates of A549 cells treated with or without wilforol A for 6h. (C) A549 cells were treated with 50 μM Cel-Biotin for 6h, and the lysates were subjected to immunoprecipitation with streptavidin agarose followed by western blotting using an anti-CIP2A antibody. (D) A549 cells were treated without or with Cel-Biotin (50 μM) for 6h, and lysates were immunoprecipitated with CIP2A antibody. Western blot was performed using an anti-CIP2A antibody or horseradish peroxidase-conjugated streptavidin (HRP-S). (E) A549 cell lysates were incubated with Cel-Biotin (50 μM) in the presence or absence of celastrol (100 or 200 μM) for 2h and subjected to streptavidin agarose affinity analysis. Captured proteins were inmmunoblotted using an anti-CIP2A antibody. (F) Flag-CIP2A purified by anti-Flag M2 beads from 293T cells transfected with Flag-CIP2A was incubated with Cel-Biotin (50 μM) for 1.5h, and western blot was conducted using indicated antibodies. (G) Anti-Flag M2 beads with Flag-CIP2A were incubated with celastrol (0.5mM) at 4°C for 2h and then washed stringently to remove free celastrol before in vitro ubiquitination assay.
Celastrol exhibits potent antilung cancer activity in vitro and in vivo
Inhibition of CIP2A protein has been shown to be associated with the anticancer effect of several compounds (6,7). Although our above data indicated that CIP2A was a direct target of celastrol, the antilung cancer effect of this compound remained unclear. We therefore examined the effect of celastrol on lung cancer cells by MTT assay and reported that, celastrol possessed the ability to inhibit proliferation of lung cancer cell lines, and the IC50 values for A549, NCI-H1975, H460, HCC827, SPC-A-1, GLC-82, L78 and 95D cells were 2.78, 1.66, 2.20, 1.30, 1.80, 1.08, 1.48 and 1.80 μM, respectively. By annexin V/flow cytometry analysis, we found that treatment with celastrol at 2–5 μM induced apoptosis in A549 and H1975 cells (Figure 5A). To test the in vivo therapeutic efficacy of celastrol, nude mice were injected subcutaneously with H1975 or A549 cells, and when tumor reached a palpable size, the animals were randomized and treated intraperitoneally with celastrol at 1 or 3mg/kg daily or 5mg/kg twice a week for 3 weeks. We showed that compared with vehicle control, celastrol at 3 and 5mg/kg significantly inhibited tumor growth (Figure 5B and C). In samples isolated from these nude mice, we found that celastrol also induced degradation of CIP2A (Figure 5D and E).
Fig. 5.

Celastrol shows potent antilung cancer activity in vitro and in vivo. (A) A549 and H1975 cells were treated with celastrol at 2 and 5 μM for 24h and detected by annexin V/7-aminoactinomycin D (7-AAD) staining and flow cytometry. (B) H1975 cells were inoculated subcutaneously into the right flank of nude mice, which were treated with indicated concentrations of celastrol intraperitoneally. Tumor volume was estimated every 3 days. Data are shown as mean ± standard error of the mean; n = 10 for each group; qd, once per day. *P = 0.019, **P = 0.003, Student’s t-test. (C) Efficacy of celastrol on tumor growth in nude mice injected with A549 cells; n = 10 for each group; qd, once per day. **P = 0.002, Student’s t-test. (D and E) Tumor samples from mice inoculated with H1975 (D) or A549 (E) cells were harvested, and western blot was performed to test the expression of CIP2A. (F) A549 cells were transfected with negative control (NC) or CIP2A-specific siRNA (50nM) for 72h, followed by celastrol (3 and 3.5 μM) treatment for 24h, and cell proliferation was assessed by MTT analysis. The mean ± SD of three independent experiments is shown. **P < 0.01, Student’s t-test. (G) A549 cells were transfected with NC or CIP2A-specific siRNA (50nM) for 72h and then cultured for 24h in the presence (3 and 3.5 μM) or absence of celastrol. Cell lysates were subjected to immunoblotting using indicated antibodies. (H and I) A549 cells were transfected with NC or CIP2A-specific siRNA and then cultured in the presence or absence of celastrol for 9 days, followed by flat plate colony formation assay. (H) Representatives of light microscopy images. (I) Quantification of the number of colonies with a number >50 cells from three separate experiments. **P < 0.01. Percentage of colonies in CIP2A knockdown cells compared with control cells was shown. (J) A549 cells were incubated with CIP2A short hairpin RNA (shRNA) or NC shRNA stable transfectants for 24h in the presence (1 and 2.5 μM) or absence of celastrol. The apoptotic cells were detected by annexin V/flow cytometry; n = 3, Student’s t-test, *P < 0.05, **P < 0.01. Error bars, standard deviation.
We next investigated CIP2A’s role in celastrol-induced inhibitory effects on lung cancer cells. Firstly, A549 cells were transfected with CIP2A-specific siRNA for 72h, followed by celastrol treatment for additional 24h, and cell proliferation was assessed by MTT assay. We showed that knockdown of CIP2A significantly enhanced celastrol-induced inhibition of cell proliferation (Figure 5F). Also, compared with NC siRNA-treated cells, depletion of CIP2A resulted in potentiated cleavage of poly (ADP ribose) polymerase (PARP) in A549 cells upon celastrol (Figure 5G), suggestive of the enhancement of apoptotic effect. A colony formation assay was also performed in A549 cells transfected with NC or CIP2A-specific siRNA followed by celastrol treatment, and the results showed that colonies formed by CIP2A knockdown cells were significantly reduced compared with control cells (Figure 5H and I). Using short hairpin RNA lentiviral vectors targeting different regions of CIP2A mRNA transcript, we generated two CIP2A silencing cell lines that were further treated with celastrol. We showed that at low dosage (1–2.5 μM), celastrol induced more apoptosis in CIP2A knockdown cells than in control cells (Figure 5J). These results indicate that CIP2A may play an important role in celastrol-induced antiproliferative and proapoptotic effects on lung cancer cells.
Celastrol inhibits CIP2A–pAKT pathway and synergizes with cisplatin in suppression of lung cancer cells
CIP2A can modulate pAKT pathway (6,7), which is constitutively activated in lung cancer cells and promotes cellular survival and resistance to chemotherapeutic agents such as cisplatin (39,40). We showed that in A549 cells, celastrol was capable of downregulating pAKT and its downstream target pS6K, but exhibited little effect on total AKT protein (Figure 6A). Our results also showed that Casp-9 and Casp-8 were activated and Casp-3 substrate PARP was cleaved in lung cancer cells treated with celastrol (Figure 6A). In A549 cells treated with celastrol and a pAKT inhibitor LY294002, the expression of CIP2A, pAKT and pS6K was downregulated, and the cleavage of PARP proteins was enhanced (Figure 6B). We next tested the combined effect of celastrol and cisplatin on lung cancer cells. To do this, A549 cells were treated with celastrol and/or cisplatin, evaluated by MTT assay, and the results were assessed using the Calcusyn Software (Biosoft). The dose–effect curves of single or combined drug treatment were analyzed by the median-effect method, where the combination indexes <, = and >1 indicate synergistic, additive and antagonistic effects, respectively. The results showed that celastrol at 3–3.5 μM synergistically enhanced the effect of cisplatin at 10–50 μM on lung cancer cells, with the combination index values <1 (Figure 6C and 6D). In consistent with this observation, combined use of celastrol and cisplatin enhanced cleavage of PARP (Figure 6E). In nude mice bearing A549 cells, cisplatin at 1mg/kg per 2 days had little effect on tumor growth and could not downregulate CIP2A and pAKT, whereas celastrol at 1mg/kg per 2 days slightly inhibited tumor growth and reduced CIP2A and pAKT (Figure 6F and G). Importantly, when these two compounds were used in combination, the inhibitory effect was significantly enhanced (Figure 6F and G), indicating that celastrol and cisplatin exert synergistic effect in vivo.
Fig. 6.
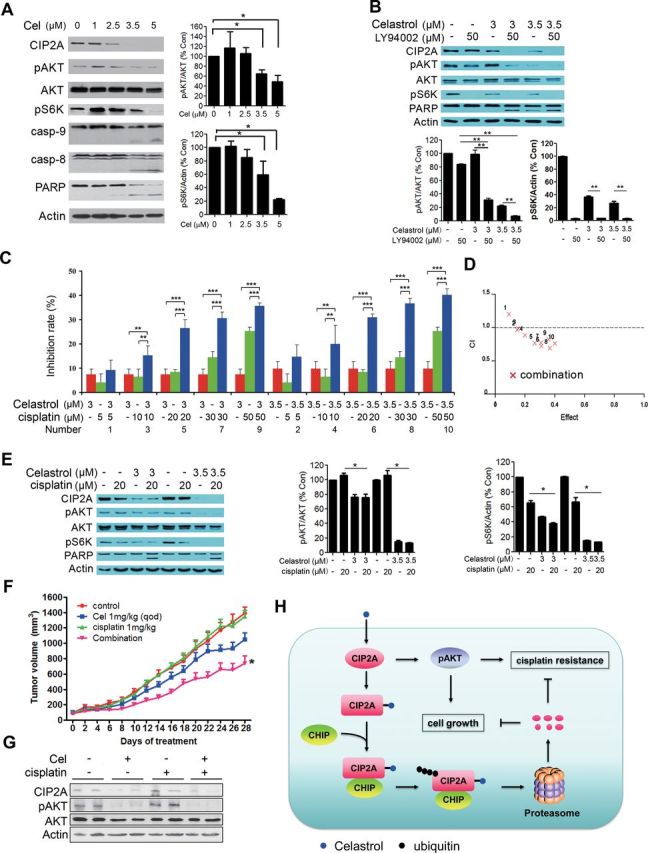
Celastrol enhances cisplatin-caused inhibitory effect on lung cancer cells. (A) A549 cells were treated with celastrol at indicated concentrations for 24h, and cell lysates were subjected to western blot using indicated antibodies (left panel). Right panels showed the densitometry analysis of western blot. The data were represented as mean ± SD of three independent experiments. *P < 0.05, Student’s t-test. (B) A549 cells were treated with celastrol and/or LY94002 for 24h, and the whole cell lysates were subjected to western blot using indicated antibodies. Results of densitometry analysis of three independent experiments were shown in bottom panel. Student’s t-test, **P < 0.01. Error bars, standard deviation. (C) A549 cells were treated with celastrol and/or cisplatin for 24h, and cell proliferation was detected by MTT assay (n = 3). Two-way analysis of variance, **P < 0.01, ***P < 0.001. Error bars, standard deviation. (D) Combination index (CI) plots corresponding to C were generated by the Chou–Talay method and Calcusyn software. The numbers 1–10 were corresponded to the number labeled in C representing different treatment combinations. The results indicate that celastrol at 3–3.5 μM synergistically enhances the inhibitory effect of cisplatin at 10–50 μM, with the CI values <1. (E) A549 cells were treated with celastrol and cisplatin for 24h, and the whole cell lysates were subjected to western blot with indicated antibodies (n = 3). Middle and right panels show the results of densitometry analysis. Student’s t-test, *P < 0.05. Error bars, standard deviation. (F) Nude mice bearing A549 cells were treated with celastrol and/or cisplatin intraperitoneally (n = 8 for each group), tumor volume was measured every 2 days. Data are shown as mean ± standard error of the mean; qod, every other day. *Combination group versus celastrol treatment group, P = 0.029; combination versus cisplatin group, P < 0.001, Student’s t-test. (G) Tumor samples from mice were harvested, homogenated and lysed for western blot using indicated antibodies. (H) Schematic representation of the mechanism of the action of celastrol in lung cancer.
Discussion
Little insight currently exists about the precise mechanisms of CIP2A catabolism in cancer cells. In fact, this represents a major unexplored area in the newly emerging CIP2A field. Here, we reported that celastrol was capable of inducing a rapid degradation of CIP2A (Figure 1), a phenomena that could not be blocked by inhibitors of caspases, calpain or lysosome (Figure 2C–E). Interestingly, proteasome inhibitors including PS-341, MG-132, PSI and epoxomycin, markedly suppressed celastrol-induced CIP2A turnover (Figure 2G), indicating that CIP2A degradation is mediated by the ubiquitin–proteasome pathway.
As a U-box-type E3 ligase, CHIP can induce ubiquitination and degradation of substrates including oncogenic proteins and act as a tumor suppressor in breast cancer (41). Recent evidence suggested a previously unexpected critical role of CHIP in the aggresome pathway (31). In this study, we found that CHIP was involved in CIP2A aggresome formation (Figure 3A–D), as shown that CHIP silencing resulted in inhibition of CIP2A aggregation in cells treated with both PS-341 and celastrol. Therefore, we speculate that CHIP may regulate CIP2A stability. In line with this, we observed that CHIP silencing resulted in reversal of CIP2A downregulation in cells treated with celastrol (Figure 3E). Interestingly, celastrol dramatically enhanced CIP2A–CHIP binding affinity (Figure 3F–H). In an in vitro ubiquitination assay, we showed that CHIP was able to induce ubiquitination of CIP2A (Figure 3I). Therefore, CHIP may play a role in determining CIP2A’s fate in cancer cells treated with celastrol (Figure 6H). Although our results indicate that celastrol is able to covalently bind CIP2A, which promotes CIP2A ablation (Figure 4), the exact binding sites of CIP2A remain to be determined. Furthermore, the mechanism underlying the potentiation of CIP2A–CHIP interaction by celastrol is also obscure. These questions warrant further investigation.
Celastrol has been shown to be a proteasome inhibitor in prostate cancer cells (42). It could accumulate ubiquitinated proteins, which was presented as evidence for proteasome inhibition. However, others argued that celastrol-caused accumulation of ubiquitinated proteins might be a result of HSP90 inhibition and stress response (43), and the activity of celastrol in inhibition of proteasome is much weaker than MG-132 and lactacystin (44,45). We tested the effects of celastrol on proteasome activities in intact lung cancer cells, and found that at 5 μM celastrol only reduced the chymotrypsin-like activity by 7% at 2h (Supplementary Figure S1A, available at Carcinogenesis Online), at which timepoint the CIP2A protein was dramatically downregulated (Figure 1D). In U2OS cells stably expressing G76V-ubiquitin that are able to identify proteasome inhibitors by measuring green fluorescent protein fluorescence (46), treatment with PS-341 at 100nM for 12h significantly accumulated green fluorescent protein. However, celastrol treatment (5 μM for 12h) did not obviously enhance the green fluorescent protein fluorescence (Supplementary Figure S1B, available at Carcinogenesis Online). In addition, celastrol did not markedly potentiate the inhibitory effect of PS-341 on chymotrypsin-like (Supplementary Figure S1C, available at Carcinogenesis Online) and peptidylglutamyl peptide-hydrolyzing (Supplementary Figure S1D, available at Carcinogenesis Online) activities in intact lung cancer cells. These results suggest that celastrol is at most a weak inhibitor of proteasome, which is not the main target of celastrol in lung cancer.
Celastrol has potent anticancer effect, e.g. inhibiting proliferation and inducing apoptosis in various cancer cells. Its in vivo anticancer efficacy has also been demonstrated in animal models for prostate cancer (42), melanoma (47) and glioma (48). However, the antilung cancer effect of celastrol remains unclear. In this study, we showed that celastrol suppressed proliferation and induced apoptosis in a variety of non-small-cell lung cancer cells (Figure 5A). In nude mice inoculated with A549 or H1975 cells, celastrol significantly inhibited tumor growth (Figure 5B and 5C). In samples isolated from these nude mice, celastrol also induced degradation of CIP2A (Figure 5D and 5E), pointing to the possibility that the antilung cancer effect of celastrol may be associated with the downregulation of CIP2A. This possibility was confirmed by our data that upon celastrol treatment, CIP2A knockdown cells underwent enhanced inhibition of cell proliferation and colony-forming activity, and potentiated apoptosis and cleavage of PARP, compared with control cells (Figure 5F–J).
It has been shown that celastrol can modulate nuclear factor-kappaB, topoisomerase II and heat shock response (49,50). However, direct targets and the mechanism of action of celastrol have not been fully defined. We reported that CIP2A was a direct celastrol-binding protein and could be rapidly decreased by this compound. Previous works showed that celastrol had the ability to inhibit AKT/mTOR/p70S6K pathway, and this effect correlated with its anticancer effect (51,52). However, the mechanisms underlying are still obscure. We showed here that the AKT pathway inhibition might result from the downregulation of CIP2A protein (Figure 6A and B), which is in consistent with our earlier studies that CIP2A can stabilize pAKT. AKT amplification and the mammalian target of rapamycin pathway have been shown to play an important role in human lung cancer cells acquiring cisplatin resistance (53). We found that celastrol synergized with cisplatin in suppressing lung cancer cells in vitro and in vivo (Figure 6C–G), which may provide a new therapeutic regimen for lung cancer. Therefore, our results shed new lights on the application of CIP2A-targeting agent for lung cancer treatment. Since the thunder god vine from which celastrol is extracted has been used widely and successfully for centuries in treating inflammatory diseases, a potential clinical trial could be conducted to test celastrol’s efficacy in patients with lung cancer.
Supplementary material
Supplementary Figure S1 can be found at http://carcin.oxfordjournals.org/
Funding
The National Key Program for Basic Research (2012CB910800, 2010CB529200); the National Natural Science Foundation (81071930, 81171925, 81201577); the Special Foundation of President and Strategic Priority Research Programme of Chinese Academy of Sciences.
Supplementary Material
Acknowledgement
The authors thank Prof. Jukka Westermarck at University of Turku, Finland for providing the pcDNA3.1-CIP2A construct.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations:
- CHIP
carboxyl terminus of Hsp70-interacting protein
- CIP2A
cancerous inhibitor of protein phosphatase 2A
- mRNA
messenger RNA
- MTT
3-(4,5)-dimethylthiahiazo (-z-y1)-3, 5-di- phenytetrazoliumromide
- NC
negative control
- pAkt
phospho-Akt
- PARP
poly (ADP ribose) polymerase
- PBS
phosphate-buffered saline
- PSI, Z-Ile-Glu(OtBu)-Ala-Leu-CHO; SDS
sodium dodecyl sulfate
- siRNA
small interfering RNA.
References
- 1. Govindan R., et al. (2012). Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell, 150, 1121–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Esteller M. (2008). Epigenetics in cancer. N. Engl. J. Med., 358, 1148–1159. [DOI] [PubMed] [Google Scholar]
- 3. Paez J.G., et al. (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science, 304, 1497–1500. [DOI] [PubMed] [Google Scholar]
- 4. Herbst R.S., et al. (2008). Lung cancer. N. Engl. J. Med., 359, 1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hoo L.S., et al. (2002). Cloning and characterization of a novel 90kDa ‘companion’ auto-antigen of p62 overexpressed in cancer. Oncogene, 21, 5006–5015. [DOI] [PubMed] [Google Scholar]
- 6. Chen K.F., et al. (2010). CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene, 29, 6257–6266. [DOI] [PubMed] [Google Scholar]
- 7. Ma L., et al. (2011). Overexpression and small molecule-triggered downregulation of CIP2A in lung cancer. Plos One, 6, e20159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Junttila M.R., et al. (2007). CIP2A inhibits PP2A in human malignancies. Cell, 130, 51–62. [DOI] [PubMed] [Google Scholar]
- 9. Côme C., et al. (2009). CIP2A is associated with human breast cancer aggressivity. Clin. Cancer Res., 15, 5092–5100. [DOI] [PubMed] [Google Scholar]
- 10. Khanna A., et al. (2009). MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J. Natl Cancer Inst., 101, 793–805. [DOI] [PubMed] [Google Scholar]
- 11. Dong Q.Z., et al. (2011). CIP2A is overexpressed in non-small cell lung cancer and correlates with poor prognosis. Ann. Surg. Oncol., 18, 857–865. [DOI] [PubMed] [Google Scholar]
- 12. Böckelman C., et al. (2011). Prognostic role of CIP2A expression in serous ovarian cancer. Br. J. Cancer, 105, 989–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lucas C.M., et al. (2011). Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood, 117, 6660–6668. [DOI] [PubMed] [Google Scholar]
- 14. Laine A., et al. (2013). Senescence sensitivity of breast cancer cells is defined by positive feedback loop between CIP2A and E2F1. Cancer Discov., 3, 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choi Y.A., et al. (2011). Increase in CIP2A expression is associated with doxorubicin resistance. FEBS Lett., 585, 755–760. [DOI] [PubMed] [Google Scholar]
- 16. Khanna A., et al. (2011). ETS1 mediates MEK1/2-dependent overexpression of cancerous inhibitor of protein phosphatase 2A (CIP2A) in human cancer cells. PLoS One, 6, e17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mathiasen D.P., et al. (2012). Identification of a c-Jun N-terminal kinase-2-dependent signal amplification cascade that regulates c-Myc levels in ras transformation. Oncogene, 31, 390–401. [DOI] [PubMed] [Google Scholar]
- 18. Liu Y., et al. (2011). Small compound 6-O-angeloylplenolin induces mitotic arrest and exhibits therapeutic potentials in multiple myeloma. Plos One, 6, e21930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Y.Q., et al. (2012). Identification of an annonaceous acetogenin mimetic, AA005, as an AMPK activator and autophagy inducer in colon cancer cells. PLoS One, 7, e47049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu X.J., et al. (2012). Gambogenic acid induces G1 arrest via GSK3β-dependent cyclin D1 degradation and triggers autophagy in lung cancer cells. Cancer Lett., 322, 185–194. [DOI] [PubMed] [Google Scholar]
- 21. Zhen T., et al. (2012). Targeting of AML1-ETO in t(8;21) leukemia by oridonin generates a tumor suppressor-like protein. Sci. Transl. Med., 4, 127ra38. [DOI] [PubMed] [Google Scholar]
- 22. Zhou G.B., et al. (2007). Oridonin, a diterpenoid extracted from medicinal herbs, targets AML1-ETO fusion protein and shows potent antitumor activity with low adverse effects on t(8;21) leukemia in vitro and in vivo . Blood, 109, 3441–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou G.S., et al. (2011). Biologic activity of triptolide in t(8;21) acute myeloid leukemia cells. Leuk. Res., 35, 214–218. [DOI] [PubMed] [Google Scholar]
- 24. Corson T.W., et al. (2007). Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell, 130, 769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu Z., et al. (2009). Synergy between proteasome inhibitors and imatinib mesylate in chronic myeloid leukemia. PLoS One, 4, e6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Esser C., et al. (2005). The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem., 280, 27443–27448. [DOI] [PubMed] [Google Scholar]
- 27. Harwood S.M., et al. (2005). Caspase and calpain function in cell death: bridging the gap between apoptosis and necrosis. Ann. Clin. Biochem., 42(Pt 6), 415–431. [DOI] [PubMed] [Google Scholar]
- 28. Ciechanover A. (2005). Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol., 6, 79–87. [DOI] [PubMed] [Google Scholar]
- 29. Adams J. (2004). The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer, 4, 349–360. [DOI] [PubMed] [Google Scholar]
- 30. Johnston J.A., et al. (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol., 143, 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sha Y., et al. (2009). A critical role for CHIP in the aggresome pathway. Mol. Cell. Biol., 29, 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ballinger C.A., et al. (1999). Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol., 19, 4535–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sreeramulu S., et al. (2009). Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew. Chem. Int. Ed. Engl., 48, 5853–5855. [DOI] [PubMed] [Google Scholar]
- 34. Trott A., et al. (2008). Activation of heat shock and antioxidant responses by the natural product celastrol: transcriptional signatures of a thiol-targeted molecule. Mol. Biol. Cell, 19, 1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morota T., et al. (1995). D-a-Friedo-24-Noroleanane triterpenoids from Tripterigium-Wilfordii. Phytochemistry, 39, 1159–1163. [Google Scholar]
- 36. Klaić L., et al. (2012). Celastrol analogues as inducers of the heat shock response. Design and synthesis of affinity probes for the identification of protein targets. ACS Chem. Biol., 7, 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cohen M.S., et al. (2005). Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science, 308, 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Usui T., et al. (2004). The anticancer natural product pironetin selectively targets Lys352 of alpha-tubulin. Chem. Biol., 11, 799–806. [DOI] [PubMed] [Google Scholar]
- 39. David O., et al. (2004). Phospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantage. Clin. Cancer Res., 10, 6865–6871. [DOI] [PubMed] [Google Scholar]
- 40. Brognard J., et al. (2001). Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res., 61, 3986–3997. [PubMed] [Google Scholar]
- 41. Chen C., et al. (2006). Genetic and expression aberrations of E3 ubiquitin ligases in human breast cancer. Mol. Cancer Res., 4, 695–707. [DOI] [PubMed] [Google Scholar]
- 42. Yang H., et al. (2006). Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res., 66, 4758–4765. [DOI] [PubMed] [Google Scholar]
- 43. Hieronymus H., et al. (2006). Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell, 10, 321–330. [DOI] [PubMed] [Google Scholar]
- 44. Mu T.W., et al. (2008). Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell, 134, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chapelsky S., et al. (2008). Inhibition of anthrax lethal toxin-induced cytolysis of RAW264.7 cells by celastrol. PLoS One, 3, e1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lindsten K., et al. (2003). A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol., 21, 897–902. [DOI] [PubMed] [Google Scholar]
- 47. Abbas S., et al. (2007). Preclinical studies of celastrol and acetyl isogambogic acid in melanoma. Clin. Cancer Res., 13(22 Pt 1), 6769–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang Y., et al. (2008). Celastrol inhibits the growth of human glioma xenografts in nude mice through suppressing VEGFR expression. Cancer Lett., 264, 101–106. [DOI] [PubMed] [Google Scholar]
- 49. Liu Z., et al. (2011). The main anticancer bullets of the Chinese medicinal herb, thunder god vine. Molecules, 16, 5283–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salminen A., et al. (2010). Celastrol: molecular targets of Thunder God Vine. Biochem. Biophys. Res. Commun., 394, 439–442. [DOI] [PubMed] [Google Scholar]
- 51. Pang X., et al. (2010). Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res., 70, 1951–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kannaiyan R., et al. (2011). Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3K/Akt signaling pathways. Apoptosis, 16, 1028–1041. [DOI] [PubMed] [Google Scholar]
- 53. Liu L.Z., et al. (2007). AKT1 amplification regulates cisplatin resistance in human lung cancer cells through the mammalian target of rapamycin/p70S6K1 pathway. Cancer Res., 67, 6325–6332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.