

Abstract
Ocular trauma affects 20% of Americans in their lifetime and can cause permanent visual system damage. We have used a mouse model of ocular trauma (exposure to an air blast from a paintball gun) to examine pathways that trigger the resulting retinal damage and to develop treatment strategies that might ameliorate the deleterious effects of trauma on retinal tissue. Our previous studies have shown that ocular blast causes an increase in protein levels of inflammatory mediators and apoptotic factors, including tumor necrosis factor alpha (TNFα) and interleukin-1-beta (IL-1β), as well as the apoptotic markers, Bax, cytochrome C, and cleaved caspase 3. Furthermore, topical treatment by eye drop application of a β-adrenergic receptor agonist, Compound 49b, was shown to decrease these inflammation/apoptosis markers and thus ameliorate the effects of blast trauma. We postulate that the protective effect of Compound 49b may be linked to its demonstrated ability to activate the β-adrenergic receptor and in turn trigger production of insulin-like growth factor binding protein 3 (IGFBP-3). In the current study, we tested this hypothesis using mice with minimal IGFBP-3 activity (IGFBP-3 knockdown mouse) vs. wildtype mice. We found that ocular blast alone did not affect IGFBP-3 levels in retinas of wild type or knockdown mice and surprisingly, the lower levels of IGFBP-3 in knockdown animals did not exacerbate the blast-induced increase in protein levels of inflammation/apoptosis markers. Nevertheless, the levels of IGFBP-3 were significantly increased in knockdown mouse retina by treatment with Compound 49b 24 hours post-trauma and as expected, the increase in IGFBP-3 was linked to a decrease in inflammation/apoptosis markers. We conclude that while lowered IGFBP-3 may not make the retina more vulnerable to blast injury, an increase in IGFBP-3 post-trauma may play an important role in limiting trauma-induced inflammatory and apoptotic pathways leading to retinal damage. Eye drop application of the β-adrenergic receptor agonist, Compound 49b, provides a promising treatment strategy for increasing IGFBP-3 levels to promote recovery from retinal inflammation and apoptosis after ocular blast.
1.0 Introduction
Ocular trauma affects 20% of Americans during their lifetime and up to 1% may experience retinal damage [1]. The most commonly affected people are young males, manual workers, and members of the military [2]. As many of 13% soldiers experience eye injuries in the battlefield, with 80% of the eye injuries related to blast exposure [2]. A study that included a representative sample of 46 Iraq and Afghanistan war veterans showed up to 28% had changes in the posterior eye causing visual damage [3]. While it is clear that exposure to ocular blast produces damage, an animal model to determine the cellular mechanisms was lacking until recently.
In order to dissect potential pathways involved in retinal damage after exposure to blast, multiple approaches have recently been developed. Work in a shock wave tube using moderate open-field blast waves showed damage to multiple organs, including a significant effect on long axon tracts of the central nervous system [4]. Blanch et al. (2012) developed two different animal models of blast: a weight drop model and a low-velocity ballistic trauma model [1]. While weight drop did not cause retinal injury, the low-velocity ballistic trauma model, in which ball bearings are shot from an air gun at a known pressure and velocity, did show apoptosis of retinal cells and disruption of photoreceptor cells, as well as changes in the electroretinogram [1]. A modification of the low-velocity ballistic trauma model was developed by Rex et al. [5], where an air gun is set to a specific pressure to induce a closed globe injury to the mouse eye. While Rex et al. only found some visual acuity reductions at pressures of 26psi, we recently reported increased protein levels of key inflammatory markers including tumor necrosis factor alpha (TNFα), and interleukin-1-beta (IL-1β), as well as apoptotic markers in retinal lysates from mice exposed to 26psi blast [6]. We also reported that a novel β-adrenergic receptor agonist, Compound 49b, could reduce the levels of inflammatory and apoptotic markers after exposure to 26psi blast.
In order to fully evaluate the treatment potential for Compound 49b as a promising therapy for individuals such as warriors, who are vulnerable to ocular blast, we wanted to ascertain a potential mechanism of action. We have previously reported that Compound 49b can regulate TNFα in other retinal damage models, specifically the streptozotocin-induced type 1 diabetic retinopathy model and retinal endothelial cells cultured in high glucose [7]. In those models, it was observed that another protein is down-regulated in response to high glucose, namely insulin-like growth factor-1-binding protein 3 (IGFBP-3). We recently showed that treatment of diabetic rats with IGFBP-3 plasmid could significantly reduce TNFα levels, as well as apoptotic markers [8]. Because IGFBP-3 can reduce both inflammatory and apoptotic markers in models of diabetic retinopathy, we hypothesize that IGFBP-3 may play a similar protective role in retinal damage after ocular blast.
To investigate the role of IGFBP-3 in ocular blast, we exposed IGFBP-3 knockdown (KD) mice to 26psi blast using the method previously described [6]. At 4, 24, and 72 hours post blast exposure, mice were treated with topical Compound 49b, to determine if the treatment increased endogenous IGFBP-3 levels concomitant with decreases in inflammatory and apoptotic markers.
2.0 Methods
2.1 Mice
IGFBP-3 KD mice were generously provided by Dr. John Pintar (Rutgers University). Although we and others originally designated these mice as IGFBP-3 knockout, our current data demonstrates low, but measurable levels of endogenous IGFBP-3 protein levels despite no measureable mRNA levels [9]. Thus we recognize that the IGFPB-3 gene is still minimally active in these animals. All mice were blasted and treated with Compound 49b at 2 months of age. All animal studies confirm to ARVO Guidelines for Use of Animals in Research.
2.2 Blast Procedure
Both eyes of the mice were exposed to the ocular blast. In one subset of mice, eyes were collected at 4 hours post-blast, 24 hours post-blast, 72-hours post blast or at 72 hours without exposure to the blast. In the second subset of mice, both eyes were blasted; however, a novel β-adrenergic receptor agonist, Compound 49b (1mM), was applied topically within 4 hours, 24 hours or 72 hours post-blast. For the Compound 49b-treated mice, mice received daily Compound 49b treatment for up to 3 days. For example, for the 4 hour treatment group, the mice received the first treatment within 4 hours post-blast, then another treatment 24 hours, 48 hours, and 72 hours post-blast (for a total of 4 treatments), while 72 hour post-blast mice only received 1 treatment of Compound 49b prior to sacrifice. All mice were sacrificed 3 days post-blast for the Compound 49b treated mice. Six mice were used at each time point for all experiments.
2.3 Western Blotting
At the appropriate time after blast or Compound 49b treatment, both eyes were removed for analyses. For Western blot analyses, eyes were used for retinal lysates and collected into lysis buffer containing protease and phosphatase inhibitors, followed by sonication. Equal amounts of protein from tissue extracts were separated on the pre-cast tris-glycine gel (Invitrogen, Carlsbad, CA), blotted onto a nitrocellulose membrane. After blocking in TBST (10mM Tris-HCl buffer, pH 8.0, 150 mM NaCl, 0.1% Tween 20) and 5% (w/v) BSA, the membrane was treated with appropriate primary antibodies followed by incubation with secondary antibodies labeled with horseradish peroxidase. Antigen-antibody complexes were detected by chemilluminescence reagent kit (Thermo Scientific). Primary antibodies used were Cytochrome C, Bax, and Bcl-xL, Akt, phosphorylated AktSer473 (all purchased from Cell Signaling, Danvers, MA). β-actin antibody was purchased from Santa Cruz (Santa Cruz, CA).
2.4 ELISA Analysis
A cleaved caspase 3 ELISA (Cell Signaling, Danvers, MA) was used to measure levels of the active apoptotic marker in whole retinal lysates. Equal protein was loaded to allow for statistics based upon optical density measurements. TNFα and IL-1β protein concentrations were measured using a TNFα and IL-1β ELISA, respectively with equal protein loaded into each well to eliminate changes in protein due to amount of protein added (ThermoFisher, Pittsburgh, PA). All ELISAs were done according to manufacturer’s instructions.
2.5 Statistics
For all analyses, all experiments were done in triplicate. Data is presented as mean ± SEM, with statistical analyses using Kruskal-Wallis non-parametric testing, followed by Dunn’s test.
3.0 Results
3.1 Blast did not affect IGFBP-3 levels, while Compound 49b increased IGFBP-3 in IGFBP-3 KD mice at 4 and 24 hours post-blast
We have previously reported diabetes significantly reduces IGFBP-3 levels [7], leading to increased TNFα levels and apoptosis in whole retina and in retinal endothelial cells (REC). Additionally, we have shown that ocular blast le ads to a significant increase in TNFα, IL-1β and a number of apoptotic markers [6], which was negated by treatment with Compound 49b. This led us to hypothesize that Compound 49b protected the retina through increasing IGFBP-3 levels. In Figure 1, we demonstrate that IGFBP-3 KD mice expressed very low levels of endogenous IGFBP-3 and that exposure to ocular blast did not affect these levels. Nevertheless, Compound 49b treatment of the IGFBP-3 KD mice was able to significantly increase protein levels if administered within 24 hours post-blast. Compound 49b given 72 hours after blast had no effect on IGFBP-3 levels. This suggests that despite very low levels of endogenous IGFBP-3 in these knockdown mice, Compound 49b is still able to substantially increase protein levels of IGFBP-3.
Figure 1.
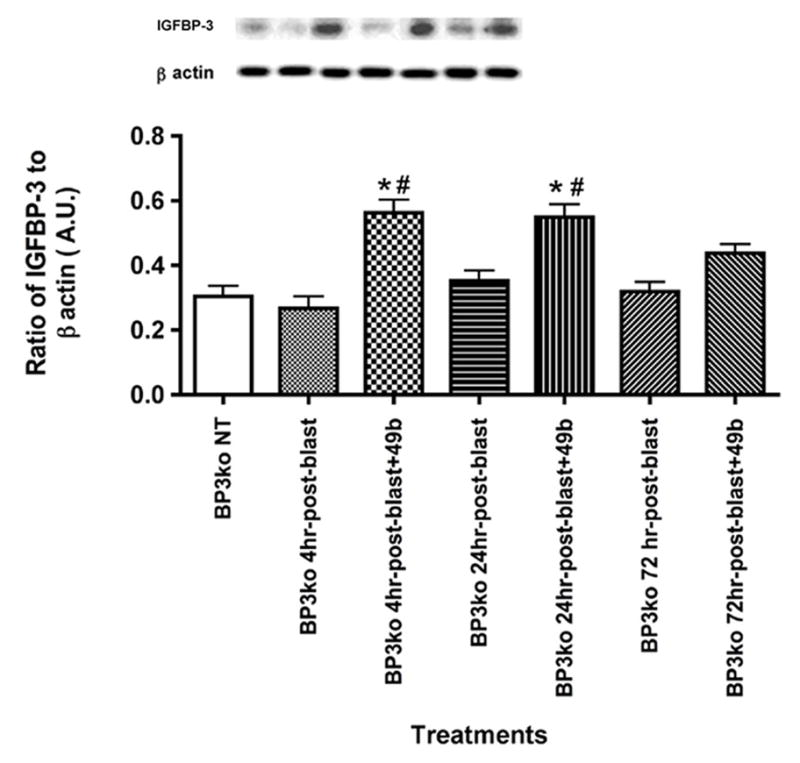
IGFBP-3 protein levels after ocular blast and Compound 49b treatment. Western blot results of IGFBP-3 KD mice without exposure to blast, IGFBP-3 KD mice exposed to blast for 4, 24, and 72 hours, and IGFBP-3 KD mice exposed to ocular blast + topical 1mM Compound 49b within 4, 24, or 72 hours after blast exposure. *P<0.05 vs. BP3 NT (not treated); # P<0.05 vs. BP3 KD+blast only at the same time point. N=6 mice in each group.
3.2 TNFα and IL-1β were increased after blast exposure in IGFBP-3 KD mice, which was negated by treatment with Compound 49b
Exposure of IGFBP-3 KD mice to ocular blast increased TNFα levels by approximately 40% (Figure 2A) and IL-1β by approximately 3-fold (Figure 2B), which is somewhat similar to data obtained after ocular blast exposure to wild type mice [6]. However, the increase in IL-1β after blast exposure in IGFBP-3 KD mice was greater than that noted in wildtype mice, suggesting that IGFBP-3 is a particularly potent regulator of IL-1β. For both TNFα and IL-1β, Compound 49b was able to significantly reduce protein levels of both inflammatory mediators, TNFα and IL-1β, when administered within 24 hours of blast. Unlike the effects of Compound 49b on wildtype mice [6], Compound 49b treatment did not reduce TNFα and IL-1β levels in IGFBP-3 KD mice at 72 hours post-blast (Figure 2).
Figure 2.
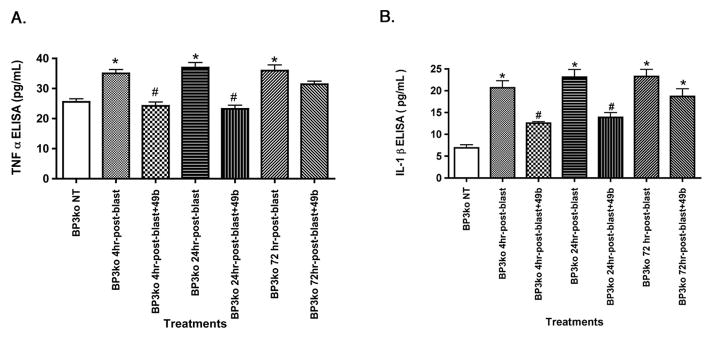
Ocular blast increased TNFα and IL-1β. ELISA results for TNFα (left) and IL-1β (right) in IGFBP-3 KD mice without exposure to blast, IGFBP-3 KD mice exposed to blast for 4, 24, and 72 hours, and IGFBP-3 KD mice exposed to ocular blast + topical 1mM Compound 49b within 4, 24, or 72 hours after blast exposure. *P<0.05 vs. BP3 NT (not treated); # P<0.05 vs. BP3 KD+blast only at the same time point. N=6 mice in each group.
3.3 Ocular blast increased pro-apoptotic factors, which were reduced with Compound 49b if given by 24 hours after blast exposure
In wildtype rats, exposure to ocular blast increased pro-apoptotic factors, while reducing anti-apoptotic factors [6]. Similar results were obtained in IGFBP-3 KD mice (Figure 3). Unlike the data in wildtype mice, Compound 49b was only effective in IGFBP-3 KD mice if administered within 24 hours of blast exposure. In wildtype mice, cleaved caspase 3 levels were still reduced at 72 hours post-blast with Compound 49b treatment [6]. This is not the case in IGFBP-3 KD mice, as Compound 49b has no effect on either pro- or anti-apoptotic factors when administered at 72 hours post-blast, with the exception of Bax (which was reduced with Compound 49b at 72 hours; however baseline Bax levels were not increased 72 hours after blast).
Figure 3.

Ocular blast increased apoptotic factors in IGFBP-3 KD mice. Western blot results for key pro-apoptotic proteins Cytochrome C (A) Bax (B) and anti-apoptotic proteins BcL-xL (D) and Akt (E). ELISA results for cleaved caspase 3 (C) in IGFBP-3 knockdown mice without exposure to blast, IGFBP-3 KD mice exposed to blast for 4, 24, and 72 hours, and IGFBP-3 KD mice exposed to ocular blast + topical 1mM Compound 49b within 4, 24, or 72 hours after blast exposure. *P<0.05 vs. BP3 NT (not treated); # P<0.05 vs. BP3 KD+blast only at the same time point. N=6 mice in each group.
4.0 Discussion
Exposure to ocular blast and the resulting trauma puts our soldiers and other vulnerable individuals at risk for long-term visual damage. In order to better understand the retinal damage caused by closed-globe blast injury, multiple models have been developed to examine the mechanism for the retinal changes, as well as test novel therapies [4, 10]. We recently reported that ocular blast produced a significant increase in TNFα, IL-1β, and apoptotic factors, which could be reduced by treatment with a novel β-adrenergic receptor agonist, Compound 49b, when administered within 24 hours [6]. Blast produced a much greater increase in IL-1β levels (>3-fold) compared to TNFα levels (1.5-fold), but Compound 49b was more effective in reducing TNFα levels than IL-1β in both wildtype mice [6] or IGFBP-3 KD mice exposed to ocular blast (Figure 2).
One potential pathway by which Compound 49b may reduce inflammatory and apoptotic factors is through increasing IGFBP-3. Despite very low endogenous IGFBP-3 levels in the IGFBP-3 KD mice, Compound 49b was still highly effective in increasing IGFBP-3 levels (Figure 1). Based upon on previous work in retinal endothelial cells, Compound 49b likely increases IGFBP-3 through activation of DNA-PK pathways [11]. Increased levels of IGFBP-3 in the knockdown mice can significantly reduce TNFα levels through activation of the Jun pathway (Zhang et al, in revision). Additionally, Compound 49b-induced increases in IGFBP-3 levels after ocular blast likely reduced pro-apoptotic factors, Bax, cytochrome C, and cleaved caspase 3, through activation of its receptor [12]. Thus, we conclude that Compound 49b is effective in protecting the retina against blast-induced injury through increasing IGFBP-3 levels, as well as reducing TNFα signaling. Based on the work by Jeschke et al., as well as our own findings in diabetic retina [8], it appears that IGFBP-3 lies upstream of TNFα and increasing IGFBP-3 levels can antagonize any responses induced by increased TNFα levels. These findings are in agreement with those from other injury models including a thermal model of total body surface burn in which increasing IGFBP-3 is protective to gut mucosal tissue [13]. Similar to our findings in the blast model, in the thermal model IGF-1/IGFBP-3 reduced the effects of thermal burn on gut homeostasis, which was also associated with decreased TNFα and IL-1β levels [13],
We have previously reported in a mouse model for diabetic retinopathy, that the increased expression of retinopathy markers including TNFα, occurred concomitantly with a decrease in endogenous IGFBP-3 in whole retinal lysates and in retinal endothelial cells [7, 9]. We hypothesize in this case, that the insult of high glucose could have direct effects on reducing β-adrenergic receptor signaling, which in turn decreases IGFBP-3 levels to trigger downstream effects on inflammation and apoptosis in diabetes. Such is not the case in ocular blast injury where IGFBP-3 levels were not affect by blast treatment either in wild type or in IGFBP-3 KD mice. Furthermore, the degree of blast injury, as measured by inflammatory and apoptotic markers, was not more severe in the knockdown mice, suggesting that low levels of IGFBP-3 does not make the retinal more vulnerable to blast injury. We assume that the minimal levels of IGFBP-3 produced by the knockdown are sufficient to maintain homeostasis under normal conditions. However, an increase in IGFBP-3 resulting from after blast treatment with Compound 49b was very beneficial in lowering blast induced increases in inflammation and apoptosis. Unlike diabetic retinopathy, blast injury does not specifically target the β-adrenergic receptor/IGFBP-3 pathway. Nevertheless, stimulation of this pathway after injury promotes recovery. These results suggest that Compound 49b could be an effective treatment for blast-induced injury and thus merits further study as a treatment option for vulnerable patient populations.
Highlights.
Ocular Blast did not affect IGFBP-3 levels
Compound 49b significantly increased IGFBP-3 levels
Ocular blast increased TNFα and IL-1β levels in IGFBP-3 KD mice
Blast increased apoptotic proteins, which were reduced by Compound 49b treatment
Acknowledgments
This work is supported by a grant from U.S. Army Medical Research and Material Command (W81XWH-12-1-0318), NIH R01 (EY022045 to JJS), JDRF Priority Research Grant (2-2011-597 to JJS); Oxnard Foundation (JJS); Research to Prevent Blindness Award (PI:James C. Fleming); and NEI Vision Core Grant: PHS 3P30 EY013080 (PI: Dianna Johnson).
Footnotes
Department of Defense Non-endorsement disclaimer: The views, opinions, and/or findings contained in this research presentation are those of the authors and do not necessarily reflect the views of the Department of Defense and should not be construed as an official DoD/Army position, policy, or decision unless so designated by other documentation. No official endorsement should be made.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blanch RJ, Ahmed Z, Sik A, Snead DR, Good PA, O’Neill J, et al. Neuroretinal cell death in a murine model of closed globe injury: pathological and functional characterization. Invest Ophthalmol Vis Sci. 2012;53:7220–6. doi: 10.1167/iovs.12-9887. [DOI] [PubMed] [Google Scholar]
- 2.Blanch RJ, Ahmed Z, Berry M, Scott RA, Logan A. Animal models of retinal injury. Invest Ophthalmol Vis Sci. 2012;53:2913–20. doi: 10.1167/iovs.11-8564. [DOI] [PubMed] [Google Scholar]
- 3.Cockerham GC, Rice TA, Hewes EH, Cockerham KP, Lemke S, Wang G, et al. Closed-eye ocular injuries in the Iraq and Afghanistan wars. N Engl J Med. 2011;364:2172–3. doi: 10.1056/NEJMc1010683. [DOI] [PubMed] [Google Scholar]
- 4.Koliatsos VE, Cernak I, Xu L, Song Y, Savonenko A, Crain BJ, et al. A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. Journal of neuropathology and experimental neurology. 2011;70:399–416. doi: 10.1097/NEN.0b013e3182189f06. [DOI] [PubMed] [Google Scholar]
- 5.Hines-Beard J, Marchetta J, Gordon S, Chaum E, Geisert EE, Rex TS. A mouse model of ocular blast injury that induces closed globe anterior and posterior pole damage. Exp Eye Res. 2012;99:63–70. doi: 10.1016/j.exer.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang Y, Liu L, Pagadala J, Miller DD, Steinle JJ. Compound 49b protects against blast-induced retinal injury. J Neuroinflammation. 2013;10:96. doi: 10.1186/1742-2094-10-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Q, Guy K, Pagadala J, Jiang Y, Walker RJ, Liu L, et al. Compound 49b Prevents Diabetes-Induced Apoptosis through Increased IGFBP-3 Levels. Invest Ophthalmol Vis Sci. 2012;53:3004–13. doi: 10.1167/iovs.11-8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang Y, Zhang Q, Steinle JJ. Intravitreal Injection of IGFBP-3 Restores Normal Insulin Signaling in Diabetic Rat Retina. PLoS One. 2014;9:e93788. doi: 10.1371/journal.pone.0093788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Jiang Y, Miller MJ, Peng B, Liu L, Soderland C, et al. IGFBP-3 and TNF-alpha Regulate Retinal Endothelial Cell Apoptosis. Invest Ophthalmol Vis Sci. 2013;54:5376–84. doi: 10.1167/iovs.13-12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cernak I. The importance of systemic response in the pathobiology of blast-induced neurotrauma. Frontiers in neurology. 2010;1:151. doi: 10.3389/fneur.2010.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Q, Steinle JJ. DNA-PK phosphorylation of IGFBP-3 is required to Prevent Apoptosis in Retinal Endothelial cells cultured in High Glucose. Invest Ophthalmol Vis Sci. 2013 doi: 10.1167/iovs.12-11533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Q, Soderland C, Steinle JJ. Regulation of retinal endothelial cell apoptosis through activation of the IGFBP-3 receptor. Apoptosis. 2013;18:361–8. doi: 10.1007/s10495-012-0793-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeschke MG, Bolder U, Chung DH, Przkora R, Mueller U, Thompson JC, et al. Gut mucosal homeostasis and cellular mediators after severe thermal trauma and the effect of insulin-like growth factor-I in combination with insulin-like growth factor binding protein-3. Endocrinology. 2007;148:354–62. doi: 10.1210/en.2006-0883. [DOI] [PubMed] [Google Scholar]
- 14.Kim HS, Park CK. Retinal ganglion cell death is delayed by activation of retinal intrinsic cell survival program. Brain Res. 2005;1057:17–28. doi: 10.1016/j.brainres.2005.07.005. [DOI] [PubMed] [Google Scholar]