

Abstract
Primary hepatocellular carcinoma (HCC) is a quite frequent tumor which results in high mortality and most often exhibits a poor response to present drug therapies. Clearly, a thorough understanding of the biological bases of this malignancy might suggest new strategies for its treatment. Here we examine the evidences that both “pharmacological” mechanisms (e.g. drug transporter or detoxification enzyme over-expression) and alterations in other critical factors, including the IAPs (Inhibitory of Apoptosis Proteins), involved in enhancement of cell survival and proliferation may determine the therapeutic resistance of HCC; we also underline the possible role in the process of the activation of transcription factors, like NF-κB, capable of contemporaneously up-regulating the mechanisms discussed. On this basis, we finally comment on the possible use of natural multi-targeted antitumoral agents like plant polyphenols to achieve sensitization to treatments in HCC.
Keywords: Hepatocellular carcinoma, Drug resistance, Drug transporters, Inhibition of cell death, IAPs, NF-κB, Plant polyphenols
INTRODUCTION
Drug resistance, either innate or acquired and especially in its multiple form (multidrug resistance, MDR), remains a major and difficult problem to resolve in the therapy of many cancer types. This process has previously been interpreted mainly in a “pharmacological” manner focused on the ability of tumor cells to extrude or inactivate the cytotoxic agents or to modify their targets of action: much attention has been drawn to the over-expression of multidrug efflux transporters such as P-glycoprotein (P-gp), Multidrug Resistance Related Proteins (MRPs) and others. Nevertheless, available evidence suggests that the sole reversion of such mechanisms has clinical success in few situations. On the other hand, today it is recognized that clinical MDR is often a multifactorial and heterogeneous process; many other different molecular alterations, known to be involved in the malignant transformation and progression, may also be responsible for tumor drug resistance[1]. For example, induction of tumor cell killing is fundamental for the effectiveness of anticancer drugs and a relevant mechanism of cellular protection from their attacks is represented by the loss of pro-apoptotic factors (e.g. functional p53 or Bax) or the over-expression of anti-apoptotic ones, like Bcl-2, Bcl-XL or IAPs (Inhibitory of Apoptosis Proteins)[1,2]. This interferes with the process of cell death at the level of different steps, including distal ones where there may be a convergence of diverse mortal pathways: IAPs, which in humans include c-IAP-1, c-IAP-2, XIAP, NAIP, survivin and livin-α, beside other anti-apoptotic mechanisms, possess the ability of inhibiting the terminal cell death effector proteases, caspase-3 and -7. They can indeed block execution of cell death triggered from many non-related pharmacologic, immunologic and irradiation stimuli[3,4].
In this dimension, primary HCC is a frequent tumor, which results in high mortality and most often exhibits a poor response to current drug therapies[5,6]. Clearly, a thorough understanding of the biological bases of this malignancy might suggest new strategies for its treatment. Here we examine the evidences that both drug transporter or detoxification enzyme expression and alterations in other critical factors, including the IAPs, involved in enhancement of cell survival and proliferation may determine the therapeutic resistance of HCC; we underline also the possible role in the process of the activation of transcription factors, like NF-κB, capable of contemporaneously up-regulating the mechanisms discussed. On this basis, we finally comment on the possible use of natural multi-targeted agents such as plant polyphenols for achieving sensitization to treatments in HCC.
ROLE OF ABC TRANSPORTERS, CYTOCHROME P450 ENZYMES AND GLUTATHIONE S-TRANSFERASES
HCC develops from hepatocytes, which physiologically express different multidrug transporters, including P-gp, and are rich of other drug elimination systems, like the phaseIor II biotransformation enzymes. Thus, preserved or increased levels of these factors might contribute to mediate the intrinsic chemoresistance of HCC.
P-gp (MDR1, ABCB1), the prototypical member of the ATP binding cassette (ABC) family of proteins, is capable of extruding from cells various unrelated antitumoral agents, including anthracyclines, taxanes, Vinca alkaloids and podophyllotoxin derivatives; though with different substrate specificities, the other transporters can do the same (Table 1). Further, at least in some cell models, P-gp may also inhibit specific mechanisms of apoptotic cell death from drugs and other stimuli[7]. P-gp has been quite often examined in HCC, but there are contrasting results on whether its over-expression is actually frequent in untreated HCCs compared to non-neoplastic liver tissues[8-12], possibly representing an adverse prognostic factor correlated with reduced survival[9,10]. According to several studies[8,11], P-gp content is significantly higher in cirrhotic liver tissues, either from patients with overt HCC or not, than in normal liver suggesting that the factor may play a role in the development of the tumoral process. For example, increased levels of the drug transporters in the pre-malignant hepatocytes might provide them with a selection advantage in a toxic environment of carcinogens or endogenous metabolic and inflammatory products. It has been also proposed that the development of P-gp over-expression and of an angiogenic phenotype are linked to each other in the multidrug resistant HCC cells[13]. Instead, p53 mutation would not appear to be a major determinant of P-gp expression[14].
Table 1.
Major drug transporters possibly involved in the drug resistance of HCC
| Transporter | Drugs interested |
| P-glycoprotein (MDR1, ABCB1) | Anthracyclines, Mitoxantrone, Podophyllotoxin derivatives, Taxanes, Vinca alkaloids |
| MRP2 (ABCC2, cMOAT) | Anthracyclines, Cisplatin, Methotrexate, Irinotecan metabolites, Vinca alkaloids |
| MRP3 (ABCC3) | Etoposide, Methotrexate |
| LRP (Major vault protein, MVP) | Cisplatin, Doxorubicin, Etoposide, 5-fluorouracil, Paclitaxel, Vincristine |
| TAP (Transporter associated with antigen processing) | Doxorubicin, Etoposide, Mitoxantrone, Vincristine |
In normal hepatocytes, P-glycoprotein and the members of the MRP family play an important role in biliary excretory function. Also vaults have a transport function, mediating bidirectional nucleo-cytoplasmic exchange and vesicular transport of compounds, including cytostatic drugs. Physiologically, TAP translocates short peptides, mostly generated by proteasome-mediated antigenic protein degradation in the cytosol, into the lumen of the endoplasmic reticulum. All these factors may mediate chemoresistance of various malignancies: the spectrum of the drugs that they may transport does not necessarily derive from specific studies on HCC.
Overall, it would appear that the presence of high P-gp levels in HCC correlates with a poorer response to chemotherapy[9,14]; nevertheless, in a clinical study the addition of the P-gp inhibitor verapamil did not improve the response of HCC to therapy with systemic doxorubicin[15].
For the other drug transporters, on the basis of analyses on clinical samples, MRP2 (ABCC2), TAP (Transporter associated with Antigen Processing) and, in individual cases, MRP3 (ABCC3) and LRP (Lung Resistance-related Protein) are other possible candidates for mediating the chemoresistance of HCC[12,16-18]. Importantly, co-expressions can occur, as those of P-gp and MRP2 documented in HCC cell line models[19]. In a study on a limited number of patients, phospholipid flippase MDR3 (ABCB4) and bile salt export pump (also known as the sister of P-glycoprotein/ABCB11) showed instead a trend for decreased levels in HCC[12]. The drug resistance of HCC, beside to increased drug export, might also be due to insufficient drug uptake, because of the tendency to decrease important factors involved in this process, like the organic anion-transport polypeptides 2 (OATP2) and 8 (OATP8) and the concentrative nucleoside transporter CNT1[12,20].
Further, an immunohistochemistry study on biotransformation enzymes in HCC suggested, that although complex, their expression may contribute to its drug resistance[21]. There was a consistent high content of microsomal epoxide hydrolase, and a variable expression of cytochromes P450 and cytosolic glutathione S-transferases: cytochromes P450 1A and 3A stained in 64.5% and 41.9% of the 31 HCCs studied, respectively. Glutathione S-transferase types alpha, mu and pi were identified in 48.4%, 38.7% and 74.2% of the samples, respectively[21]. Variable expressions of the broad substrate specificity cytochrome P450 3A enzymes and of glutathione S-transferases alpha or pi, which may increase cell resistance to alkylating agents or cisplatin, have been reported in other immunohistochemical studies on HCC[22-25].
THE IAPS ARE ABUNDANT IN HCC
As anticipated, there is a large body of evidence that an imbalance between unrestrained cell proliferation and the low ability to perform apoptosis, either spontaneous or induced from pharmacologic or immunologic agents, is another critical feature of HCC. Like in other tumors, a major factor, which can determine such behaviour, is the re- or over-expression in HCC of inhibitors of apoptosis like the proteins of the Bcl-2 family[26] and the IAPs.
It must be said that the majority of the studies related to IAPs in HCC have focused mainly on survivin. Unlike other IAPs, which can be expressed also in normal adult tissues, survivin is detected predominantly in fetal or neoplastic tissues. Importantly, it is also recognized that survivin not only inhibits apoptosis, but also, as a component of the chromosomal passenger complex, it favours cancer cell proliferative activity[27]. Several studies[28-40] have indicated that, especially nuclear[30,34,38], expression of survivin is frequent in HCC, higher than in corresponding non-malignant tissues, and correlates with increases in cell proliferation indexes, possibly lowered apoptosis, as well as with adverse histological and clinical features. In addition, some survivin alternative splice variants (Survivin-2B, Survivin-deltaEx3 and Survivin-3B), differing in their anti-apoptotic properties, have been identified. Of these, at least survivin-2B and survivin-ΔEx3 can be expressed in HCC, where the levels of survivin–ΔEx3, but not of survivin-2B, correlate with high proliferative activity[33,37,38].
IAPs are present in chronic hepatitis and cirrhosis possibly contributing to carcinogenesis[30-33,38,39]. With regard to this, it has been shown that the hepatitis B virus X protein (HBX) can up-regulate survivin in hepatoma tissues as well as bind to survivin-HBX-interacting protein (HBXIP) complexes to suppress caspase activation[39,41]; other authors have found increases in c-IAP-1 or c-IAP-2, but not in the other IAPs, in a variant of the HepG2 HCC cell line persistently expressing hepatitis B virus by integrated HBV genome[42], so that, collectively, the available data suggest a link between the IAP family and an important viral pathogen involved in hepatocellular carcinogenesis.
Indeed, focusing on the behaviour of other IAPs besides survivin in HCC, an immunohistochemical study[43] has indicated that XIAP is another principal factor over-expressed in this tumor, which inversely correlates with apoptosis, without an impact on proliferation. Others have reported the expression of XIAP, c-IAP-1 and c-IAP-2 in HCC, although less frequently than survivin[39]. Our personal observations, all on HCCs of hepatitis C origin, have corroborated these last findings, emphasizing, in particular, the possible role that the coordinated expression of different IAPs (noticeably including NAIP) and of their splice variants may play in the biology and resistance to treatment of the tumor[33]. In relation to the particular context of this paper, it should also be stressed that targeting of survivin or XIAP with specific approaches has determined chemo-, immuno- or radio-sensitization in different tumor types, including HCC[44,45].
NF-κB: AN UNIFYING PILLAR IN HCC RESISTANCE?
The regulatory mechanisms of IAP expression are not completely defined yet, but it is known that their expression (at least of c-IAP-1, c-IAP-2 and XIAP) can be promoted by the transcription factor NF-κB[46,47]. The activation of this signalling system is indeed emerging now as a possible critical mechanism of treatment resistance in the tumors. Further, owing to different mechanisms, NF-κB is frequently abnormally present in clinical HCC[48-50]; its persistent and inappropriate activation in hepatocytes is implicated in tumor initiation and progression following events like viral infection (HBV or HCV), exposure to carcinogens, growth factor stimulation (TGF-α, HGF/SF and TGF-β) and inflammation, as recently reviewed by Arsura and Cavin[51]. Beside IAPs, the transactivating forms of NF-κB (e.g. dimers containing the p65/RelA subunit) may up-regulate the expression of several other genes involved in anti-apoptosis, cell proliferation and invasion, and drug resistance (e.g., Bcl-2, Bcl-XL, cyclin D1, c-myc, IL-6, COX-2, iNOS, MMPs and MDR1/P-gp). The links between NF-κB activation and P-gp[52] or IAPs , like XIAP[52], expression have been specifically evidenced for HCC. Accordingly, NF-κB mostly results anti-apoptotic, an activity to which may contribute also post-trascriptional effects, but, depending on certain cell types and stimuli, it can also promote cell death. Undoubtedly, in many studies, interference with NF-κB by different independent approaches has been shown to increase tumor cell response to different NF-κB activating anticancer drugs, including doxorubicin[46,47,53]; nevertheless, other results have suggested that activation of NF-κB may be required for the cytotoxicity of doxorubicin and its analogs[54]. With reference to this, interestingly, recent studies have shown that in some circumstances the p65 subunit can, non-canonically, repress the transcription of anti-apoptotic genes; doxorubicin treatment may produce p65 which is defective in post-translational modifications and blocks NF-κB signaling[55,56], corroborating that in some experimental models doxorubicin may rely on NF-κB to exert its antitumoral activity.
SINGLE- VERSUS MULTI-TARGETED AGENTS TO FACE HCC RESISTANCE
Here we have critically discussed only some selected critical mechanisms of drug resistance in HCC, but there are many others that could be highlighted for their responsibility in the process. The multiplicity of the possible drug resistance determinants poses the question of the optimal strategies to contrast them and achieve sensitization to treatments. During the last years, many novel molecular therapies have been developed, represented for example by monoclonal antibodies or tyrosine kinase inhibitors directed against specific important targets. However, the targeted agents currently in clinical use have not generally shown to lead to cures or long-term survival for most intractable cancers; whatever relevant the blocked mechanism is, the genetic instability of cancer cells enables them to devise adaptation changes and alternative signaling pathways that stimulate cell proliferation and survival, so that resistance develops. Reasonably, multi-targeted agents might be less likely to encounter problems of drug resistance than single-targeted ones; this might apply also to HCC.
On the other hand, from the inspection of the literature there emerges different natural compounds endowed with a remarkable number of different antitumoral activities and mechanisms, yet accompanied by a limited toxicity for the host; their properties may be exploited for chemopreventive purposes and possibly also for the treatment of already established tumors. Among these “privileged” structures[57], are the dietary polyphenols; there are evidences[58,59] that representative members of this family, like resveratrol (or its metabolite piceatannol), curcumin and epigallocatechin-3-gallate (EGCG), in general share the ability of: inhibiting the ligand binding to, or phosphorylation of growth factor receptors, including the EGFR family. This leads to down-regulation of the MAPK cascades (ERK and also JNK or p38); inhibiting the signaling through pAkt, NF-κB and STATs; inducing cell cycle arrest, with involvement of decreases in cyclin D1 and phosphorylation of Rb and of up-regulation of p21 and p27; inducing cell death through release of cytochrome c from mitochondria and activation of caspase -9 and -3; Polyphenols also down-regulate Bcl-2, Bcl-XL and IAP family members and up-regulate Bax; interfering with telomerase, the enzyme that immortalize cancer cells, thus releasing programs of apoptosis or senescence-related cell growth arrest; down-regulating targets relevant to angiogenesis, invasion and metastasis, like VEGF, matrix metalloproteinases (MMPs), β-catenin, inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX-2); inhibiting the function of P-gp, other ABC transporters (MRPs and BRCP), glutathione S-transferases (GSTP1 or GSTM1 type enzymes) and certain cytochrome P450 isoenzymes. Importantly, it has to be underlined that, at least curcumin, does not appear to be itself transported by P-gp; indeed, its antitumor activity is not affected in cells over-expressing such a mechanism[60,61].
POLYPHENOLS AS THERAPEUTIC AND CHEMOSENSITIZER AGENTS IN HCC?
Personally, our attention to these agents has been attracted by the very simple consideration that examining the effects of curcumin and other polyphenols in the HL-60 myeloid leukemia and MCF-7 breast cancer cell lines and in their variants endowed with different mechanisms (over-expression of P-gp and IAPs) of drug resistance together, we could not observe any reduction in the antitumoral activity of these compounds in the MDR cells (personal unpublished observations). In addition, in many studies the polyphenols have been shown to increase tumor cell sensitivity to therapeutic agents and this being the result of different mechanisms, including counteraction of the drug transporters or of the detoxifying enzymes[62-64] (Figure 1). However, one of the mechanisms which has been particularly frequently put forth to explain these synergies is the interference of the polyphenols with the signaling of NF-κB. Curcumin and other polyphenols inhibit the transcription factor, generally by impeding the phosphorylation of the IκB factor by IκB kinase and thus preventing NF-κB nuclear translocation; a study has suggested that curcumin may also directly interfere with DNA binding by NF-κB[65]. Polyphenols may also inhibit Akt, which abrogates cell death signals through phosphorylation of Bad, GSK3 and caspase-9, elevation of different IAPs and other anti-apoptotic factors, and activation of transcriptional factors such as Forkhead and NF-κB itself. In turn, NF-κB may increase both the expression and phosphorylation of Akt[66]; thus the inhibition of each pathway may reinforce that of the other.
Figure 1.
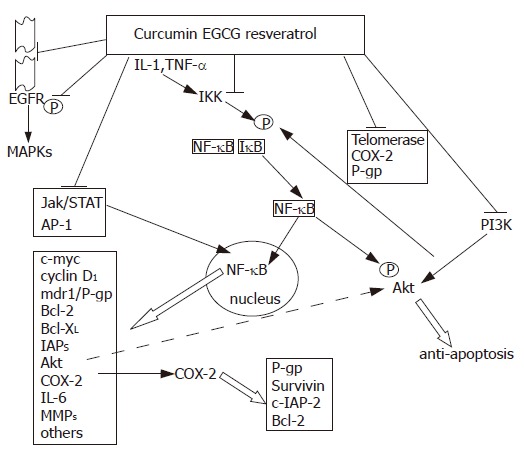
Some of the principal tumor targets of polyphenols discussed in the text. The agents may directly interfere with the affected molecules (e.g. Telomerase, COX-2 and P-gp) and/or influence their activity or expression due to modifications in an upstream regulatory signal or in factors (e.g. NF-κB) which regulate gene transcription.
With reference to the points above discussed, we have studied the effects of curcumin in the human HCC HA22T/VGH cells, which constitutively express activated NF-κB, the different IAPs and P-gp all together[67]. Curcumin exerted cell growth inhibitory and apoptotic effects, related, at least in part, to free radical generation and mainly dependent on caspase -9 and -3 activation. Curcumin sensitized the cells to the antitumoral effects of cisplatin, while the results were only additive in combination with doxorubicin. As reported in similar settings[53], curcumin reduced (only modestly) the basal levels of nuclear activated NF-κB (p65 subunit), but, when combined with cisplatin or doxorubicin, it blunted their increases induced from the two drugs, which were slight for cisplatin and very remarkable in the case of doxorubicin.
In the same cells, curcumin determined early reductions in COX-2, c-myc, P-gp and IL-6 mRNAs. Later it decreased Bcl-XL mRNA and increased Bcl-XS and c-IAP-2 mRNAs. Cisplatin and doxorubicin exerted distinct effects on gene expression and the interactions of these agents with curcumin were accompanied by synergistic (in particular with cisplatin) or additive reductions in the levels of different mRNAs, including those of c-myc, Bcl-XL, c-IAP-2, NAIP, XIAP and P-gp. Overall, the effects of curcumin on HA22T/VGH cells did not show a simple relationship to its influences on NF-κB activation, clearly implying the involvement of additional mechanisms; strikingly, curcumin affected above all the strong NF-κB activation from doxorubicin, but its antitumor effects were more favorable when it was combined with cisplatin. Further, at a molecular level, the effects of curcumin were in part different from those of a pure NF-κB inhibitor, dehydroxymethylepoxyquinomicin (DHMEQ), which also sensitized the cells to cisplatin[68].
In a study on ovarian cancer cells, in which increases in response to cisplatin induced by curcumin also occurred, this result was attributed, at least in part, to the ability of the agent in interfering with the autologous production of interleukin 6 (IL-6) by the cells[69]. Cancer cells may indeed often support their own growth, survival and drug resistance by autocrine/paracrine or also intracrine loops based on the production of different factors; similar IL-6-based loops, possibly driven by constitutive NF-κB activation, are operative in tumors like multiple myeloma and prostate or renal cancer[69-71].
Nevertheless, on the basis of different, also personal, observations on the inhibitory effect of IL-6 on the growth of HCC cell lines[72,73], we can propose that drug resistance mediated by autologous IL-6 is less likely to occur in HCC with respect to other cancers; further, even though curcumin or DHMEQ efficiently inhibited the abundant release of IL-6 by HA22T/VGH cells, this result, due to the lack of the IL-6 receptor alpha subunit in the cells, could not explain the direct antitumor effects of these agents, alone or in combination with cisplatin, in this model of HCC[68]. Nevertheless, since release of IL-6 by tumor cells is frequent in the more advanced stages of HCC[74], the use of curcumin or DHMEQ in this tumor might be beneficial also to contrast the adverse systemic effects (e.g. cachexia) of the cytokine.
CONCLUSIONS AND FUTURE DIRECTIONS
Overall, the findings of our and other groups support the possible use of natural multi-targeted agents like the polyphenols, alone or in combination with conventional chemotherapeutic agents, in the treatment of HCC[67,75-79]. Nevertheless, one of the principal limitations to the in vivo use of such compounds is their low oral bioavalaibility; in addition, free curcumin is highly hydrophobic and difficult to be administered systemically. Several efforts are now addressed to development of analogues, pro-drugs or delivery systems (liposomes, nanoparticles and others) to improve the pharmacokinetic characteristics of the polyphenols and enhance their disposition to the target tissues and tumors.
At the moment, however, different polyphenols are now already undergoing clinical trials, which ultimately will confirm or not the expectations regarding the efficacy and safety of their use in advanced cancers, including HCC.
Footnotes
Supported by PRIN MIUR 2005051211 2005
S- Editor Wang J L- Editor Karam SM E- Editor Chen GJ
References
- 1.D'Alessandro N, Flugy A, Borsellino N, Travali S. Biology versus pharmacology in drug resistance. Oncol Rep. 1997;4:207–210. [Google Scholar]
- 2.Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol. 1999;17:2941–2953. doi: 10.1200/JCO.1999.17.9.2941. [DOI] [PubMed] [Google Scholar]
- 3.LaCasse EC, Baird S, Korneluk RG, MacKenzie AE. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998;17:3247–3259. doi: 10.1038/sj.onc.1202569. [DOI] [PubMed] [Google Scholar]
- 4.Notarbartolo M, Cervello M, Dusonchet L, Cusimano A, D'Alessandro N. Resistance to diverse apoptotic triggers in multidrug resistant HL60 cells and its possible relationship to the expression of P-glycoprotein, Fas and of the novel anti-apoptosis factors IAP (inhibitory of apoptosis proteins) Cancer Lett. 2002;180:91–101. doi: 10.1016/s0304-3835(01)00834-5. [DOI] [PubMed] [Google Scholar]
- 5.Falkson JM, MacIntyre JM, Moertel CG. Primary liver cancer. Cancer. 1994;54:977–980. doi: 10.1002/1097-0142(19840915)54:6<970::aid-cncr2820540604>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 6.Montalto G, Cervello M, Giannitrapani L, Dantona F, Terranova A, Castagnetta LA. Epidemiology, risk factors, and natural history of hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:13–20. doi: 10.1111/j.1749-6632.2002.tb04090.x. [DOI] [PubMed] [Google Scholar]
- 7.Smyth MJ, Krasovskis E, Sutton VR, Johnstone RW. The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc Natl Acad Sci USA. 1998;95:7024–7029. doi: 10.1073/pnas.95.12.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chenivesse X, Franco D, Bréchot C. MDR1 (multidrug resistance) gene expression in human primary liver cancer and cirrhosis. J Hepatol. 1993;18:168–172. doi: 10.1016/s0168-8278(05)80243-0. [DOI] [PubMed] [Google Scholar]
- 9.Ng IO, Liu CL, Fan ST, Ng M. Expression of P-glycoprotein in hepatocellular carcinoma. A determinant of chemotherapy response. Am J Clin Pathol. 2000;113:355–363. doi: 10.1309/AC1M-4TY4-U0TN-EN7T. [DOI] [PubMed] [Google Scholar]
- 10.Kato A, Miyazaki M, Ambiru S, Yoshitomi H, Ito H, Nakagawa K, Shimizu H, Yokosuka O, Nakajima N. Multidrug resistance gene (MDR-1) expression as a useful prognostic factor in patients with human hepatocellular carcinoma after surgical resection. J Surg Oncol. 2001;78:110–115. doi: 10.1002/jso.1129. [DOI] [PubMed] [Google Scholar]
- 11.Grudé P, Conti F, Mennecier D, Louvel A, Houssin D, Weill B, Calmus Y. MDR1 gene expression in hepatocellular carcinoma and the peritumoral liver of patients with and without cirrhosis. Cancer Lett. 2002;186:107–113. doi: 10.1016/s0304-3835(02)00155-6. [DOI] [PubMed] [Google Scholar]
- 12.Zollner G, Wagner M, Fickert P, Silbert D, Fuchsbichler A, Zatloukal K, Denk H, Trauner M. Hepatobiliary transporter expression in human hepatocellular carcinoma. Liver Int. 2005;25:367–379. doi: 10.1111/j.1478-3231.2005.01033.x. [DOI] [PubMed] [Google Scholar]
- 13.Lasagna N, Fantappiè O, Solazzo M, Morbidelli L, Marchetti S, Cipriani G, Ziche M, Mazzanti R. Hepatocyte growth factor and inducible nitric oxide synthase are involved in multidrug resistance-induced angiogenesis in hepatocellular carcinoma cell lines. Cancer Res. 2006;66:2673–2682. doi: 10.1158/0008-5472.CAN-05-2290. [DOI] [PubMed] [Google Scholar]
- 14.Chou YY, Cheng AL, Hsu HC. Expression of P-glycoprotein and p53 in advanced hepatocellular carcinoma treated by single agent chemotherapy: clinical correlation. J Gastroenterol Hepatol. 1997;12:569–575. doi: 10.1111/j.1440-1746.1997.tb00487.x. [DOI] [PubMed] [Google Scholar]
- 15.Lai EC, Choi TK, Cheng CH, Mok FP, Fan ST, Tan ES, Wong J. Doxorubicin for unresectable hepatocellular carcinoma. A prospective study on the addition of verapamil. Cancer. 1990;66:1685–1687. doi: 10.1002/1097-0142(19901015)66:8<1685::aid-cncr2820660805>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 16.Nies AT, König J, Pfannschmidt M, Klar E, Hofmann WJ, Keppler D. Expression of the multidrug resistance proteins MRP2 and MRP3 in human hepatocellular carcinoma. Int J Cancer. 2001;94:492–499. doi: 10.1002/ijc.1498. [DOI] [PubMed] [Google Scholar]
- 17.Raidl M, Berger W, Schulte-Hermann R, Kandioler-Eckersberger D, Kappel S, Wrba F, Micksche M, Grasl-Kraupp B. Expression of the lung resistance-related protein in human and rat hepatocarcinogenesis. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1117–G1124. doi: 10.1152/ajpgi.00195.2002. [DOI] [PubMed] [Google Scholar]
- 18.Deng XL, Chen W, Cai MY, Wei DP. Expression of class I MHC molecule, HSP70 and TAP in human hepatocellular carcinoma. World J Gastroenterol. 2003;9:1853–1855. doi: 10.3748/wjg.v9.i8.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minemura M, Tanimura H, Tabor E. Overexpression of multidrug resistance genes MDR1 and cMOAT in human hepatocellular carcinoma and hepatoblastoma cell lines. Int J Oncol. 1999;15:559–563. doi: 10.3892/ijo.15.3.559. [DOI] [PubMed] [Google Scholar]
- 20.Vavricka SR, Jung D, Fried M, Grützner U, Meier PJ, Kullak-Ublick GA. The human organic anion transporting polypeptide 8 (SLCO1B3) gene is transcriptionally repressed by hepatocyte nuclear factor 3beta in hepatocellular carcinoma. J Hepatol. 2004;40:212–218. doi: 10.1016/j.jhep.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Murray GI, Paterson PJ, Weaver RJ, Ewen SW, Melvin WT, Burke MD. The expression of cytochrome P-450, epoxide hydrolase, and glutathione S-transferase in hepatocellular carcinoma. Cancer. 1993;71:36–43. doi: 10.1002/1097-0142(19930101)71:1<36::aid-cncr2820710107>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 22.Hayes PC, May L, Hayes JD, Harrison DJ. Glutathione S-transferases in human liver cancer. Gut. 1991;32:1546–1549. doi: 10.1136/gut.32.12.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fritz P, Behrle E, Beaune P, Eichelbaum M, Kroemer HK. Differential expression of drug metabolizing enzymes in primary and secondary liver neoplasm: immunohistochemical characterization of cytochrome P4503A and glutathione-S-transferase. Histochemistry. 1993;99:443–451. doi: 10.1007/BF00274096. [DOI] [PubMed] [Google Scholar]
- 24.Yusof YA, Yan KL, Hussain SN. Immunohistochemical expression of pi class glutathione S-transferase and alpha-fetoprotein in hepatocellular carcinoma and chronic liver disease. Anal Quant Cytol Histol. 2003;25:332–338. [PubMed] [Google Scholar]
- 25.Shen LJ, Zhang HX, Zhang ZJ, Li JY, Chen MQ, Yang WB, Huang R. Detection of HBV, PCNA and GST-pi in hepatocellular carcinoma and chronic liver diseases. World J Gastroenterol. 2003;9:459–462. doi: 10.3748/wjg.v9.i3.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frommel TO, Yong S, Zarling EJ. Immunohistochemical evaluation of Bcl-2 gene family expression in liver of hepatitis C and cirrhotic patients: a novel mechanism to explain the high incidence of hepatocarcinoma in cirrhotics. Am J Gastroenterol. 1999;94:178–182. doi: 10.1111/j.1572-0241.1999.00792.x. [DOI] [PubMed] [Google Scholar]
- 27.Wheatley SP, McNeish IA. Survivin: a protein with dual roles in mitosis and apoptosis. Int Rev Cytol. 2005;247:35–88. doi: 10.1016/S0074-7696(05)47002-3. [DOI] [PubMed] [Google Scholar]
- 28.Ito T, Shiraki K, Sugimoto K, Yamanaka T, Fujikawa K, Ito M, Takase K, Moriyama M, Kawano H, Hayashida M, et al. Survivin promotes cell proliferation in human hepatocellular carcinoma. Hepatology. 2000;31:1080–1085. doi: 10.1053/he.2000.6496. [DOI] [PubMed] [Google Scholar]
- 29.Ikeguchi M, Hirooka Y, Kaibara N. Quantitative analysis of apoptosis-related gene expression in hepatocellular carcinoma. Cancer. 2002;95:1938–1945. doi: 10.1002/cncr.10898. [DOI] [PubMed] [Google Scholar]
- 30.Moon WS, Tarnawski AS. Nuclear translocation of survivin in hepatocellular carcinoma: a key to cancer cell growth? Hum Pathol. 2003;34:1119–1126. doi: 10.1053/j.humpath.2003.07.016. [DOI] [PubMed] [Google Scholar]
- 31.Shiraki K, Nakano T, Hisatomi H. Survivin in liver. Gastroenterology. 2003;124:1565–1566; author reply 1565-1566;. doi: 10.1016/s0016-5085(03)00359-7. [DOI] [PubMed] [Google Scholar]
- 32.Morinaga S, Nakamura Y, Ishiwa N, Yoshikawa T, Noguchi Y, Yamamoto Y, Rino Y, Imada T, Takanashi Y, Akaike M, et al. Expression of survivin mRNA associates with apoptosis, proliferation and histologically aggressive features in hepatocellular carcinoma. Oncol Rep. 2004;12:1189–1194. [PubMed] [Google Scholar]
- 33.Notarbartolo M, Cervello M, Giannitrapani L, Meli M, Poma P, Dusonchet L, Montalto G, D'Alessandro N. Expression of IAPs and alternative splice variants in hepatocellular carcinoma tissues and cells. Ann N Y Acad Sci. 2004;1028:289–293. doi: 10.1196/annals.1322.033. [DOI] [PubMed] [Google Scholar]
- 34.Fields AC, Cotsonis G, Sexton D, Santoianni R, Cohen C. Survivin expression in hepatocellular carcinoma: correlation with proliferation, prognostic parameters, and outcome. Mod Pathol. 2004;17:1378–1385. doi: 10.1038/modpathol.3800203. [DOI] [PubMed] [Google Scholar]
- 35.Zhu H, Chen XP, Zhang WG, Luo SF, Zhang BX. Expression and significance of new inhibitor of apoptosis protein survivin in hepatocellular carcinoma. World J Gastroenterol. 2005;11:3855–3859. doi: 10.3748/wjg.v11.i25.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan XX, Ou JS, Li Y, Su JJ, Ou C, Yang C, Yue HF, Ban KC. Dynamic expression of apoptosis-related genes during development of laboratory hepatocellular carcinoma and its relation to apoptosis. World J Gastroenterol. 2005;11:4740–4744. doi: 10.3748/wjg.v11.i30.4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kannangai R, Wang J, Liu QZ, Sahin F, Torbenson M. Survivin overexpression in hepatocellular carcinoma is associated with p53 dysregulation. Int J Gastrointest Cancer. 2005;35:53–60. doi: 10.1385/IJGC:35:1:053. [DOI] [PubMed] [Google Scholar]
- 38.Takashima H, Nakajima T, Moriguchi M, Sekoguchi S, Nishikawa T, Watanabe T, Katagishi T, Kimura H, Minami M, Itoh Y, et al. In vivo expression patterns of survivin and its splicing variants in chronic liver disease and hepatocellular carcinoma. Liver Int. 2005;25:77–84. doi: 10.1111/j.1478-3231.2004.0979.x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Dong N, Yin L, Cai N, Ma H, You J, Zhang H, Wang H, He R, Ye L. Hepatitis B virus X protein upregulates survivin expression in hepatoma tissues. J Med Virol. 2005;77:374–381. doi: 10.1002/jmv.20466. [DOI] [PubMed] [Google Scholar]
- 40.Guo RP, Zhong C, Shi M, Zhang CQ, Wei W, Zhang YQ, Li JQ. Clinical value of apoptosis and angiogenesis factors in estimating the prognosis of hepatocellular carcinoma. J Cancer Res Clin Oncol. 2006;132:547–555. doi: 10.1007/s00432-006-0097-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I, Reed JC. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003;22:2729–2740. doi: 10.1093/emboj/cdg263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu X, Lee M, Tran T, Block T. High level expression of apoptosis inhibitor in hepatoma cell line expressing Hepatitis B virus. Int J Med Sci. 2005;2:30–35. doi: 10.7150/ijms.2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shiraki K, Sugimoto K, Yamanaka Y, Yamaguchi Y, Saitou Y, Ito K, Yamamoto N, Yamanaka T, Fujikawa K, Murata K, et al. Overexpression of X-linked inhibitor of apoptosis in human hepatocellular carcinoma. Int J Mol Med. 2003;12:705–708. [PubMed] [Google Scholar]
- 44.Yamaguchi Y, Shiraki K, Fuke H, Inoue T, Miyashita K, Yamanaka Y, Saitou Y, Sugimoto K, Nakano T. Targeting of X-linked inhibitor of apoptosis protein or survivin by short interfering RNAs sensitize hepatoma cells to TNF-related apoptosis-inducing ligand- and chemotherapeutic agent-induced cell death. Oncol Rep. 2005;14:1311–1316. [PubMed] [Google Scholar]
- 45.Nakao K, Hamasaki K, Ichikawa T, Arima K, Eguchi K, Ishii N. Survivin downregulation by siRNA sensitizes human hepatoma cells to TRAIL-induced apoptosis. Oncol Rep. 2006;16:389–392. [PubMed] [Google Scholar]
- 46.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 47.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, Hideshima T, Munshi N, Treon SP, Anderson KC. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood. 2002;99:4079–4086. doi: 10.1182/blood.v99.11.4079. [DOI] [PubMed] [Google Scholar]
- 48.Liu P, Kimmoun E, Legrand A, Sauvanet A, Degott C, Lardeux B, Bernuau D. Activation of NF-kappa B, AP-1 and STAT transcription factors is a frequent and early event in human hepatocellular carcinomas. J Hepatol. 2002;37:63–71. doi: 10.1016/s0168-8278(02)00064-8. [DOI] [PubMed] [Google Scholar]
- 49.Chiao PJ, Na R, Niu J, Sclabas GM, Dong Q, Curley SA. Role of Rel/NF-kappaB transcription factors in apoptosis of human hepatocellular carcinoma cells. Cancer. 2002;95:1696–1705. doi: 10.1002/cncr.10829. [DOI] [PubMed] [Google Scholar]
- 50.Chan CF, Yau TO, Jin DY, Wong CM, Fan ST, Ng IO. Evaluation of nuclear factor-kappaB, urokinase-type plasminogen activator, and HBx and their clinicopathological significance in hepatocellular carcinoma. Clin Cancer Res. 2004;10:4140–4149. doi: 10.1158/1078-0432.CCR-03-0574. [DOI] [PubMed] [Google Scholar]
- 51.Arsura M, Cavin LG. Nuclear factor-kappaB and liver carcinogenesis. Cancer Lett. 2005;229:157–169. doi: 10.1016/j.canlet.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 52.Kuo MT, Liu Z, Wei Y, Lin-Lee YC, Tatebe S, Mills GB, Unate H. Induction of human MDR1 gene expression by 2-acetylaminofluorene is mediated by effectors of the phosphoinositide 3-kinase pathway that activate NF-kappaB signaling. Oncogene. 2002;21:1945–1954. doi: 10.1038/sj.onc.1205117. [DOI] [PubMed] [Google Scholar]
- 53.Chuang SE, Yeh PY, Lu YS, Lai GM, Liao CM, Gao M, Cheng AL. Basal levels and patterns of anticancer drug-induced activation of nuclear factor-kappaB (NF-kappaB), and its attenuation by tamoxifen, dexamethasone, and curcumin in carcinoma cells. Biochem Pharmacol. 2002;63:1709–1716. doi: 10.1016/s0006-2952(02)00931-0. [DOI] [PubMed] [Google Scholar]
- 54.Ashikawa K, Shishodia S, Fokt I, Priebe W, Aggarwal BB. Evidence that activation of nuclear factor-kappaB is essential for the cytotoxic effects of doxorubicin and its analogues. Biochem Pharmacol. 2004;67:353–364. doi: 10.1016/j.bcp.2003.08.039. [DOI] [PubMed] [Google Scholar]
- 55.Campbell KJ, Rocha S, Perkins ND. Active repression of antiapoptotic gene expression by RelA(p65) NF-kappa B. Mol Cell. 2004;13:853–865. doi: 10.1016/s1097-2765(04)00131-5. [DOI] [PubMed] [Google Scholar]
- 56.Ho WC, Dickson KM, Barker PA. Nuclear factor-kappaB induced by doxorubicin is deficient in phosphorylation and acetylation and represses nuclear factor-kappaB-dependent transcription in cancer cells. Cancer Res. 2005;65:4273–4281. doi: 10.1158/0008-5472.CAN-04-3494. [DOI] [PubMed] [Google Scholar]
- 57.Costantino L, Barlocco D. Privileged structures as leads in medicinal chemistry. Curr Med Chem. 2006;13:65–85. [PubMed] [Google Scholar]
- 58.Manson MM. Inhibition of survival signalling by dietary polyphenols and indole-3-carbinol. Eur J Cancer. 2005;41:1842–1853. doi: 10.1016/j.ejca.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 59.D'Incalci M, Steward WP, Gescher AJ. Use of cancer chemopreventive phytochemicals as antineoplastic agents. Lancet Oncol. 2005;6:899–904. doi: 10.1016/S1470-2045(05)70425-3. [DOI] [PubMed] [Google Scholar]
- 60.Anuchapreeda S, Leechanachai P, Smith MM, Ambudkar SV, Limtrakul PN. Modulation of P-glycoprotein expression and function by curcumin in multidrug-resistant human KB cells. Biochem Pharmacol. 2002;64:573–582. doi: 10.1016/s0006-2952(02)01224-8. [DOI] [PubMed] [Google Scholar]
- 61.Bielak-Mijewska A, Piwocka K, Magalska A, Sikora E. P-glycoprotein expression does not change the apoptotic pathway induced by curcumin in HL-60 cells. Cancer Chemother Pharmacol. 2004;53:179–185. doi: 10.1007/s00280-003-0705-x. [DOI] [PubMed] [Google Scholar]
- 62.Garg AK, Buchholz TA, Aggarwal BB. Chemosensitization and radiosensitization of tumors by plant polyphenols. Antioxid Redox Signal. 2005;7:1630–1647. doi: 10.1089/ars.2005.7.1630. [DOI] [PubMed] [Google Scholar]
- 63.HemaIswarya S, Doble M. Potential synergism of natural products in the treatment of cancer. Phytother Res. 2006;20:239–249. doi: 10.1002/ptr.1841. [DOI] [PubMed] [Google Scholar]
- 64.Sarkar FH, Li Y. Using chemopreventive agents to enhance the efficacy of cancer therapy. Cancer Res. 2006;66:3347–3350. doi: 10.1158/0008-5472.CAN-05-4526. [DOI] [PubMed] [Google Scholar]
- 65.Brennan P, O'Neill LA. Inhibition of nuclear factor kappaB by direct modification in whole cells--mechanism of action of nordihydroguaiaritic acid, curcumin and thiol modifiers. Biochem Pharmacol. 1998;55:965–973. doi: 10.1016/s0006-2952(97)00535-2. [DOI] [PubMed] [Google Scholar]
- 66.Meng F, D'Mello SR. NF-kappaB stimulates Akt phosphorylation and gene expression by distinct signaling mechanisms. Biochim Biophys Acta. 2003;1630:35–40. doi: 10.1016/j.bbaexp.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 67.Notarbartolo M, Poma P, Perri D, Dusonchet L, Cervello M, D'Alessandro N. Antitumor effects of curcumin, alone or in combination with cisplatin or doxorubicin, on human hepatic cancer cells. Analysis of their possible relationship to changes in NF-kB activation levels and in IAP gene expression. Cancer Lett. 2005;224:53–65. doi: 10.1016/j.canlet.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 68.Poma P, Notarbartolo M, Labbozzetta M, Sanguedolce R, Alaimo A, Carina V, Maurici A, Cusimano A, Cervello M, D'Alessandro N. Antitumor effects of the novel NF-kappaB inhibitor dehydroxymethyl-epoxyquinomicin on human hepatic cancer cells: analysis of synergy with cisplatin and of possible correlation with inhibition of pro-survival genes and IL-6 production. Int J Oncol. 2006;28:923–930. [PubMed] [Google Scholar]
- 69.Chan MM, Fong D, Soprano KJ, Holmes WF, Heverling H. Inhibition of growth and sensitization to cisplatin-mediated killing of ovarian cancer cells by polyphenolic chemopreventive agents. J Cell Physiol. 2003;194:63–70. doi: 10.1002/jcp.10186. [DOI] [PubMed] [Google Scholar]
- 70.Borsellino N, Bonavida B, Ciliberto G, Toniatti C, Travali S, D'Alessandro N. Blocking signaling through the Gp130 receptor chain by interleukin-6 and oncostatin M inhibits PC-3 cell growth and sensitizes the tumor cells to etoposide and cisplatin-mediated cytotoxicity. Cancer. 1999;85:134–144. [PubMed] [Google Scholar]
- 71.Xiao W, Hodge DR, Wang L, Yang X, Zhang X, Farrar WL. NF-kappaB activates IL-6 expression through cooperation with c-Jun and IL6-AP1 site, but is independent of its IL6-NFkappaB regulatory site in autocrine human multiple myeloma cells. Cancer Biol Ther. 2004;3:1007–1017. doi: 10.4161/cbt.3.10.1141. [DOI] [PubMed] [Google Scholar]
- 72.Cervello M, Notarbartolo M, Landino M, Cusimano A, Virruso L, Montalto G, D'Alessandro N. Downregulation of wild-type beta-catenin expression by interleukin 6 in human hepatocarcinoma HepG2 cells: a possible role in the growth-regulatory effects of the cytokine? Eur J Cancer. 2001;37:512–519. doi: 10.1016/s0959-8049(00)00421-4. [DOI] [PubMed] [Google Scholar]
- 73.Moran DM, Mayes N, Koniaris LG, Cahill PA, McKillop IH. Interleukin-6 inhibits cell proliferation in a rat model of hepatocellular carcinoma. Liver Int. 2005;25:445–457. doi: 10.1111/j.1478-3231.2005.01083.x. [DOI] [PubMed] [Google Scholar]
- 74.Giannitrapani L, Cervello M, Soresi M, Notarbartolo M, La Rosa M, Virruso L, D'Alessandro N, Montalto G. Circulating IL-6 and sIL-6R in patients with hepatocellular carcinoma. Ann N Y Acad Sci. 2002;963:46–52. doi: 10.1111/j.1749-6632.2002.tb04093.x. [DOI] [PubMed] [Google Scholar]
- 75.Lin LI, Ke YF, Ko YC, Lin JK. Curcumin inhibits SK-Hep-1 hepatocellular carcinoma cell invasion in vitro and suppresses matrix metalloproteinase-9 secretion. Oncology. 1998;55:349–353. doi: 10.1159/000011876. [DOI] [PubMed] [Google Scholar]
- 76.Jung EM, Lim JH, Lee TJ, Park JW, Choi KS, Kwon TK. Curcumin sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through reactive oxygen species-mediated upregulation of death receptor 5 (DR5) Carcinogenesis. 2005;26:1905–1913. doi: 10.1093/carcin/bgi167. [DOI] [PubMed] [Google Scholar]
- 77.Yoysungnoen P, Wirachwong P, Bhattarakosol P, Niimi H, Patumraj S. Effects of curcumin on tumor angiogenesis and biomarkers, COX-2 and VEGF, in hepatocellular carcinoma cell-implanted nude mice. Clin Hemorheol Microcirc. 2006;34:109–115. [PubMed] [Google Scholar]
- 78.Wu SL, Sun ZJ, Yu L, Meng KW, Qin XL, Pan CE. Effect of resveratrol and in combination with 5-FU on murine liver cancer. World J Gastroenterol. 2004;10:3048–3052. doi: 10.3748/wjg.v10.i20.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nishikawa T, Nakajima T, Moriguchi M, Jo M, Sekoguchi S, Ishii M, Takashima H, Katagishi T, Kimura H, Minami M, et al. A green tea polyphenol, epigalocatechin-3-gallate, induces apoptosis of human hepatocellular carcinoma, possibly through inhibition of Bcl-2 family proteins. J Hepatol. 2006;44:1074–1082. doi: 10.1016/j.jhep.2005.11.045. [DOI] [PubMed] [Google Scholar]