

Abstract
Renal ammonia metabolism and transport mediates a central role in acid-base homeostasis. In contrast to most renal solutes, the majority of renal ammonia excretion derives from intrarenal production, not from glomerular filtration. Renal ammoniagenesis predominantly results from glutamine metabolism, which produces 2 NH4+ and 2 HCO3− for each glutamine metabolized. The proximal tubule is the primary site for ammoniagenesis, but there is evidence for ammoniagenesis by most renal epithelial cells. Ammonia produced in the kidney is either excreted into the urine or returned to the systemic circulation through the renal veins. Ammonia excreted in the urine promotes acid excretion; ammonia returned to the systemic circulation is metabolized in the liver in a HCO3−-consuming process, resulting in no net benefit to acid-base homeostasis. Highly regulated ammonia transport by renal epithelial cells determines the proportion of ammonia excreted in the urine versus returned to the systemic circulation. The traditional paradigm of ammonia transport involving passive NH3 diffusion, protonation in the lumen and NH4+ trapping due to an inability to cross plasma membranes is being replaced by the recognition of limited plasma membrane NH3 permeability in combination with the presence of specific NH3-transporting and NH4+-transporting proteins in specific renal epithelial cells. Ammonia production and transport are regulated by a variety of factors, including extracellular pH and K+, and by several hormones, such as mineralocorticoids, glucocorticoids and angiotensin II. This coordinated process of regulated ammonia production and transport is critical for the effective maintenance of acid-base homeostasis.
Introduction
Acid-base homeostasis is critical for normal health, growth, and development. Acid-base disorders lead to a wide variety of clinical problems, including growth retardation in neonates and children, nausea and vomiting, electrolyte disturbances, increased susceptibility to cardiac arrhythmias, decreased catecholamine sensitivity, particularly in the cardiovascular system, osteoporosis and osteomalacia, recurrent nephrolithiasis, skeletal muscle atrophy, paresthesias, and coma.
The kidneys have two major functions in acid-base homeostasis, reabsorption of filtered bicarbonate and generation of new bicarbonate. First, daily filtered bicarbonate averages approximately 4200 mmol/d for the adult with normal renal function, and is reabsorbed almost completely by renal epithelial cells. Second, both intracellular and extracellular buffers are used to buffer endogenous and exogenous acid loads and must be replenished to maintain acid-base homoeostasis. The kidneys produce “new bicarbonate” to do so, and the primary mechanism of new bicarbonate generation involves renal ammonia metabolism. This manuscript’s purpose is to review the mechanisms and the regulation of renal ammonia metabolism and transport. Ammonia exists in two different molecular forms, NH3 and NH4+. In this manuscript, we use the terms “ammonia” and “total ammonia” to refer to the combination of NH3 and NH4+. When referring specifically to NH3, we state “NH3.” When discussing NH4+ we state “NH4+” specifically.
Role of Ammonia Metabolism in Acid-Base Homeostasis
The major acid-base disturbance that mammals face on a daily basis reflects the acid load resulting from metabolism of proteins. For the average human, this acid load approximates 0.8 mEq/kg/d, or approximately 56 mEq for a 70 kg individual. The correlation between endogenous acid production and body mass presumably occurs because protein metabolism often parallels body mass, and endogenous acid production results from endogenous protein metabolism. This acid load is buffered by intracellular and extracellular buffers, primarily bicarbonate. Long-term maintenance of acid-base homeostasis requires production of new alkaline buffers from the process of net acid excretion to replace those “consumed” in buffering this daily acid load. In humans and in rodents, urinary ammonia excretion comprises approximately 50% to 70% of basal net acid excretion (15, 36, 92, 132, 177).
Understanding the role of renal ammonia metabolism in acid-base homeostasis involves consideration of the effects of renal and extrarenal ammonia metabolism. Renal ammoniagenesis, utilizing glutamine as the primary metabolic substrate, generates equivalent amounts of NH4+ and HCO3−. The HCO3− produced is preferentially transported across the basolateral plasma membrane, absorbed into peritubular capillaries and returned to the systemic circulation. Renal NH4+ excretion thereby quantitatively indicates renal HCO3− production. Although a substantial component of renal NH4+ that is produced is returned to the systemic circulation via the renal vein, this NH4+is metabolized in the liver, predominantly via the urea cycle, in a process that utilizes HCO3−. Accordingly, the component of NH4+ produced by the kidney that is then metabolized by the liver does not contribute to “new” HCO3− production.
Regulation of renal ammoniagenesis, transport, and excretion are integral to acid-base homeostasis. In response to metabolic acidosis (Fig. 1), increases in urinary ammonia excretion are the primary component of the increase in net acid excretion (36, 92, 177); indeed in studies in the mouse using HCl addition to food to induce metabolic acidosis, we observed no change in titratable acid excretion; increased ammonia excretion was almost entirely responsible for the increase in net acid excretion (15, 92). In hypokalemia, urinary ammonia excretion increases despite concurrent metabolic alkalosis (60), and may be a major factor in the development of metabolic alkalosis. Thus, in many of the most common clinical conditions changes in urinary ammonia excretion are the predominant mechanism through which the kidneys regulate net acid excretion.
Figure 1.
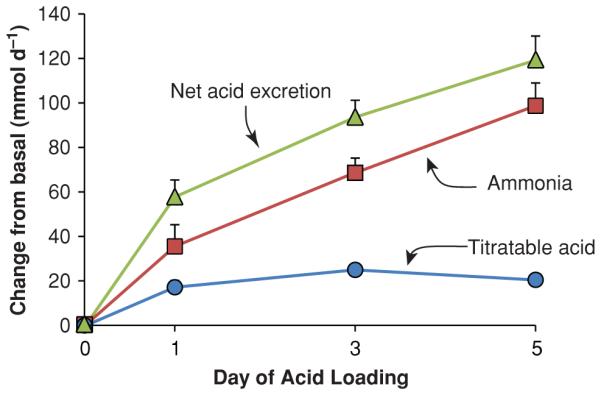
Relative responses of titratable acid and ammonia excretion in the response to metabolic acidosis. Normal human volunteers were acid loaded with approximately 2 mmol/kg/d of ammonium chloride and changes in urinary ammonia and titratable acid excretion were quantified. Data recalculated, with permission, from reference (36).
Overview of Ammonia Metabolism
Ammonia metabolism involves integrated function of multiple portions of the kidney. Only a minimal amount of urinary ammonia derives from glomerular filtration, making ammonia unique among the major compounds present in the urine. Quantitatively, assuming a plasma ammonia concentration of 20 μmol/L and a GFR of 120 mL/min/1.73 m2, the filtered load of ammonia in a normal individual is approximately 3 to 4 mmol/d, 60 to 80% of which is reabsorbed prior to the collecting duct, leading to filtered ammonia contributing only approximately 1 mmol/d of total renal ammonia excretion. In most individuals, the normal urinary ammonia excretion is 30 to 40 mmol/d, indicating that filtered ammonia is only a minor component of total excreted ammonia. In response to metabolic acidosis, where urinary ammonia can increase to more than 200 mmol/d, plasma ammonia levels, and consequently the filtered load of ammonia, do not change and may even decrease slightly (126). Renal ammonia excretion almost entirely reflects intrarenal ammoniagenesis and selective renal epithelial cell ammonia transport into either the urine or into the circulation.
Ammonia’s selective transport into either the tubule fluid, where it can be excreted, or into the peritubular fluid, when it can be returned to the systemic circulation, determines its effect on acid-base homeostasis. Ammonia excreted into the urine results in equimolar new bicarbonate formation. Ammonia returned to the systemic circulation is metabolized by the liver; this process utilizes equimolar amounts of bicarbonate, thereby consuming the bicarbonate produced during renal ammoniagenesis. This process of selective, directional transport is a critical component of the renal regulation of acid-base homeostasis. Under basal conditions, approximately 50% of ammonia generated by the kidney is excreted in the urine and approximately 50% returns to the renal vein (126). With metabolic acidosis, total renal ammoniagenesis increases, urinary ammonia excretion increases, but ammonia return to the circulation does not change proportionately, and may not even change at all (126). Selective transport of ammonia into either the urine or the circulation involves integrated transport in the proximal tubule, thick ascending limb of the loop of Henle, and the collecting duct (Fig. 2).
Figure 2.

Summary of renal ammonia metabolism. The proximal tubule produces ammonia, as NH4+, from glutamine. NH4+ is then secreted preferentially into the luminal fluid, primarily by NHE-3, and, in addition, there is a component of NH3 secretion. Ammonia is then reabsorbed in the thick ascending limb, resulting in ammonia delivery to the distal nephron accounting for approximately 20% to 40% of final urinary ammonia. The remaining approximately 60% to 80% of urinary ammonia is secreted in the collecting duct through parallel NH3 and H+ transport. Numbers in red indicate the proportion of total urinary ammonia present at the indicated sites under baseline conditions. Figure modified from reference (180) with permission.
Ammonia Chemistry
Ammonia exists in two molecular forms, NH3 and NH4+. The relative amounts of each are governed by the buffer reaction: NH3 + H+ ↔ NH4+. This reaction occurs essentially instantaneously and has a pKa’ under biologically relevant conditions of approximately 9.15. Accordingly, the majority of ammonia is present as NH4+; at pH 7.4 only approximately 1.7 % of total ammonia is present as NH3. The pH of most biological fluids is substantially below the pKa’ of this buffer reaction; consequently changes in pH cause exponential changes in NH3 concentration, but almost no change in NH4+ concentration (Fig. 3). Because there are two molecular forms of ammonia, one needs to consider the biophysical characteristics of each to understand the differences in transport of each.
Figure 3.
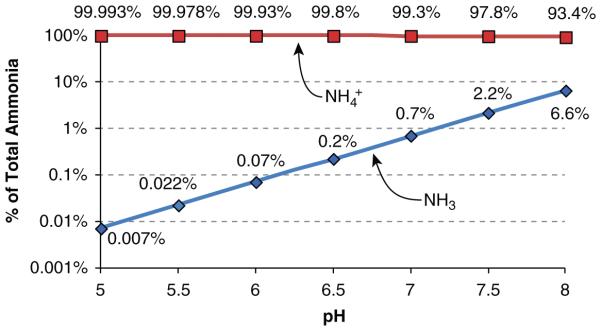
Relative changes in NH3 and NH4+ in solution as a function of solution pH. NH3 and NH4+ contributions to total ammonia were determined from the buffer reaction, NH3 + H+ ↔ NH4+. A pKa′ of 9.15 was used for calculations. Amounts shown are proportion of total ammonia present as NH3 and as NH4+. Note that the Y-axis is log transformed.
NH3 is a small, uncharged molecule. These characteristics led initially to the belief that NH3 is highly lipid permeable and essentially in diffusion equilibrium across lipid membranes. However, a substantial body of evidence indicates that this is not the case. In particular, NH3, although uncharged, has an asymmetric arrangement of positively charged hydrogen nuclei surrounding a central nitrogen, which makes NH3 a relatively polar molecule (Fig. 4). Quantitatively, NH3 has a molecular dipole moment, a measure of the degree of separation of positive and negative charge, of 1.47 D (73). For comparison, the molecular dipole moments of HCl and H2O are 1.08 and 1.85, respectively. Functionally, this charge polarity results in NH3 actually having only a limited lipid permeability (14,124), which leads to limited, sometimes only minimal, plasma membrane NH3 permeability (18,152,168,193). Importantly, limited plasma membrane NH3 permeability causes the development of transepithelial NH3 gradients, which have been demonstrated to be present in the kidney (50).
Figure 4.
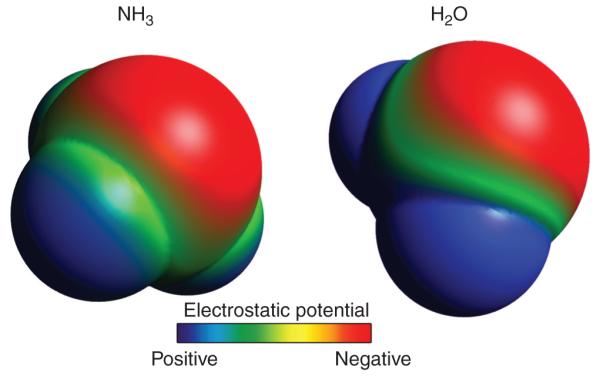
Space-filling model of NH3 showing molecular polarity. Space-filling model was created using Avogadro, v1.0.3, software. Surface is pseudocolored to demonstrate surface charge characteristics (red—negative, blue—positive). A similar process was used to generate space-filling model of H2O. Molecules are not drawn to scale.
NH4+ also has limited permeability across lipid bilayers in the absence of specific transport proteins. However, the model of ammonia transport with “NH4+ trapping” due to lack of NH4+ transport across plasma membranes is now known to not be fully correct. In the absence of specific transporters, NH4+ has a very low permeability across plasma membranes. Thus, in some tissues, such as the apical membrane of collecting duct segments, there is essentially no detectable NH4+ permeability. However, in many tissues, specific proteins are able to transport NH4+ across plasma membranes, and this process is critical to renal ammonia excretion. The ability of these proteins to transport NH4+ is due, in large part, to the specific biophysical characteristics of hydrated NH4+ ions. When examined in aqueous solutions, NH4+ and K+ have nearly identical biophysical characteristics (177, 180). This effective molecular mimicry enables NH4+ to be transported at the K+-transport site of essentially every known K+-transporting protein (177). As will be discussed below, NH4+ transport by specific membrane proteins contributes importantly to renal ammonia excretion.
Ammonia Production
Ammonia is produced by almost all renal epithelial cells, but the proximal tubule is the primary site for physiologically relevant ammoniagenesis. Studies examining dissected renal segments, including the glomeruli, S1, S2, and S3, descending thin limb of the loop of Henle, medullary and cortical thick ascending limb of the loop of Henle, distal convoluted tubule (DCT), cortical collecting duct (CCD), outer medullary collecting duct (OMCD), and inner medullary collecting duct (IMCD), show that all have the capability to synthesize ammonia using glutamine as their primary metabolic substrate (49). Quantitatively, metabolism of each glutamine molecule leads to generation of 2 NH4+ and 2 HCO3− ions when glutamine is metabolized through phosphate-dependent glutaminase (PDG), glutamate dehydrogenase (GDH), and phosphoenolpyruvate carboxykinase (PEPCK) during metabolic acidosis and hypokalemia. Although essentially all renal epithelial cells have the capability of producing ammonia, most studies have shown that metabolic acidosis only increases the rates of ammoniagenesis in the S1 and S2 proximal tubule segments (49, 190), and under certain in vitro conditions also in S3 segments (116). Taking into consideration the production rates per unit length and the relative lengths of the different nephron segments, the proximal tubule accounts for 60% to 70% of total renal ammonia production under basal conditions and at least 70 to 80% under conditions of metabolic acidosis (49) (Fig. 5).
Figure 5.

Ammonia production in various renal segments. Ammonia production rates in different renal components measured in microdissected tubule segments from rats on control diets and after inducing metabolic acidosis. All segments produced ammonia. Metabolic acidosis increases total renal ammoniagenesis, but only through increased production in proximal tubule segments (S1, S2, and S3). Rates calculated from measured ammonia production rates and mean length per segment as described in (49). Size of pie graph is proportional to total renal ammoniagenesis rates. Abbreviations: DTL, descending thin limb of Henle’s loop; MAL, medullary thick ascending limb of Henle’s loop; CAL, cortical thick ascending limb of Henle’s lop; DCT, distal convoluted tubule; CCD, cortical collecting duct; OMCD, outer medullary collecting duct; IMCD, inner medullary collecting duct.
Multiple pathways for ammoniagenesis are present in the proximal tubule (Fig. 6), but the predominant pathway involves the enzyme, PDG. PDG is present inside mitochondria and metabolizes glutamine to glutamate, producing NH4+. Glutamate then undergoes further metabolism through multiple pathways. The major pathway involves GDH, resulting in production of α-ketoglutarate (α-KG) and NH4+. GDH-mediated metabolism of glutamate to form α-KG is the quantitatively predominant mechanism, and is regulated in parallel with changes in total renal ammoniagenesis. Although transamination of glutamate via glutamic-oxaloacetic transaminase (GOT) does not release NH4+, the aspartate produced can be metabolized through the purine nucleotide cycle (PNC), forming fumarate and releasing NH4+. Quantitatively, the PNC pathway mediates only a minor role in overall ammoniagenesis (159). Glutamate can also be metabolized through glutamate decarboxylase to form γ-aminobutyric acid (GABA), and this may account for as much as 25% of glutamate oxidation in the renal cortex (89), but this pathway does not alter net ammoniagenesis since GABA metabolism to succinate semialdehyde via the enzyme γ-aminobutyrate transaminase results in regeneration of glutamate.
Figure 6.

Mechanisms of ammoniagenesis. Multiple pathways for enzymatic ammonia production originating from glutamine metabolism are present in the proximal tubule. Glutamine metabolism through phosphate-dependent glutaminase (PDG) and glutamate dehydrogenase (GDH) and involving phosphoenolpyruvate carboxykinase (PEPCK) is the quantitatively most significant component of renal ammoniagenesis and the primary pathway stimulated in response to metabolic acidosis.
α-KG produced from GDH is then sequentially metabolized resulting in HCO3− production. α-KG is metabolized through the enzymes α-KG-dehydrogenase and succinate dehydrogenase to form oxaloacetic acid (OAA). OAA then serves as a substrate for PEPCK to form PEP, which can then be utilized for gluconeogenesis (157). Thus, ammoniagenesis results in renal gluconeogenesis and renal gluconeogenesis is increased by many conditions which increase ammoniagenesis, including metabolic acidosis, respiratory acidosis, and hypokalemia (4).
Glutamate can also be converted back to glutamine via the enzyme glutamine synthetase. Because this reaction utilizes NH4+ as a cosubstrate, it results in decreased net NH4+ formation. Glutamine synthetase is expressed primarily in the proximal tubule of the rat and mouse (23, 130). Metabolic acidosis in the rat decreases glutamine synthetase activity (95), but not protein expression (161), and in the mouse decreases both activity and expression (15, 29, 92, 93).
Glutamine can also be metabolized by γ-glutamyl transpeptidase (γ-GT), also known as phosphate-independent glutaminase, although this is unlikely to be a major mechanism of renal ammoniagenesis. The glutamate resulting from γ-GT-dependent glutamine metabolism is then transported into the cell, where it can undergo further metabolism through GDH, resulting in further NH4+ production. Another pathway involves sequential metabolism through glutamine ketoacid aminotransferase and ω-amidase, forming α-KG and releasing NH4+. Quantitative assessments suggest this pathway does not contribute substantially to net ammoniagenesis either under basal conditions or in response to either acute or chronic metabolic acidosis (159).
Glutamine Transport in Ammoniagenesis
Glutamine is the primary metabolic source for renal ammoniagenesis (161,166). Under conditions of normal acid-base balance, renal glutamine uptake is low, less than 3% of delivered glutamine. During acute metabolic acidosis there is a rapid, approximately 2×-fold increase in plasma glutamine levels, primarily from increased skeletal muscle glutamine release (161). There is a parallel increase in renal glutamine uptake, to approximately 20% of delivered glutamine (68, 161). With chronic metabolic acidosis, renal glutamine uptake increases substantially, to as much as 50% of delivered glutamine, even though plasma glutamine levels often decrease (68). Increased renal glutamine utilization is balanced by increased skeletal muscle and liver glutamine release (157, 161). As will be discussed below, renal glutamine uptake involves both apical and basolateral transport processes.
Plasma glutamine is effectively 100% filtered by the glomerulus and the filtered glutamine is almost completely reabsorbed by the proximal convoluted tubules (145). The mechanism(s) of proximal tubule glutamine transport remain incompletely characterized, and there is a degree of contradictory evidence about the specific proteins involved in this process. Apical glutamine transport, when assessed in brush border membrane vesicles, is Na+ dependent and has inhibitor characteristics similar to those identified for SNAT3/SN1 (SLC38A3) (76) and SNAT3 has been identified by immunoblot analysis in brush border membrane vesicles (76). However, immunohistochemistry has only identified basolateral SNAT3 immunoreactivity in the proximal tubule (25). Thus, there is conflicting evidence regarding SNAT3’s role in apical glutamine transport. Other glutamine transporters expressed in the apical membrane in the proximal tubule include the Na+-dependent neutral amino acid transporters B0AT1 (SLC6A19) and B0AT3 (SLC6A18). Under basal conditions, relatively little of the glutamine reabsorbed in the proximal tubule is metabolized, and instead is transported across the basolateral membrane into the peritubular space, resulting in net glutamine reabsorption. The mechanism of basolateral glutamine exit has not been well characterized, but may involve the y+L amino acid-transport system.
Whether apical uptake of glutamine is enhanced during acidosis is not clear. Some studies have found that chronic metabolic acidosis increases both apical glutamine transport and SNAT3 protein expression in brush border membrane vesicles (76). Other studies have found no change in apical Na+-dependent glutamine transport (186). Chronic metabolic acidosis does not alter expression of the other apical glutamine transporters, B0AT1 (SLC6A19) and B0AT3 (SLC6A18) (107). However, because filtered glutamine is essentially completely reabsorbed in the proximal tubule under basal conditions, increased rates of apical glutamine transport with metabolic acidosis do not increase the net amount of uptake.
Basolateral glutamine uptake is also present. Mechanistically, basolateral glutamine uptake appears to be mediated by SNAT3 expression, at least in the proximal tubule S3 segment under basal conditions and throughout the entire proximal tubule in response to metabolic acidosis (76, 153). Basolateral glutamine uptake by proximal tubule cells is regulated by a variety of conditions. Both metabolic and respiratory acidosis increases basolateral glutamine uptake (76, 186, 187). Immunohistochemistry shows that multiple conditions that increase ammoniagenesis, that is, chronic metabolic acidosis, hypokalemia and high protein intake, also increase basolateral SNAT3 immunolabel in the S3 proximal tubule, and also induce detectable expression in the basolateral plasma membrane in the proximal tubule S1 and S2 segments (25, 107). Increased basolateral SNAT3 expression presumably mediates increased glutamine uptake from the peritubular fluid, enabling increased ammoniagenesis and explaining the observation that renal glutamine extraction can exceed glutamine filtration.
Multiple other transporters may also contribute to glutamine transport, but their quantitative contribution is probably less. A system ASC glutamine transporter is present, at least in the renal cortex, and may contribute to glutamine transport under basal conditions (76). The basolateral y+L exchange transport system, which is formed by y+-LAT1 (SLC7A7) and 4F2hc subunits, is present in the basolateral plasma membrane of proximal tubule cells (17, 127) and mediates Na+-dependent glutamine uptake and cationic amino acid efflux. Metabolic acidosis decreases y+-LAT1 expression, but does not alter 4F2hc protein expression (107). If this results in decreased transport activity, it would be predicted to decrease y+L-mediated glutamine uptake, and thus would not contribute to increased glutamine uptake in the proximal tubule. However, this would decrease y+L-mediated arginine basolateral export, which could increase arginine-dependent ammoniagenesis.
Because the initial enzymatic step in ammoniagenesis involves the inner mitochondrial enzyme, PDG, glutamine transport into mitochondria is a critical step. Conditions associated with increased ammoniagenesis and with increased urinary ammonia excretion, such as metabolic acidosis, hypokalemia, and high-protein diets, stimulate mitochondrial glutamine uptake (1, 3, 46). Mitochondrial glutamine uptake involves a specific transporter-mediated mechanism (1, 46, 133, 151), which may have an apparent molecular weight of 41.5 kDa (71). The specific molecular identity of this transporter, however, has not been identified.
Regulation of Ammoniagenesis
Multiple conditions stimulate renal ammoniagenesis. Both acute and chronic metabolic acidosis stimulate ammoniagenesis (159); this involves increases in the PDG pathway. Although acute respiratory acidosis stimulates ammoniagenesis, the effect of chronic respiratory acidosis is unclear as different studies have found either no change or increased ammoniagenesis (158). Chronic metabolic acidosis is also associated with increases in PDG, GDH, and PEPCK expression and activity (159), all of which contribute to increased ammoniagenesis. Because changes in extracellular cell pH cause parallel changes in mitochondrial pH, these changes are generally thought to be an important mechanism in the regulation of ammoniagenesis (117, 135). In addition, a separate, pH-independent mechanism involving a small, nondialyzable factor may also regulate ammoniagenesis (5). Many pH sensors, including GPR4, InsR-RR, Pyk2, Erb1/2, pH sensitive ion channels, such as acid-sensing ion channel (ASIC), Tandem of P domains in a Weak Inward rectifying K+ channel (TWIK)-related acid-sensitive K+ channel (TASK), and Rat Outer Medullary K+ (ROMK), and bicarbonate-stimulated adenylyl cyclase, have been identified over the past few years (21). Studies examining whether any of these directly regulate renal ammoniagenesis have not been reported to date.
Chronic hypokalemia increases ammoniagenesis through mechanisms involving increased enzyme expression and activity similar to those identified for chronic metabolic acidosis. Acute changes in extracellular K+ also regulate proximal tubule ammoniagenesis; these effects are directly mediated by extracellular K+ and, because they can be observed within 45 min of altering extracellular K+, likely do not require changes in protein expression (113). How hypokalemia signals the proximal tubule to increase ammoniagenesis is incompletely understood. In vivo studies, measuring intracellular pH with 31P-NMR, have shown hypokalemia causes intracellular acidification, which could explain the stimulation of ammoniagenesis (2). However, in vitro studies demonstrated that hypokalemia induces only transient intracellular acidification, maximal at approximately 27 min, that reverts to no change in intracellular pH by 8 h (8). Moreover, dietary K+ deficiency increases both urinary ammonia excretion and expression of ammoniagenic enzymes prior to detectable changes in extracellular K+ concentration, suggesting that factors other than extracellular K+ concentration regulate ammoniagenesis in response to K+ deficiency (67). In particular, in view of evidence that changes in dietary K+ intake alter renal K+ excretion prior to detectable changes in serum K+ (67) and that gastrointestinal tract-derived factors can regulate renal K+ excretion (91), it is intriguing to speculate that intestinal tract-derived factors might regulate renal ammonia excretion in response to changes in K+ intake.
Because hypokalemia-induced increases in ammonia excretion often induces an acid-base disorder, metabolic alkalosis, rather than protecting against extracellular pH changes, other physiologic benefits of changes in ammonia metabolism have been considered. One possibility is that ammonia regulates renal potassium transport, that is, that it serves as a signaling molecule regulating K+ transport. Studies in both humans and rodents have shown that stimulating ammoniagenesis, by administering exogenous glutamine, simultaneously increases urinary ammonia excretion and decreases potassium excretion, and that the time course of changes are similar (72, 134, 160). Because changes in K+ excretion occur despite no significant change in extracellular K+ concentration, these findings suggest changes in ammonia metabolism and/or transport can regulate renal K+ transport. These effects on K+ transport have been localized to sites distal to the late micropuncturable tubule, suggesting regulation of collecting duct K+ transport (72). In vitro microperfusion studies show ammonia decreases both net and unidirectional K+ secretion in the collecting duct (54), and that ammonia stimulates H+-K+-ATPase-mediated H+ secretion (43). These effects of ammonia on H+-K+-ATPase occur independent of changes in intracellular pH and involve microtubule formation, intracellular calcium, and SNARE-protein-mediated vesicular trafficking (41-43). Thus, changes in proximal tubule ammoniagenesis in response to hypokalemia may facilitate changes in intrarenal ammonia concentrations, which then function as signaling molecules to regulate collecting duct K+ transport and net renal K+ excretion.
Ammoniagenesis is also modulated through mechanisms independent of acid base and potassium. These include tricarboxylic acid cycle intermediates, prostaglandin F2 alpha, insulin, growth hormone, angiotensin II (Ang II), corticosteroids, aldosterone, and tubular flow rate (159). Ang II likely mediates an important role regulating ammoniagenesis, with evidence that both luminal and peritubular Ang II stimulate ammonia production rates, but that more of the ammonia that is produced enters the lumen with luminal Ang II while more exits across the basolateral membrane with peritubular Ang II (114).
Ammonia Transport
Proximal tubule
Ammonia produced in the proximal tubule is secreted preferentially into the tubule lumen, although there is some transport across the basolateral plasma membrane. Preferential apical secretion dominates due to multiple factors, including luminal acidification which binds to secreted NH3, forming NH4+ and decreasing luminal NH3 concentration, thereby maintaining the gradient for NH3secretion, and apical NHE-3-mediated Na+/NH4+ exchange (112,149). An apical Ba+2-sensitive K+ channel and a diffusive NH3-transport component also appear to be involved in ammonia secretion (57, 149). H+ secretion via H+-ATPase as well as by NHE-3 titrates this secreted NH3to form NH4+. Figure 7 summarizes the model of our current understanding of proximal tubule ammonia secretion.
Figure 7.

Ammonia transport in the proximal tubule. Ammonia is produced in the proximal tubule primarily from metabolism of glutamine, and occurs primarily in the mitochondria. The enzymatic details of ammoniagenesis are not shown. Three transport mechanisms appear to mediate preferential apical ammonia secretion. These include Na+/NH4+ exchange via NHE-3, parallel NH3 secretion and NHE-3-mediated Na+/H+ exchange, and a Ba2+-sensitive NH4+ conductance likely mediated by apical K+ channels. Secreted NH3 is titrated to NH4+ by reaction with H+, which is secreted either by NHE-3 or by apical H+-ATPase. HCO3− is produced in equimolar amounts as NH4+ in the process of ammoniagenesis, and is primarily transported across the basolateral plasma membrane by NBCe1. Minor components of basolateral NH4+ uptake via Na+-K+-ATPase and by basolateral K+ channels are not shown. Figure modified from reference (180) with permission.
The proximal tubule also can reabsorb luminal ammonia; this appears to occur primarily in the late proximal tubule (56). The proximal tubule expresses glutamine synthetase, which catalyzes the reaction of NH4+ with glutamate to form glutamine (23). Metabolic acidosis converts late proximal tubule ammonia transport from net reabsorption to net secretion (56). The molecular mechanism underlying this conversion includes, at least in part, decreased glutamine synthetase-mediated reaction of NH4+ with glutamate to form glutamine (29,129). Because this glutamine synthetase-accelerated reaction consumes NH4+ decreased glutamine synthetase activity results in increased net ammonia production.
Loop of Henle
The thick ascending limb of the loop of Henle (TAL) is an important site for reabsorbing luminal ammonia. The apical Na+-K+-2Cl− cotransporter, NKCC2, transports NH4+ at the K+-binding site, and is the primary protein responsible for ammonia reabsorption (10). The relatively low permeability of the TAL apical plasma membrane to NH3 relative to its permeability to NH4+ is critical in preventing back leak of NH3(78). Ammonia exit across the basolateral plasma membrane involves basolateral Na+/NH4+ exchange mediated by NHE-4 (19). Although NHE-1 is also present in the TAL basolateral plasma membrane (27), pharmacologic inhibition studies do not support a significant role for NHE-1 in TAL ammonia reabsorption (19). A second mechanism of basolateral ammonia transport may involve dissociation of cytosolic NH4+ to NH3 and H+, with basolateral NH3 exit and buffering of the intracellular H+ load by basolateral bicarbonate uptake mediated by the NBCn1 cotransporter (87, 94, 125). Finally, there is a Cl−-dependent NH4+-transport mechanism, likely in the basolateral membrane and likely to be mediated by one of the isoforms of the KCC family (9). Figure 8 summarizes this model of TAL ammonia reabsorption.
Figure 8.

Ammonia transport in the thick ascending limb (TAL). The primary mechanism of ammonia reabsorption in the TAL is via substitution of NH4+ for K+ and transport by Na+-K+-2Cl− cotransporter 2 (NKCC-2). Electroneutral K+/NH4+ exchange and conductive K+transport are also present, but quantitatively less significant components of apical K+ transport. Diffusive NH3 transport across the apical plasma membrane is present, but not quantitatively significant. Cytosolic NH4+ can exit via basolateral NHE-4. A second mechanism of basolateral NH4+ exit may involve dissociation to NH3 and H+, with NH3 exit via an uncharacterized, presumably diffusive, mechanism, and buffering of intracellular H+ released via NBCn1-mediated HCO3− entry. Figure modified from reference (180) with permission.
Some of the ammonia absorbed by the medullary thick ascending limb (mTAL) of the loop of Henle undergoes recycling into the descending thin limb of the loop of Henle (DTL), resulting in countercurrent amplification of medullary interstitial ammonia concentration. Ammonia recycling predominantly involves NH3 transport, with a smaller component of NH4+ transport (39). The molecular mechanisms of DTL NH3 and NH4+ transport have not been well characterized to date.
Ammonia reabsorption by the thick ascending limb of the loop of Henle has two important consequences. First, passive ammonia secretion into the thin descending limb of the loop of Henle causes development of axial ammonia concentration gradient in the medullary interstitium that parallels the hypertonicity gradient. Second, ammonia absorption by the mTAL results in ammonia delivery to the distal tubule accounting for only approximately 20% to 40% of final urinary ammonia content (34, 55). Consequently, urinary ammonia excretion requires a major component of ammonia secretion at sites distal to the loop of Henle, that is, distal convolute tubule, connecting segment and the collecting duct.
Distal convoluted tubule and connecting segment
A small component of ammonia secretion occurs in the regions of the distal tubule prior to the collecting duct, that is, the DCT and CNT. Micropuncture studies in the rat kidney have generally shown net ammonia secretion between the early and late portions of the micropuncturable distal tubule, accounting for approximately 10% to 15% of total urinary ammonia excretion under basal conditions (147, 185). The specific mechanism of ammonia transport in these segments has not been specifically determined, but in view of multiple similarities with the collecting duct in the expression of H+ and ammonia transporters it is likely that similar mechanisms are utilized.
Collecting duct
Ammonia secretion by the collecting duct appears to account for the majority, 60% to 80%, of urinary ammonia content. Mechanistically, collecting duct ammonia secretion involves parallel NH3 and H+ secretion, and there is no significant component of transepithelial NH4+ transport (34, 83). H+ secretion likely involves both H+-ATPase- and H+-K+-ATPase-mediated transport. Excellent reviews of H+-ATPase and H+-K+-ATPase are available for the interested reader (53, 165). Our current model of collecting duct ammonia transport involves the integrated action of a large number of proteins (Fig. 9). These include proteins involved in basolateral ammonia uptake (Rhbg, Rhcg, and Na+-K+-ATPase), cytosolic H+ production (carbonic anhydrase II), apical ammonia transport (Rhcg), apical H+ secretion (H+-ATPase, H+-K+-ATPase), and basolateral HCO3− transport (Cl−/HCO3− exchange).
Figure 9.

Ammonia transport in the collecting duct. Interstitial NH4+ is in equilibrium with NH3 and H+. NH3 is transported across the basolateral membrane, predominantly by Rhcg, but also possibly partly by Rhbg. In the inner medullary collecting duct, basolateral Na+-K+-ATPase transports NH4+. Intracellular NH3 is secreted across the apical membrane by apical Rhcg. H+-ATPase and H+-K+-ATPase secrete H+, which combines with luminal NH3 to form NH4+ which is “trapped” in the lumen. Intracellular H+ is generated by CA II-accelerated CO2 hydration that forms carbonic acid, which dissociates to H+ and HCO3−. Basolateral Cl−/HCO3− exchange transports HCO3− across the basolateral membrane; HCO3− combines with H+ released from NH4+ to form carbonic acid, which dissociates to CO2 and water. This CO2 can recycle into the cell, supplying the CO2 used for cytosolic H+ production. The net result is NH4+ transport from the peritubular space into the luminal fluid. Figure modified from reference (180) with permission.
NH3 movement across collecting duct cell apical and basolateral membranes appears to involve both diffusion- and transporter-mediated NH3 transport. The transporter-mediated component of NH3 is Na+ and K+ independent and is electroneutral (62, 63). Studies using cultured collecting duct cells show that transporter-mediated NH3 movement is the predominant route of ammonia transport across both the apical and basolateral plasma membranes at the ammonia concentrations present in the cortex and outer medulla (62, 63).
The Rh glycoprotein, Rhcg, mediates a critical role in renal ammonia excretion. Rhcg is an ammonia-specific transporter, with no identifiable affinity for solutes other than ammonia and its methyl-derivative, methylammonia (13, 102, 106, 108). Rhcg is expressed in the DCT, CNT, initial collecting tubule (ICT), CCD, OMCD, and IMCD (35, 58, 167). Rhcg is expressed both in intercalated cells and in principal cells, DCT cells and CNT cells in these tubule segments, with the exception that it is not expressed in non-intercalated cells in the IMCD or in the B-type intercalated cell (58, 167). In the human kidney, Rhcg is expressed in the same distribution, with the exception that principal cells lack Rhcg expression (58). Although originally Rhcg expression was only detected in the apical plasma membrane, more recent studies have shown both apical and basolateral Rhcg expression (20, 58, 80, 141, 142).
A variety of studies, using mice with intact Rhcg expression and with global and cell-specific Rhcg gene deletions, have shown Rhcg’s essential role in renal ammonia excretion. Metabolic acidosis stimulates Rhcg expression in the cortex and the outer medulla, and it induces changes in Rhcg’s subcellular distribution, with decreased relative intracellular expression and increased expression in the apical plasma membrane (92, 141, 142). Gene-deletion studies, utilizing both global and collecting duct-specific Rhcg deletion, decrease basal urinary ammonia excretion, and impair the normal increase in urinary ammonia excretion in response to metabolic acidosis (16, 92); Rhcg deletion only from intercalated cells does not alter net basal ammonia excretion, but does significantly inhibit ammonia excretion in response to metabolic acidosis (93).
Studies using in vitro microperfused collecting duct segments have also shown that Rhcg has a critical role in ammonia secretion. These studies examined cortical and OMCD segments from mice with chronic metabolic acidosis and found global Rhcg deletion decreases apical NH3 permeability by 60% to 70% (16). The mechanism of the remaining apical NH3 permeability was not determined, but studies using cultured collecting duct cells have shown that apical ammonia permeability involves components of both transporter-mediated, likely via Rhcg, and diffusive NH3 movement (63).
Rhbg is another member of the Rh glycoprotein family which, like Rhcg, is an ammonia-specific transporter. The majority of studies examining the molecular form of ammonia transported by Rhbg have identified NH3 transport (101, 102, 194), but other studies have identified NH4+ transport (120-122). Rhbg is expressed in the same cells as Rhcg, with the significant difference that Rhbg has only basolateral expression (128, 167). Whether Rhbg expression is necessary for renal ammonia excretion has been the subject of two separate studies. An initial report using mice with global Rhbg deletion did not find altered ammonia excretion either under basal conditions or after acid loading (26). In contrast, a more recent study using a different model of acid loading, producing greater stimulation of renal ammonia excretion, found that intercalated cell-specific Rhbg deletion inhibited ammonia excretion (15).
A second mechanism of basolateral ammonia uptake involves basolateral Na+-K+-ATPase. In the IMCD, basolateral Na+-K+-ATPase has been shown to contribute to basolateral NH4+ uptake (169, 174). Intracellular NH4+ dissociates to H+ and NH3, which are then secreted across the apical plasma membrane. However, in the CCD basolateral Na+-K+-ATPase does not contribute to ammonia secretion (84), and its role in the OMCD has not been reported.
Carbonic anhydrase has several important roles in collecting duct ammonia secretion. First, cytosolic carbonic anhydrase activity is necessary for ammonia secretion, probably through a role in supplying cytosolic H+ for secretion (170). Second, the presence or absence of luminal carbonic anhydrase activity, mediated by apical carbonic anhydrase IV, has an important impact on the rate of collecting duct ammonia secretion. In the absence of luminal carbonic anhydrase activity, the luminal [H+] increases above equilibrium levels due to delayed dissociation of H2CO3; this is termed a luminal “disequilibrium pH.” Because the H+ + NH3 ↔ NH4+ reaction occurs rapidly, this shifts the ammonia buffer reaction toward NH4+ decreasing the luminal NH3 concentration and thereby increasing the transepithelial gradient for NH3 secretion. The importance of this is underscored by experiments showing that addition of carbonic anhydrase to the luminal fluid reduces ammonia secretion (85). Detailed studies of the collecting duct have demonstrated a luminal disequilibrium pH in the rat in the inner stripe of the OMCD and the terminal IMCD (40, 172), and in the rabbit in the CCD (84, 156) and outer stripe of OMCD (155), but not in the inner stripe of OMCD (155).
Specific proteins involved in renal ammonia metabolism
Phosphate-dependent glutaminase
PDG is the initial enzymatic step in renal ammoniagenesis. It is located in mitochondria and catalyzes the reaction, L-glutamine + H2O → L-glutamate− + NH4+. In the kidney, PDG activity has been localized to the proximal straight and convoluted tubules (32, 77). However, enzymatic assays have also identified PDG activity in many other sites, including the descending thin limb of the loop of Henle (DTL), mTAL, DCT, and throughout the collecting duct (32,77,190). Whether activity in these sites is stimulated by conditions associated with increased ammonia excretion is unclear. Some studies have found that metabolic acidosis increases PDG activity in the DTL, mTAL in the outer stripe, and DCT (77), while others have found no change in activity in sites other than the proximal tubule (190).
PDG is also present in many extrarenal tissues, including liver and brain. In humans, the gene for the kidney-type isoform has 19 exons, and gives rise to at least two transcripts, a KGA mRNA resulting from joining exons 1-14 and 16-19 and GAC mRNA product which uses only exons 1-15 (104). A separate gene gives rise to an LGA isoform. The KGA protein is ubiquitously expressed, including the renal proximal tubule. The GAC isoform is expressed primarily in the heart and pancreas, at least based on mRNA expression, and the LGA isoform is widely expressed, including liver, brain, pancreas and breast cancer (104). The KGA isoform is increased in response to metabolic acidosis and appears to be the source of the majority of renal PDG activity.
Metabolic acidosis increases proximal tubule PDG activity; these increases derive from multiple mechanisms. There is increased protein expression, and this appears due to increased mRNA expression (162, 163). Increased PDG mRNA results from stabilization of the mRNA product, and does not involve increased transcription rates (69). A direct repeat of an 8-nt AU sequence functions as a pH-response element, and is both necessary and sufficient to generate pH-response gene stabilization. This response element binds multiple RNA-binding proteins, including ζ-crystallin, AU-factor 1 and HuR. Acidosis also causes an endoplasmic reticulum stress response that causes formation of cytoplasmic stress granules, ζ-crystallin undergoes transient movement to stress granules and simultaneously HuR is translocated from the nucleus to the cytoplasm (70). A second regulatory mechanism likely involves changes in intramitochondrial glutamate. Glutamate is a competitive inhibitor of PDG (143), and decreases in intramitochondrial glutamate concentration, as can occur during metabolic acidosis, result in increased PDG activity (4).
Glutamate dehydrogenase
GDH is a mitochondrial enzyme that catalyzes the reaction, L-glutamate− + H2O + NAD+ (or NADP+) → α-KG−2+ NH4+ + NADH (or NADPH) + H+. Two GDH isoforms exist, and are products of two different genes; GLUD1 is widely expressed, whereas GLUD2 appears to be a neural and testicular specific isoform (105,144). GDH activity is high in liver, brain, kidney, heart, pancreas, ovaries, and testis in addition to the kidney. Brain and testicular GDH reflects products of GLUD1 and GLUD2, whereas in most other tissues, including the kidney, GDH is a product of the GLUD1 mRNA.
Metabolic acidosis stimulates renal GDH activity (189), both by altering its affinity for glutamate (4) and by increasing protein (33) and mRNA (GLUD1) expression (31). GDH activity is increased in part through metabolic acidosis-induced decreases in intramitochondrial α-KG concentration (143). This decrease in α-KG both accelerates GDH activity, by relieving α-KG-mediated competitive inhibition of the enzymatic reaction, and it inhibits the reverse reaction (136, 137). The increased mRNA expression appears related to increased mRNA stability, not transcription (75). Four AU-rich elements are present in the 3′-UTR, bind ζ-cystallin and appear to confer pH-responsive stabilization of GDH mRNA (138).
Phosphoenolpyruvate carboxykinase
Renal PEPCK is a cytosolic enzyme and is the product of the PCK1 gene. In the kidney, as in extrarenal sites, including liver, adipose tissue, and small intestine, PEPCK is a key enzyme in gluconeogenesis through its role in conversion of oxaloacetate into PEP and HCO3−. It also mediates an important role in the renal response to metabolic acidosis (24) by contributing to renal bicarbonate synthesis and by promoting ammoniagenesis by degrading α-KG, an end product of glutamine degradation. The adaptive increase in PEPCK activity and protein expression results from increased protein synthesis and mRNA expression (31). Increased PEPCK mRNA expression appears to result both from increased gene transcription and from mRNA stabilization (65,109). Acidosis-induced increases in gene transcription appear to involve binding to the P3(II) and CRE-1 elements in the PCK1 promoter (31). The 3’-nontranslated region of the PEPCK mRNA also contains an instability element that facilitates its rapid turnover and contributes to the regulation of PEPCK gene expression (90). Metabolic acidosis also stimulates PEPCK gene expression through p38 MAPK phosphorylation of ATF-2, which binds to the CRE-1 element and either recruits or interacts with additional transcription factors that activate gene transcription (31).
γ-GT
γ-GT accounts for phosphate-independent glutaminase activity identified in many early enzymatic studies of renal ammoniagenesis. Its quantitative role, however, in ammoniagenesis remains unclear. γ-GT is primarily expressed in the proximal straight tubule (32), and micropuncture studies suggest that glutamine is completely reabsorbed by the PCT, suggesting PST ammoniagenesis via γ-GT is unlikely to contribute significantly to renal ammoniagenesis. Studies examining the late proximal convoluted tubule have shown that γ-GT activity contributes only a minor component of total ammoniagenesis (150), and chronic metabolic acidosis does not alter γ-GT activity (32). However, although filtered glutamine is essentially entirely reabsorbed prior to reaching the downstream γ-GT-containing proximal tubule segments, there is evidence that glutamine may back flux into the lumen and support ammonia production via this enzyme (146, 182).
NHE-3
The majority of current evidence suggests that the apical Na+/H+ exchanger, NHE-3, mediates a substantial component of proximal tubule ammonia secretion, through substitution of NH4+ for H+, resulting in functional Na+/NH4+ exchange activity. This data include evidence that proximal tubule brush border membrane vesicles exhibit NH4+/Na+ exchange activity (82), that combining a low luminal Na+ concentration with Na+/H+ exchange inhibitor, amiloride, decreases ammonia secretion (111) and that the Na+/H+ exchange inhibitor, 5-(N-Ethyl-N-isopropyl)amiloride (EIPA), blunts ammonia secretion when alternative secretory pathways are blocked (149). Although NHE-3 is the predominant apical Na+/H+ exchanger in the proximal tubule, direct studies examining the effect of proximal tubule-specific NHE-3 deletion are necessary to confirm its central role in Na+/NH4+ exchange activity. In particular, some studies suggest that facilitated NH3 transport in parallel with H+ secretion is a primary mechanism of proximal tubule ammonia secretion (148) and that a Ba2+-sensitive transporter, most likely an apical K+ channel, also contributes to NH4+ secretion (149).
Nonetheless, NHE-3 appears to be important in the regulation of proximal tubule ammonia transport. In response to chronic metabolic acidosis, changes in extracellular potassium and Ang II, changes in NHE-3 expression, and activity parallel changes in ammonia secretion (6, 37, 117). In both the S2 and S3 segments, chronic metabolic acidosis increases AT1 receptor-mediated stimulation of NHE-3 (115,116,118). Other studies show that increased endothelin-1 expression with subsequent activation of the endothelin-B receptor mediates an important role in increasing NHE-3 expression and renal ammonia excretion in metabolic acidosis (88).
Apical NHE-3 is also present in the TAL (7). However, since this transporter secretes NH4+ and the TAL reabsorbs NH4+ NHE-3 is unlikely to mediate an important role in TAL ammonia transport.
Potassium channels
Although molecular K+ and NH4+ have different molecular weights and atomic radii (243 pm for K+ and 137 pm for NH4+), hydration in aqueous solutions alters these characteristics, such that they have nearly identical hydrodynamic radii (180). When examined, almost every K+ transporter examined also transports NH4+; in general, the relative rate of NH4+ transport is 10% to 20% of that observed for K+ (177). The primary evidence that K+ channels contribute to proximal tubule ammonia transport comes from in vitro microperfusion studies showing that barium, a nonspecific K+ channel inhibitor, can inhibit proximal tubule ammonia transport (149). Multiple K+ channels are present in the apical membrane of the proximal tubule, including KCNA10, KCNQ1/KCNE1, and TWIK-1; which of these mediate ammonia transport is not currently known.
In the TAL, apical K+ channels are present and can contribute to luminal NH4+ uptake when apical Na+-K+-2Cl− cotransport is inhibited. Quantitatively, they may mediate 14% to 30% of luminal NH4+ uptake, with the exact proportion dependent on luminal total ammonia concentration and on the specific assay used to quantify apical NH4+ transport (175). However, NKCC2 inhibitors almost completely inhibit TAL transepithelial ammonia reabsorption (51, 175), suggesting that apical K+ channels are unlikely to mediate a quantitatively important role in TAL ammonia reabsorption.
Na+-K+-2Cl− cotransport
The Na+-K+-2Cl−-cotransporters are widely expressed proteins that transport Na+, K+ and2 Cl− ions in an electroneutral manner across plasma membranes. Two isoforms have been identified, NKCC-1 (SLC12A1) and NKCC-2 (SLC12A2). They have distinct expression patterns and differing roles in renal ammonia transport.
NKCC1 is a widely expressed isoform that in the kidney is present in the basolateral membrane of A-type intercalated cells in the outer and IMCD cells (45). However, addition of bumetanide, an NKCC1/NKCC2 inhibitor, to the peritubular solution does not alter OMCD ammonia secretion (170). In the IMCD, NH4+ and K+ compete for a common binding site on NKCC1, but inhibiting NKCC1 does not alter peritubular NH4+ uptake significantly (169). Thus, NKCC1 appears unlikely to mediate a substantial role in renal ammonia secretion.
NKCC-2 is a kidney-specific isoform expressed selectively in the apical plasma membrane of the TAL. It is the major mechanism of ammonia reabsorption across the TAL apical membrane through coupled NH4+ Na+, and 2 Cl− transport (48, 175). Competition of luminal NH4+ with K+ for the K+-transport site has several significant implications. Most importantly, changes in luminal K+ in hypokalemia and hyperkalemia alter net NH4+ transport and thus medullary interstitial ammonia concentration, which likely alters collecting duct ammonia secretion and thus net renal ammonia excretion. Transport of NH4+ at the K+-binding site may also contribute to TAL NaCl transport, particularly in forms of Bartter’s syndrome where abnormal ROMK expression prevents K+ recycling (181). Metabolic acidosis increases TAL ammonia reabsorption (47) through transcriptionally mediated increases in NKCC2 expression (11) due to metabolic acidosis-induced elevation in systemic glucocorticoids (12).
Na+-K+-ATPase
Na+-K+-ATPase is a P-type ATPase present in the basolateral plasma membrane of renal epithelial cells, and its expression is greatest in the mTAL of the loop of Henle, with lesser expression in the cortical thick ascending limb, DCT, CCD, MCD, and the proximal tubule (74). NH4+ competes with K+ at the K+-binding site of Na+-K+-ATPase, enabling net Na+-NH4+ exchange (86, 173). However, the relative affinities of Na+-K+-ATPase for NH4+ approximately 11 mmol/L, and K+, approximately 1.9 mmol/L, have important implications for Na+-K+-ATPase-mediated NH4+ transport (86). In the cortex, interstitial ammonia concentrations are less than 1 mmol/L, suggesting NH4+ is unlikely to be transported to a significant extent by basolateral Na+-K+-ATPase. In addition, even in the presence of high concentrations of peritubular ammonia, Na+-K+-ATPase inhibitors do alter CCD ammonia secretion (84). In contrast, interstitial ammonia concentrations in the inner medulla are high, and Na+-K+-ATPase-mediated basolateral NH4+ uptake is critical for IMCD ammonia and acid secretion (86, 173). In hypokalemia, the decreased interstitial K+ concentration, by facilitating increased NH4+ uptake by Na+-K+-ATPase, enables increased rates of NH4+ secretion despite an absence of changes in Na+-K+-ATPase expression (171).
H+-K+-ATPase
H+-K+-ATPase proteins are other members of the P-type ATPase family that transport NH4+. Potassium deficiency increases expression of the HKα1 and HKα2 isoforms of H+-K+-ATPase (53). This has been postulated to mediate increased NH4+ secretion via binding of NH4+ to and transport at the H+-binding site (119). Other studies, however, have suggested that NH4+ binds to and is transported at the K+-binding site (44). Additional studies are needed to define more clearly the specific role(s) of H+-K+-ATPase in collecting duct ammonia secretion.
In addition to being a substrate for H+-K+-ATPase transport, ammonia also appears to regulate H+-K+-ATPase H+ secretion. In studies examining in vitro microperfused CCD segments, ammonia causes concentration-dependent stimulation of net H+-K+-ATPase, but not H+-ATPase-mediated, H+ secretion (43). In the OMCD, ammonia may stimulate net H+ secretion by as much as approximately 50% (40). Ammonia’s stimulation of CCD H+-K+-ATPase is independent of changes in intracellular pH and involves changes in intracellular calcium, microtubules and SNARE protein-mediated vesicle fusion (41-43). This stimulation of H+-K+-ATPase activity, by increasing unidirectional K+ reabsorption, likely contributes to ammonia’s ability to regulate CCD net K+ secretion (54).
K+/NH4+(H+) exchange
An electroneutral, Ba+2- and verapamil-inhibitable apical K+/NH4+ (H+) activity is present in the apical membrane of the TAL (10). The gene product and the protein that correlate with this transport activity have not yet been identified. However, the observation that NKCC2 inhibitors nearly completely inhibit transepithelial TAL ammonia transport (51, 175) suggests that K+ for NH4+ exchange activity may not mediate a major role in transepithelial TAL ammonia reabsorption.
Aquaporins
The aquaporins belong to an extended family of proteins that facilitate water transport. Because both H2O and NH3 have similar molecular sizes and charge distribution, several studies have examined the role of aquaporins in NH3 transport. Importantly, many, but not all aquaporins transport ammonia (110).
AQP1 was the first aquaporin shown to transport ammonia. Studies using AQP1 expression in Xenopus oocytes demonstrated AQP1 expression increases NH3 transport (110,123). However, not all studies have confirmed NH3 transport by AQP1 (66). AQP1 is present in the descending thin limb of the loop of Henle, suggesting it may contribute to ammonia permeability in this segment. It is also present in both the apical and basolateral plasma membranes in the proximal tubule; whether it contributes to proximal tubule, ammonia transport has not been determined experimentally, but it is certainly ideally located to mediate a component of the Ba+2- and NHE-3-independent apical NH3 secretion.
AQP3 is present in the basolateral plasma membrane of collecting duct principal cells and transports NH3, when expressed in the Xenopus oocytes, transports NH3 (66). Whether AQP3 contributes to renal principal cell basolateral NH3 transport has not been determined.
AQP8 is expressed in intracellular sites in the proximal tubule, CCD and OMCD in the kidney, but not in the plasma membrane (38). AQP8’s specific intracellular site in mammalian cells has not been determined, but it localizes to the inner mitochondrial membrane when expressed in yeast (154). AQP8’s role in renal ammonia metabolism is unclear. Genetic deletion slightly decreases hepatic and increases renal ammonia concentrations, but does not alter systemic pH, serum chloride concentration, urine ammonia concentration or urine pH either under basal conditions or in response to acid loading (191, 192). Thus, AQP8 does not appear to mediate a major role in renal ammonia transport.
Carbonic anhydrase
Carbonic anhydrase, in addition to its role in bicarbonate reabsorption, also contributes to ammonia secretion. Studies examining isolated perfused OMCD segments have shown that carbonic anhydrase inhibition, presumably through effects on CA II, block ammonia secretion (170).
The membrane-bound isoform, CA IV, likely contributes to the regulation of both bicarbonate reabsorption and ammonia secretion. Collecting duct CA IV, although functioning to increase bicarbonate reabsorption, likely decreases collecting duct ammonia secretion by preventing a luminal disequilibrium pH. Apical CA IV expression has been demonstrated in the rabbit CCD-type A intercalated cell, in the rabbit OMCD and IMCD (140), and in the human CCD and OMCD (99), but has not been found in the rat collecting duct (22). This pattern is consistent with the evidence of luminal disequilibrium pH in the rabbit CCD and OMCD outer stripe segments (155, 156). Whether collecting duct CA IV expression is regulated by physiologic stimuli has not been rigorously examined. Studies examining tissue homogenates have identified that metabolic acidosis increases CA IV mRNA expression (164,188). This observation is consistent with the observation of basolateral CA IV expression in the proximal tubule, where it may contribute to bicarbonate reabsorption (139).
Rh glycoproteins
Rh glycoproteins are mammalian orthologs of Mep/AMT proteins, ammonia transporter family proteins present in yeast, plants, bacteria, and many other organisms. Three mammalian Rh glycoproteins have been identified to date, Rh A glycoprotein (RhAG/Rhag), Rh B Glycoprotein (RhBG/Rhbg), and Rh C Glycoprotein (RhCG/Rhcg). By convention, Rh A glycoprotein is termed RhAG in human tissues and is termed Rhag in nonhumans; a similar terminology is used for RhBG/Rhbg and RhCG/Rhcg.
RhAG/Rhag
Rh A glycoprotein (RhAG/Rhag) is a component of the Rh complex, which consists of the nonglycosylated Rh proteins, RhD and RhCE in humans and Rh30 in nonhuman mammals, in association with RhAG/Rhag. RhAG mediates electroneutral, NH3 transport (103, 131, 183, 184). Because studies in human (176) and mouse (unpublished observations) kidneys have found no evidence of renal RhAG expression, RhAG/Rhag appears unlikely to contribute to renal ammonia metabolism.
RhBG/Rhbg
RhBG/Rhbg is expressed in a wide variety of organs involved in ammonia metabolism, including kidneys, liver, skin, lung, stomach, and gastrointestinal tract (61, 64, 98, 128, 167, 178). The kidneys express basolateral Rhbg immunoreactivity in the DCT, CNT, ICT, CCD, OMCD, and IMCD (128, 167). In general, both intercalated and principal cells express Rhbg, with greater expression in intercalated cells than in principal cells. The exceptions to this rule are the IMCD, where only intercalated cells express Rhbg, and the B-type intercalated cell, which does not express Rhbg detectable with immuno-histochemistry (167). Rhbg’s basolateral expression appears due to basolateral stabilization through specific interactions of its cytoplasmic carboxy-terminus with ankyrin-G (100). Although, the human kidney expresses high amounts of RhBG mRNA (98), a recent study using a variety of antibodies did not identify detectable RhBG protein expression (20).
RhBG/Rhbg transports ammonia and the ammonia analogue, methylammonia, but does not appear to have other known substrates. Different studies have reached different conclusions regarding the exact ammonia species, NH3 or NH4+ transported. Most studies have shown that Rhbg mediates electroneutral, Na+- and K+-independent, NH3 transport (101, 102, 194), while other studies identified electrogenic NH4+ transport (122). Recent studies also indicated that Rhbg might transport both NH3 and NH4+ (121). In all of these studies, the affinity for ammonia was approximately 2 to 4 mmol/L. The explanation underlying the seemingly differing molecular substrates transported in different studies is not evident at present. From a physiologic perspective, both electroneutral NH3 transport and electrogenic NH4+ transport result in net basolateral ammonia uptake in renal collecting duct cells.
Rhbg’s specific role in renal ammonia metabolism has been the subject of several studies. In the mouse, one study showed increased mRNA expression with metabolic acidosis, but the effect on Rhbg protein expression was not reported (28). In another study, genetic deletion of pendrin, an apical Cl−/HCO3− exchanger present in the B-type intercalated cell and non-A, non-B intercalated cell, decreased Rhbg expression (81). Since pendrin deletion increases urine acidification, the decreased Rhbg expression may have been a compensatory mechanism to maintain normal ammonia excretion. Genetic deletion of Rhcg from acid-loaded mice increased Rhbg expression, suggesting increased Rhbg protein expression may contribute to ammonia excretion under these conditions (92, 93).
Two studies have used mice with genetic deletion of Rhbg to determine Rhbg’s role in renal ammonia transport. In the first, mice with global Rhbg deletion had normal basal acid-base parameters, normal responses to acid loading with addition of NH4Cl to the drinking water, and normal basolateral NH3 and NH4+ transport in microperfused CCD segments (26). Studies examining mice with intercalated cell-specific Rhbg deletion, however, came to different conclusions. Mice with intercalated cell-specific Rhbg deletion that were acid loaded by addition of HCl to food had impaired ammonia excretion in response to acid loading (15). Moreover, although basal ammonia excretion was normal, adaptive responses to Rhbg deletion were identified that appeared to compensate for Rhbg’s absence, indicating a role for Rhbg in basal ammonia excretion (15).
RhCG/Rhcg
RhCG/Rhcg is expressed in multiple ammonia transporting/metabolizing organs, including kidneys, CNS, testes, lung, liver, and throughout the gastrointestinal tract (61, 64, 97, 178). In the kidney, Rhcg expression parallels Rhbg’s expression, that is, in the DCT, CNT, ICT, CCD, OMCD, and IMCD (35, 167). Intercalated cell expression exceeds principal cell expression in the DCT, CNT, ICT, CCD, and OMCD, and in the IMCD Rhcg expression has only been detected in intercalated cells (35, 167).
Rhcg’s subcellular location differs from the exclusive basolateral location observed for Rhbg. In the human, rat, and mouse kidney, Rhcg immunoreactivity is both apical and basolateral (20, 58, 80, 141, 142). Immunogold electron microscopy has shown Rhcg immunoreactivity in the apical plasma membrane, basolateral plasma membrane and, additionally, in sub-apical vesicles (142). Basolateral Rhcg expression is approximately 20% of total cellular expression, at least in the rat OMCD in the inner stripe (80, 142). In the mouse there are substantial strain-specific variations in the extent of basolateral Rhcg expression, and these differences correlate with differing ability to excrete ammonia in response to acid loading (80, 179).
Multiple studies have addressed the molecular ammonia species transported by RhCG/Rhcg. Most studies examining heterologous expression of Rhcg have shown that Rhcg mediates electroneutral NH3 transport (102,194), while others have reported NH4+ transport (13) or both NH3 and NH4+ transport (personal communication, T Caner and NL Nakhoul). Studies examining purified Rhcg expression in liposomes have found that Rhcg transports NH3, not NH4+ (52,108), and studies examining apical membrane NH3 and NH4+ permeability using in vitro microperfused collecting duct segments have shown that Rhcg deletion decreases NH3 permeability, and does not change NH4+ permeability (16).
Rhcg’s expression parallels ammonia excretion in several conditions. Chronic metabolic acidosis significantly increases Rhcg protein expression in both the OMCD and the IMCD, but not in the cortex (141). These changes appear to occur through posttranscriptional mechanisms, as Rhcg steady-state mRNA expression does not significantly change (141). In response to reduced renal mass, where there is increased single nephron ammonia secretion, apical Rhcg expression increases in the CCD A-type intercalated cell, CCD principal cell, OMCD intercalated cell and OMCD principal cell, and basolateral expression increases in the CCD principal cell, OMCD intercalated cell, and the OMCD principal cell (79). In ischemia-reperfusion injury, where there is decreased renal ammonia excretion despite intact interstitial ammonia concentrations, there is selective damage to Rhcg-expressing OMCD intercalated cells, with induction of apoptosis and cellular extrusion (59). Cyclosporine A nephropathy is associated with decreased Rhcg expression, which likely contributes to impaired ammonia excretion and to development of metabolic acidosis (96). Lastly, hypokalemia, which increases ammonia excretion despite development of metabolic alkalosis in many species, increases Rhcg expression significantly (60).
At least two distinct mechanisms contribute to increased Rhcg expression in these models. First, there is increased Rhcg protein expression without increased Rhcg mRNA expression, suggesting regulation through posttranscriptional mechanisms (142). Second, there are changes in Rhcg’s subcellular location. Under basal conditions, Rhcg is located in both the apical and basolateral plasma membrane and in subapical sites in both principal cells and intercalated cells. In response to chronic metabolic acidosis, particularly in the intercalated cell, apical plasma membrane expression increases and cytoplasmic expression decreases (142). Importantly, the relative magnitude of these two regulatory mechanisms differs in different renal epithelial cells; a change in the subcellular distribution is the predominant response in the OMCD intercalated cell and increased protein expression is the predominant mechanism in the OMCD principal cell (142).
Basolateral plasma membrane Rhcg expression increases in chronic metabolic acidosis, hypokalemia, and reduced renal mass (60, 79, 142). This increase does not appear to involve redistribution from cytoplasmic sites to the basolateral plasma membrane (142).
Most recently, genetic deletion studies have confirmed the key role of Rhcg in renal ammonia excretion. Global Rhcg deletion decreases basal ammonia excretion and impairs urinary ammonia excretion in response to metabolic acidosis (16). Collecting duct-specific Rhcg deletion produced identical findings, demonstrating that these effects of Rhcg deletion reflected impaired collecting duct ammonia secretion and were not indirectly mediated through mechanisms involving extrarenal Rhcg expression (92). Moreover, Rhcg deletion decreases transepithelial ammonia permeability in perfused collecting duct segments and decreases apical membrane NH3 permeability (16).
Nonglycosylated Rh proteins
The nonglycosylated Rh proteins, RhD and RhCE in humans and Rh30 in nonhumans, are present in erythrocytes in a multimeric complex with RhAG. RhCE appears to neither transport ammonia or its analogue, methylammonia, nor alter transport by RhAG (183). Structural models suggest that the arrangement of key amino acids is sufficiently different in RhCE and RhD that they either do not transport ammonia or that they do so by mechanisms different from those used by the glycosylated Rh glycoproteins and their bacterial orthologs (30).
Acknowledgements
The preparation of this manuscript was supported in part by funds from NIH (R01-DK045788) and from the Department of Veterans Affairs (1I01BX000818).
Reference
- 1.Adam W, Simpson DP. Glutamine transport in rat kidney mitochondria in metabolic acidosis. J Clin Invest. 1974;54:165. doi: 10.1172/JCI107738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adam WR, Koretsky AP, Weiner MW. 31P-NMR in vivo measurement of renal intracellular pH: Effects of acidosis and K+ depletion in rats. Am J Physiol. 1986;251:F904–F910. doi: 10.1152/ajprenal.1986.251.5.F904. [DOI] [PubMed] [Google Scholar]
- 3.Adam WR, Simpson DP. Renal mitochondrial glutamine metabolism and dietary potassium and protein content. Kidney Int. 1975;7:325–330. doi: 10.1038/ki.1975.45. [DOI] [PubMed] [Google Scholar]
- 4.Adrogue HJ. Glucose homeostasis and the kidney. Kidney Int. 1992;42:1266–1282. doi: 10.1038/ki.1992.414. [DOI] [PubMed] [Google Scholar]
- 5.Alleyne GA, Barnswell J, McFarlane-Anderson N, Alexander JE. Renal ammoniagenic factor in the plasma of rats with acute metabolic acidosis. Am J Physiol. 1981;241:F112–F116. doi: 10.1152/ajprenal.1981.241.2.F112. [DOI] [PubMed] [Google Scholar]
- 6.Ambuhl PM, Amemiya M, Danczkay M, Lotscher M, Kaissling B, Moe OW, Preisig PA, Alpern RJ. Chronic metabolic acidosis increases NHE3 protein abundance in rat kidney. Am J Physiol. 1996;271:F917–F925. doi: 10.1152/ajprenal.1996.271.4.F917. [DOI] [PubMed] [Google Scholar]
- 7.Amemiya M, Loffing J, Lotscher M, Kaissling B, Alpern RJ, Moe OW. Expression of NHE-3 in the apical membrane of rat renal proximal tubule and thick ascending limb. Kidney Int. 1996;48:1206–1215. doi: 10.1038/ki.1995.404. [DOI] [PubMed] [Google Scholar]
- 8.Amemiya M, Tabei K, Kusano E, Asano Y, Alpern RJ. Incubation of OKP cells in low-K+ media increases NHE3 activity after early decrease in intracellular pH. Am J Physiol. 1999;276:C711–C716. doi: 10.1152/ajpcell.1999.276.3.C711. [DOI] [PubMed] [Google Scholar]
- 9.Amlal H, Paillard M, Bichara M. Cl−-dependent NH4+ transport mechanisms in medullary thick ascending limb cells. Am J Physiol. 1994;267:C1607–C1615. doi: 10.1152/ajpcell.1994.267.6.C1607. [DOI] [PubMed] [Google Scholar]
- 10.Amlal H, Paillard M, Bichara M. NH4+ transport pathways in cells of medullary thick ascending limb of rat kidney. NH4+ conductance and K+/NH4+(H+) antiport. J Biol Chem. 1994;269:21962–21971. [PubMed] [Google Scholar]
- 11.Attmane-Elakeb A, Mount DB, Sibella V, Vernimmen C, Hebert SC, Bichara M. Stimulation by in vivo and in vitro metabolic acidosis of expression of rBSC-1, the Na+-K+ (NH4+)-2Cl- cotransporter of the rat medullary thick ascending limb. J Biol Chem. 1998;273:33681–33691. doi: 10.1074/jbc.273.50.33681. [DOI] [PubMed] [Google Scholar]
- 12.Attmane-Elakeb A, Sibella V, Vernimmen C, Belenfant X, Hebert SC, Bichara M. Regulation by glucocorticoids of expression and activity of rBSC1, the Na+-K+(NH4+)-2Cl- cotransporter of medullary thick ascending limb. J Biol Chem. 2000;275:33548–33553. doi: 10.1074/jbc.M006591200. [DOI] [PubMed] [Google Scholar]
- 13.Bakouh N, Benjelloun F, Hulin P, Brouillard F, Edelman A, Cherif-Zahar B, Planelles G. NH3 is involved in the NH4+ transport induced by the functional expression of the human Rh C glycoprotein. J Biol Chem. 2004;279:15975–15983. doi: 10.1074/jbc.M308528200. [DOI] [PubMed] [Google Scholar]
- 14.Bell JM, Feild AL. The distribution of ammonia between water and chloroform. J Am Chem Soc. 1911;33:940–943. [Google Scholar]
- 15.Bishop JM, Verlander JW, Lee HW, Nelson RD, Weiner AJ, Handlogten ME, Weiner ID. Role of the Rhesus glycoprotein, Rh B Glycoprotein, in renal ammonia excretion. Am J Physiol Renal Physiol. 2010;299:F1065–F1077. doi: 10.1152/ajprenal.00277.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biver S, Belge H, Bourgeois S, Van Vooren P, Nowik M, Scohy S, Houillier P, Szpirer J, Szpirer C, Wagner CA, Devuyst O, Marini AM. A role for Rhesus factor Rhcg in renal ammonium excretion and male fertility. Nature. 2008;456:339–343. doi: 10.1038/nature07518. [DOI] [PubMed] [Google Scholar]
- 17.Bode BP. Recent molecular advances in mammalian glutamine transport. J Nutr 131: 2475S-2485S, 2001. doi: 10.1093/jn/131.9.2475S. [DOI] [PubMed] [Google Scholar]
- 18.Boron WF, Waisbren SJ, Modlin IM, Geibel JP. Unique permeability barrier of the apical surface of parietal and chief cells in isolated perfused gastric glands. J Exp Biol. 1994;196:347–360. doi: 10.1242/jeb.196.1.347. [DOI] [PubMed] [Google Scholar]
- 19.Bourgeois S, Meer LV, Wootla B, Bloch-Faure M, Chambrey R, Shull GE, Gawenis LR, Houillier P. NHE4 is critical for the renal handling of ammonia in rodents. J Clin Invest. 2010;120:1895–1904. doi: 10.1172/JCI36581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown ACN, Hallouane D, Mawby WJ, Karet FE, Saleem MA, Howie AJ, Toye AM. RhCG is the major putative ammonia transporter expressed in human kidney and RhBG is not expressed at detectable levels. Am J Physiol Renal Physiol. 2009;296:F1279–F1290. doi: 10.1152/ajprenal.00013.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown D, Wagner CA. Molecular mechanisms of acid-base sensing by the kidney. J Am Soc Nephrol. 2012;23:774–780. doi: 10.1681/ASN.2012010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown D, Zhu XL, Sly WS. Localization of membrane-associated carbonic anhydrase type IV in kidney epithelial cells. Proc Natl Acad Sci USA. 1990;87:7457–7461. doi: 10.1073/pnas.87.19.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burch HB, Choi S, McCarthy WZ, Wong PY, Lowry OH. The location of glutamine synthetase within the rat and rabbit nephron. Biochem Biophys Res Commun. 1978;82:498–505. doi: 10.1016/0006-291x(78)90902-6. [DOI] [PubMed] [Google Scholar]
- 24.Burch HB, Narins RG, Chu C, Fagioli S, Choi S, McCarthy W, Lowry OH. Distribution along the rat nephron of three enzymes of gluconeogenesis in acidosis and starvation. Am J Physiol. 1978;235:F246–F253. doi: 10.1152/ajprenal.1978.235.3.F246. [DOI] [PubMed] [Google Scholar]
- 25.Busque SM, Wagner CA. Potassium restriction, high protein intake, and metabolic acidosis increase expression of the glutamine transporter SNAT3 (Slc38a3) in mouse kidney. Am J Physiol Renal Physiol. 2009;297:F440–F450. doi: 10.1152/ajprenal.90318.2008. [DOI] [PubMed] [Google Scholar]
- 26.Chambrey R, Goossens D, Bourgeois S, Picard N, Bloch-Faure M, Leviel F, Geoffroy V, Cambillau M, Colin Y, Paillard M, Houillier P, Cartron JP, Eladari D. Genetic ablation of Rhbg in mouse does not impair renal ammonium excretion. Am J Physiol Renal Physiol. 2005;289:F1281–F1290. doi: 10.1152/ajprenal.00172.2005. [DOI] [PubMed] [Google Scholar]
- 27.Chambrey R, John PL, Eladari D, Quentin F, Warnock DG, Abraham-son DR, Podevin RA, Paillard M. Localization and functional characterization of Na+/H +exchanger isoform NHE4 in rat thick ascending limbs. Am J Physiol Renal Physiol. 2001;281:F707–F717. doi: 10.1152/ajprenal.2001.281.4.F707. [DOI] [PubMed] [Google Scholar]
- 28.Cheval L, Morla L, Elalouf JM, Doucet A. Kidney collecting duct acid-base “regulon”. Physiol Genomics. 2006;27:271–281. doi: 10.1152/physiolgenomics.00069.2006. [DOI] [PubMed] [Google Scholar]
- 29.Conjard A, Komaty O, Delage H, Boghossian M, Martin M, Ferrier B, Baverel G. Inhibition of glutamine synthetase in the mouse kidney: A novel mechanism of adaptation to metabolic acidosis. J Biol Chem. 2003;278:38159–38166. doi: 10.1074/jbc.M302885200. [DOI] [PubMed] [Google Scholar]
- 30.Conroy MJ, Bullough PA, Merrick M, Avent ND. Modelling the human rhesus proteins: Implications for structure and function. Br J Haematol. 2005;131:543–551. doi: 10.1111/j.1365-2141.2005.05786.x. [DOI] [PubMed] [Google Scholar]
- 31.Curthoys NP, Gstraunthaler G. Mechanism of increased renal gene expression during metabolic acidosis. Am J Physiol Renal Physiol. 2001;281:F381–F390. doi: 10.1152/ajprenal.2001.281.3.F381. [DOI] [PubMed] [Google Scholar]
- 32.Curthoys NP, Lowry OH. The distribution of glutaminase isoenzymes in the various structures of the nephron in normal, acidotic, and alkalotic rat kidney. J Biol Chem. 1973;248:162–168. [PubMed] [Google Scholar]
- 33.Curthoys NP, Taylor L, Hoffert JD, Knepper MA. Proteomic analysis of the adaptive response of rat renal proximal tubules to metabolic acidosis. Am J Physiol Renal Physiol. 2007;292:F140–F147. doi: 10.1152/ajprenal.00217.2006. [DOI] [PubMed] [Google Scholar]
- 34.DuBose TD, Good DW, Hamm LL, Wall SM. Ammonium transport in the kidney: New physiological concepts and their clinical implications. J Am Soc Nephrol. 1991;1:1193–1203. doi: 10.1681/ASN.V1111193. [DOI] [PubMed] [Google Scholar]
- 35.Eladari D, Cheval L, Quentin F, Bertrand O, Mouro I, Cherif-Zahar B, Cartron JP, Paillard M, Doucet A, Chambrey R. Expression of RhCG, a new putative NH3/NH4+ transporter, along the rat nephron. J Am Soc Nephrol. 2002;13:1999–2008. doi: 10.1097/01.asn.0000025280.02386.9d. [DOI] [PubMed] [Google Scholar]
- 36.Elkinton JR, Huth EJ, Webster GD, Jr., McCance RA. The renal excretion of hydrogen ion in renal tubular acidosis. Am J Med. 1960;36:554–575. doi: 10.1016/0002-9343(60)90090-5. [DOI] [PubMed] [Google Scholar]
- 37.Elkjar ML, Kwon TH, Wang W, Nielsen J, Knepper MA, Frokiar J, Nielsen S. Altered expression of renal NHE3, TSC, BSC-1, and ENaC subunits in potassium-depleted rats. Am J Physiol Renal Physiol. 2002;283:F1376–F1388. doi: 10.1152/ajprenal.00186.2002. [DOI] [PubMed] [Google Scholar]
- 38.Elkjar ML, Nejsum LN, Gresz V, Kwon TH, Jensen UB, Frokiar J, Nielsen S. Immunolocalization of aquaporin-8 in rat kidney, gastrointestinal tract, testis, and airways. Am J Physiol Renal Physiol. 2001;281:F1047–F1057. doi: 10.1152/ajprenal.0158.2001. [DOI] [PubMed] [Google Scholar]
- 39.Flessner MF, Mejia R, Knepper MA. Ammonium and bicarbonate transport in isolated perfused rodent long-loop thin descending limbs. Am J Physiol. 1993;264:F388–F396. doi: 10.1152/ajprenal.1993.264.3.F388. [DOI] [PubMed] [Google Scholar]
- 40.Flessner MF, Wall SM, Knepper MA. Ammonium and bicarbonate transport in rat outer medullary collecting ducts. Am J Physiol. 1992;262:F1–F7. doi: 10.1152/ajprenal.1992.262.1.F1. [DOI] [PubMed] [Google Scholar]
- 41.Frank AE, Weiner ID. Effects of ammonia on acid-base transport by the B-type intercalated cell. J Am Soc Nephrol. 2001;12:1607–1614. doi: 10.1681/ASN.V1281607. [DOI] [PubMed] [Google Scholar]
- 42.Frank AE, Wingo CS, Andrews PM, Ageloff S, Knepper MA, Weiner ID. Mechanisms through which ammonia regulates cortical collecting duct net proton secretion. Am J Physiol Renal Physiol. 2002;282:F1120–F1128. doi: 10.1152/ajprenal.00266.2001. [DOI] [PubMed] [Google Scholar]
- 43.Frank AE, Wingo CS, Weiner ID. Effects of ammonia on bicarbonate transport in the cortical collecting duct. Am J Physiol Renal Physiol. 2000;278:F219–F226. doi: 10.1152/ajprenal.2000.278.2.F219. [DOI] [PubMed] [Google Scholar]
- 44.Garg LC, Narang N. Ouabain-insensitive K-adenosine triphosphatase in distal nephron segments of the rabbit. J Clin Invest. 1988;81:1204–1208. doi: 10.1172/JCI113436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ginns SM, Knepper MA, Ecelbarger CA, Terris J, He X, Coleman RA, Wade JB. Immunolocalization of the secretory isoform of Na-K-Cl cotransporter in rat renal intercalated cells. J Am Soc Nephrol. 1996;7:2533–2542. doi: 10.1681/ASN.V7122533. [DOI] [PubMed] [Google Scholar]
- 46.Goldstein L. Glutamine transport by mitochondria isolated from normal and acidotic rats. Am J Physiol. 1975;229:1027–1033. doi: 10.1152/ajplegacy.1975.229.4.1027. [DOI] [PubMed] [Google Scholar]
- 47.Good DW. Adaptation of HCO3− and NH4+ transport in rat MTAL: Effects of chronic metabolic acidosis and Na+ intake. Am J Physiol. 1990;258:F1345–F1353. doi: 10.1152/ajprenal.1990.258.5.F1345. [DOI] [PubMed] [Google Scholar]
- 48.Good DW. Ammonium transport by the thick ascending limb of henles loop. Annu Rev Physiol. 1994;56:623–647. doi: 10.1146/annurev.ph.56.030194.003203. [DOI] [PubMed] [Google Scholar]
- 49.Good DW, Burg MB. Ammonia production by individual segments of the rat nephron. J Clin Invest. 1984;73:602–610. doi: 10.1172/JCI111250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Good DW, DuBose TD. Concentrations of NH3 in cortex and medulla of rat kidney. Contrib Nephrol. 1988;63:16–20. doi: 10.1159/000415692. [DOI] [PubMed] [Google Scholar]
- 51.Good DW, Knepper MA, Burg MB. Ammonia and bicarbonate transport by thick ascending limb of rat kidney. Am J Physiol. 1984;247:F35–F44. doi: 10.1152/ajprenal.1984.247.1.F35. [DOI] [PubMed] [Google Scholar]
- 52.Gruswitz F, Chaudhary S, Ho JD, Schlessinger A, Pezeshki B, Ho CM, Sali A, Westhoff CM, Stroud RM. Function of human Rh based on structure of RhCG at 2.1 Ä. Proc Natl Acad Sci U S A. 2010;107:9638–9643. doi: 10.1073/pnas.1003587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gumz ML, Lynch IJ, Greenlee MM, Cain BD, Wingo CS. The renal H+-K+-ATPases: Physiology, regulation, and structure. Am J Physiol Renal Physiol. 2010;298:F12–F21. doi: 10.1152/ajprenal.90723.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamm LL, Gillespie C, Klahr S. NH4Cl inhibition of transport in the rabbit cortical collecting tubule. Am J Physiol. 1985;248:F631–F637. doi: 10.1152/ajprenal.1985.248.5.F631. [DOI] [PubMed] [Google Scholar]
- 55.Hamm LL, Simon EE. Roles and mechanisms of urinary buffer excretion. Am J Physiol. 1987;253:F595–F605. doi: 10.1152/ajprenal.1987.253.4.F595. [DOI] [PubMed] [Google Scholar]
- 56.Hamm LL, Simon EE. Ammonia transport in the proximal tubule in vivo. Am J Kidney Dis. 1989;14:253–257. doi: 10.1016/s0272-6386(89)80197-0. [DOI] [PubMed] [Google Scholar]
- 57.Hamm LL, Simon EE. Ammonia transport in the proximal tubule. Miner Electrolyte Metab. 1990;16:283–290. [PubMed] [Google Scholar]
- 58.Han KH, Croker BP, Clapp WL, Werner D, Sahni M, Kim J, Kim HY, Handlogten ME, Weiner ID. Expression of the ammonia transporter, Rh C Glycoprotein, in normal and neoplastic human kidney. J Am Soc Nephrol. 2006;17:2670–2679. doi: 10.1681/ASN.2006020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han KH, Kim HY, Croker BP, Reungjui S, Lee SY, Kim J, Handlogten ME, Adin CA, Weiner ID. Effects of ischemia-reperfusion injury on renal ammonia metabolism and the collecting duct. Am J Physiol Renal Physiol. 2007;293:F1342–F1354. doi: 10.1152/ajprenal.00437.2006. [DOI] [PubMed] [Google Scholar]
- 60.Han KH, Lee HW, Handlogten ME, Bishop J, Levi M, Kim J, Verlander JW, Weiner ID. Effect of hypokalemia on renal expression of the ammonia transporter family members, Rh B glycoprotein and Rh C glycoprotein, in the rat kidney. Am J Physiol Renal Physiol. 2011;301:F823–F832. doi: 10.1152/ajprenal.00266.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han KH, Mekala K, Babida V, Kim HY, Handlogten ME, Verlander JW, Weiner ID. Expression of the gas transporting proteins, Rh B Glycoprotein and Rh C Glycoprotein, in the murine lung. Am J Physiol Lung Cell Mol Physiol. 2009;297:L153–L163. doi: 10.1152/ajplung.90524.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Handlogten ME, Hong SP, Westhoff CM, Weiner ID. Basolateral ammonium transport by the mouse inner medullary collecting duct cell (mIMCD-3) Am J Physiol Renal Physiol. 2004;287:F628–F638. doi: 10.1152/ajprenal.00363.2003. [DOI] [PubMed] [Google Scholar]
- 63.Handlogten ME, Hong SP, Westhoff CM, Weiner ID. Apical ammonia transport by the mouse inner medullary collecting duct cell (mIMCD-3) Am J Physiol Renal Physiol. 2005;289:F347–F358. doi: 10.1152/ajprenal.00253.2004. [DOI] [PubMed] [Google Scholar]
- 64.Handlogten ME, Hong SP, Zhang L, Vander AW, Steinbaum ML, Campbell-Thompson M, Weiner ID. Expression of the ammonia transporter proteins, Rh B Glycoprotein and Rh C Glycoprotein, in the intestinal tract. Am J Physiol Gastrointest. 2005;288:G1036–G1047. doi: 10.1152/ajpgi.00418.2004. [DOI] [PubMed] [Google Scholar]
- 65.Hanson RW, Reshef L. Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev Biochem. 1997;66:581–611. doi: 10.1146/annurev.biochem.66.1.581. [DOI] [PubMed] [Google Scholar]
- 66.Holm LM, Jahn TP, Moller AL, Schjoerring JK, Ferri D, Klaerke DA, Zeuthen T. NH3 and NH4 +permeability in aquaporin-expressing Xenopus oocytes. Pflugers Arch. 2005;450:415–428. doi: 10.1007/s00424-005-1399-1. [DOI] [PubMed] [Google Scholar]
- 67.Hossain SA, Chaudhry FA, Zahedi K, Siddiqui F, Amlal H. Cellular and molecular basis of increased ammoniagenesis in potassium deprivation. Am J Physiol - Renal Physiol. 2011;301:F969–F978. doi: 10.1152/ajprenal.00010.2011. [DOI] [PubMed] [Google Scholar]
- 68.Hughey RP, Rankin BB, Curthoys NP. Acute acidosis and renal arterivenous differences of glutamine in normal and adrenalectomized rats. Am J Physiol Renal Physiol. 1980;238:F199–F204. doi: 10.1152/ajprenal.1980.238.3.F199. [DOI] [PubMed] [Google Scholar]
- 69.Hwang JJ, Perera S, Shapiro RA, Curthoys NP. Mechanism of altered renal glutaminase gene expression in response to chronic acidosis. Biochemistry. 1991;30:7522–7526. doi: 10.1021/bi00244a022. [DOI] [PubMed] [Google Scholar]
- 70.Ibrahim H, Lee YJ, Curthoys NP. Renal response to metabolic acidosis: Role of mRNA stabilization. Kidney Int. 2007;73:11–18. doi: 10.1038/sj.ki.5002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Indiveri C, Abruzzo G, Stipani I, Palmieri F. Identification and purification of the reconstitutively active glutamine carrier from rat kidney mitochondria. Biochem J. 1998;333:285–290. doi: 10.1042/bj3330285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jaeger P, Karlmark B, Giebisch G. Ammonium transport in rat cortical tubule: Relationship to potassium metabolism. Am J Physiol. 1983;245:F593–F600. doi: 10.1152/ajprenal.1983.245.5.F593. [DOI] [PubMed] [Google Scholar]
- 73.Jones K. Comprehensive Inorganic Chemistry. Oxford; Pergamon: 1973. [Google Scholar]
- 74.Jorgensen PL. Sodium and potassium ion pump in kidney tubules. Physiol Rev. 1980;60:864–917. doi: 10.1152/physrev.1980.60.3.864. [DOI] [PubMed] [Google Scholar]
- 75.Kaiser S, Hwang JJ, Smith H, Banner C, Welbourne TC, Curthoys NP. Effect of altered acid-base balance and of various agonists on levels of renal glutamate dehydrogenase mRNA. Am J Physiol Renal Physiol. 1992;262:F507–F512. doi: 10.1152/ajprenal.1992.262.3.F507. [DOI] [PubMed] [Google Scholar]
- 76.Karinch AM, Lin CM, Wolfgang CL, Pan M, Souba WW. Regulation of expression of the SN1 transporter during renal adaptation to chronic metabolic acidosis in rats. Am J Physiol Renal Physiol. 2002;283:F1011–F1019. doi: 10.1152/ajprenal.00106.2002. [DOI] [PubMed] [Google Scholar]
- 77.Karnovsky MJ, Himmelhoch SR. Histochemical localization of glutaminase I activity in kidney. Am J Physiol. 1961;201:786–790. doi: 10.1152/ajplegacy.1961.201.5.786. [DOI] [PubMed] [Google Scholar]
- 78.Kikeri D, Sun A, Zeidel ML, Hebert SC. Cell membranes impermeable to NH3. Nature. 1989;339:478–480. doi: 10.1038/339478a0. [DOI] [PubMed] [Google Scholar]
- 79.Kim HY, Baylis C, Verlander JW, Han KH, Reungjui S, Handlogten ME, Weiner ID. Effect of reduced renal mass on renal ammonia transporter family, Rh C glycoprotein and Rh B glycoprotein, expression. Am J Physiol Renal Physiol. 2007;293:F1238–F1247. doi: 10.1152/ajprenal.00151.2007. [DOI] [PubMed] [Google Scholar]
- 80.Kim HY, Verlander JW, Bishop JM, Cain BD, Han KH, Igarashi P, Lee HW, Handlogten ME, Weiner ID. Basolateral expression of the ammonia transporter family member, Rh C Glycoprotein, in the mouse kidney. Am J Physiol Renal Physiol. 2009;296:F545–F555. doi: 10.1152/ajprenal.90637.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim YH, Verlander JW, Matthews SW, Kurtz I, Shin WK, Weiner ID, Everett LA, Green ED, Nielsen S, Wall SM. Intercalated cell H+/OH− transporter expression is reduced in Slc26a4 null mice. Am J Physiol Renal Physiol. 2005;289:F1262–F1272. doi: 10.1152/ajprenal.00206.2005. [DOI] [PubMed] [Google Scholar]
- 82.Kinsella JL, Aronson PS. Interaction of NH4+ and Li+ with the renal microvillus membrane Na+-H+ exchanger. Am J Physiol. 1981;241:C220–C226. doi: 10.1152/ajpcell.1981.241.5.C220. [DOI] [PubMed] [Google Scholar]
- 83.Knepper MA. NH4+ transport in the kidney. Kidney Int. 1991;40:S95–S102. [PubMed] [Google Scholar]
- 84.Knepper MA, Good DW, Burg MB. Mechanism of ammonia secretion by cortical collecting ducts of rabbits. Am J Physiol. 1984;247:F729–F738. doi: 10.1152/ajprenal.1984.247.5.F729. [DOI] [PubMed] [Google Scholar]
- 85.Knepper MA, Good DW, Burg MB. Ammonia and bicarbonate transport by rat cortical collecting ducts perfused in vitro. Am J Physiol. 1985;249:F870–F877. doi: 10.1152/ajprenal.1985.249.6.F870. [DOI] [PubMed] [Google Scholar]
- 86.Kurtz I, Balaban RS. Ammonium as a substrate for Na+-K+-ATPase in rabbit proximal tubules. Am J Physiol. 1986;250:F497–F502. doi: 10.1152/ajprenal.1986.250.3.F497. [DOI] [PubMed] [Google Scholar]
- 87.Kwon TH, Fulton C, Wang W, Kurtz I, Frokiaer J, Aalkjaer C, Nielsen S. Chronic metabolic acidosis upregulates rat kidney Na-HCO cotransporters NBCn1 and NBC3 but not NBC1. Am J Physiol Renal Physiol. 2002;282:F341–F351. doi: 10.1152/ajprenal.00104.2001. [DOI] [PubMed] [Google Scholar]
- 88.Laghmani K, Preisig PA, Moe OW, Yanagisawa M, Alpern RJ. Endothelin-1/endothelin-B receptor-mediated increases in NHE3 activity in chronic metabolic acidosis. J Clin Invest. 2001;107:1563–1569. doi: 10.1172/JCI11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lancaster G, Mohyuddin F, Scriver CR, Whelan DT. A-aminobutyrate pathway in mammalian kidney cortex. Biochim Biophys Acta. 1973;297:229–240. doi: 10.1016/0304-4165(73)90069-x. [DOI] [PubMed] [Google Scholar]
- 90.Laterza OF, Taylor L, Unnithan S, Nguyen L, Curthoys NP. Mapping and functional analysis of an instability element in phosphoenolpyruvate carboxykinase mRNA. Am J Physiol Renal Physiol. 2000;279:F866–F873. doi: 10.1152/ajprenal.2000.279.5.F866. [DOI] [PubMed] [Google Scholar]
- 91.Lee FN, Oh G, McDonough AA, Youn JH. Evidence for gut factor in K+ homeostasis. Am J Physiol Renal Physiol. 2007;293:F541–F547. doi: 10.1152/ajprenal.00427.2006. [DOI] [PubMed] [Google Scholar]
- 92.Lee HW, Verlander JW, Bishop JM, Igarashi P, Handlogten ME, Weiner ID. Collecting duct-specific Rh C glycoprotein deletion alters basal and acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol. 2009;296:F1364–F1375. doi: 10.1152/ajprenal.90667.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee HW, Verlander JW, Bishop JM, Nelson RD, Handlogten ME, Weiner ID. Effect of intercalated cell-specific Rh C glycoprotein deletion on basal and metabolic acidosis-stimulated renal ammonia excretion. Am J Physiol Renal Physiol. 2010;299:F369–F379. doi: 10.1152/ajprenal.00120.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee S, Lee HJ, Yang HS, Thornell IM, Bevensee MO, Choi I. Na/bicarbonate cotransporter NBCn1 in the kidney medullary thick ascending limb cell line is upregulated under acidic conditions and enhances ammonium transport. Exp Physiol. 2010;95:926–937. doi: 10.1113/expphysiol.2010.053967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lemieux G, Baverel G, Vinay P, Wadoux P. Glutamine synthetase and glutamyltransferase in the kidney of man, dog, and rat. Am J Physiol. 1976;231:1068–1073. doi: 10.1152/ajplegacy.1976.231.4.1068. [DOI] [PubMed] [Google Scholar]
- 96.Lim SW, Ahn KO, Kim WY, Han DH, Li C, Ghee JY, Han KH, Kim HY, Handlogten ME, Kim J, Yang CW, Weiner ID. Expression of ammonia transporters, Rhbg and Rhcg, in chronic cyclosporine nephropathy in rats. Nephron Exp Nephrol. 2008;110:e49–e58. doi: 10.1159/000153245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu Z, Chen Y, Mo R, Hui Cc, Cheng JF, Mohandas N, Huang CH. Characterization of human RhCG and mouse Rhcg as novel nonerythroid Rh glycoprotein homologues predominantly expressed in kidney and testis. J Biol Chem. 2000;275:25641–25651. doi: 10.1074/jbc.M003353200. [DOI] [PubMed] [Google Scholar]
- 98.Liu Z, Peng J, Mo R, Hui Cc, Huang CH. Rh type B glycoprotein is a new member of the Rh superfamily and a putative ammonia transporter in mammals. J Biol Chem. 2001;276:1424–1433. doi: 10.1074/jbc.M007528200. [DOI] [PubMed] [Google Scholar]
- 99.Lonnerholm G, Wistrand PJ. Membrane-bound carbonic anhydrase CA IV in the human kidney. Acta Physiol Scand. 2008;141:231–234. doi: 10.1111/j.1748-1716.1991.tb09072.x. [DOI] [PubMed] [Google Scholar]
- 100.Lopez C, Metral S, Eladari D, Drevensek S, Gane P, Chambrey R, Bennett V, Cartron JP, Van Kim C, Colin Y. The ammonium transporter RhBG: Requirement of a tyrosine-based signal and ankyrin-G for basolateral targeting and membrane anchorage in polarized kidney epithelial cells. J Biol Chem. 2004;280:8221–8228. doi: 10.1074/jbc.M413351200. [DOI] [PubMed] [Google Scholar]
- 101.Ludewig U. Electroneutral ammonium transport by basolateral Rhesus B glycoprotein. J Physiol. 2004;559:751–759. doi: 10.1113/jphysiol.2004.067728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mak DO, Dang B, Weiner ID, Foskett JK, Westhoff CM. Characterization of transport by the kidney Rh glycoproteins, RhBG and RhCG. Am J Physiol Renal Physiol. 2006;290:F297–F305. doi: 10.1152/ajprenal.00147.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marini AM, Matassi G, Raynal V, Andre B, Cartron JP, Cherif-Zahar B. The human Rhesus-associated RhAG protein and a kidney homologue promote ammonium transport in yeast. Nat Genet. 2000;26:341–344. doi: 10.1038/81656. [DOI] [PubMed] [Google Scholar]
- 104.Marquez J, Lopez de la Oliva A, Mates JM, Segura JA, Alonso FJ. Glutaminase: A multifaceted protein not only involved in generating glutamate. Neurochem Int. 2005;48:465–471. doi: 10.1016/j.neuint.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 105.Mavrothalassitis G, Tzimagiorgis G, Mitsialis A, Zannis V, Plaitakis A, Papamatheakis J, Moschonas N. Isolation and characterization of cDNA clones encoding human liver glutamate dehydrogenase: Evidence for a small gene family. Proc Natl Acad Sci U S A. 1988;85:3494–3498. doi: 10.1073/pnas.85.10.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mayer M, Schaaf G, Mouro I, Lopez C, Colin Y, Neumann P, Cartron JP, Ludewig U. Different transport mechanisms in plant and human AMT/Rh-type ammonium transporters. J Gen Physiol. 2006;127:133–144. doi: 10.1085/jgp.200509369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moret C, Dave MH, Schulz N, Jiang JX, Verrey F, Wagner CA. Regulation of renal amino acid transporters during metabolic acidosis. Am J Physiol Renal Physiol. 2007;292:F555–F566. doi: 10.1152/ajprenal.00113.2006. [DOI] [PubMed] [Google Scholar]
- 108.Mouro-Chanteloup I, Cochet S, Chami M, Genetet S, Zidi-Yahiaoui N, Engel A, Colin Y, Bertrand O, Ripoche P. Functional reconstitution into liposomes of purified human RhCG ammonia channel. PLoS ONE. 2010;5:e8921. doi: 10.1371/journal.pone.0008921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mufti J, Hajarnis S, Shepardson K, Gummadi L, Taylor L, Curthoys NP. Role of AUF1 and HuR in the pH-responsive stabilization of phosphoenolpyruvate carboxykinase mRNA in LLC-PK1-F +cells. Am J Physiol - Renal Physiol. 2011;301:F1066–F1077. doi: 10.1152/ajprenal.00303.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Musa-Aziz R, Chen LM, Pelletier MF, Boron WF. Relative CO2/NH3 selectivities of AQP1, AQP4, AQP5, AmtB, and RhAG. Proc Natl Acad Sci U S A. 2009;106:5406–5411. doi: 10.1073/pnas.0813231106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nagami GT. Luminal secretion of ammonia in the mouse proximal tubule perfused in vitro. J Clin Invest. 1988;81:159–164. doi: 10.1172/JCI113287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nagami GT. Ammonia production and secretion by the proximal tubule. Am J Kidney Dis. 1989;14:258–261. doi: 10.1016/s0272-6386(89)80198-2. [DOI] [PubMed] [Google Scholar]
- 113.Nagami GT. Effect of bath and luminal potassium concentration on ammonia production and secretion by mouse proximal tubules perfused in vitro. J Clin Invest. 1990;86:32–39. doi: 10.1172/JCI114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nagami GT. Effect of angiotensin II on ammonia production and secretion by mouse proximal tubules perfused in vitro. J Clin Invest. 1992;89:925–931. doi: 10.1172/JCI115673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nagami GT. Enhanced ammonia secretion by proximal tubules from mice receiving NH(4)Cl: Role of angiotensin II. Am J Physiol Renal Physiol. 2002;282:F472–F477. doi: 10.1152/ajprenal.00249.2001. [DOI] [PubMed] [Google Scholar]
- 116.Nagami GT. Ammonia production and secretion by S3 proximal tubule segments from acidotic mice: Role of ANG II. Am J Physiol Renal Physiol. 2004;287:F707–F712. doi: 10.1152/ajprenal.00189.2003. [DOI] [PubMed] [Google Scholar]
- 117.Nagami GT, Sonu CM, Kurokawa K. Ammonia production by isolated mouse proximal tubules perfused in vitro: Effect of metabolic acidosis. J Clin Invest. 1986;78:124–129. doi: 10.1172/JCI112540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nagami GT. Role of angiotensin II in the enhancement of ammonia production and secretion by the proximal tubule in metabolic acidosis. Am J Physiol Renal Physiol. 2008;294:F874–F880. doi: 10.1152/ajprenal.00286.2007. [DOI] [PubMed] [Google Scholar]
- 119.Nakamura S, Amlal H, Galla JH, Soleimani M. NH4+ secretion in inner medullary collecting duct in potassium deprivation: Role of colonic H+-K+-ATPase. Kidney Int. 1999;56:2160–2167. doi: 10.1046/j.1523-1755.1999.00780.x. [DOI] [PubMed] [Google Scholar]
- 120.Nakhoul NL, Abdulnour-Nakhoul SM, Schmidt E, Doetjes R, Rabon E, Hamm LL. pH sensitivity of ammonium transport by Rhbg. Am J Physiol Cell Physiol. 2010;299:C1386–C1397. doi: 10.1152/ajpcell.00211.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nakhoul NL, Abdulnour-Nakhoul SM, Boulpaep EL, Rabon E, Schmidt E, Hamm LL. Substrate specificity of Rhbg: Ammonium and methyl ammonium transport. Am J Physiol Cell Physiol. 2010;299:C695–C705. doi: 10.1152/ajpcell.00019.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nakhoul NL, DeJong H, Abdulnour-Nakhoul SM, Boulpaep EL, Hering-Smith K, Hamm LL. Characteristics of renal Rhbg as an NH4+ transporter. Am J Physiol Renal Physiol. 2004;288:F170–F181. doi: 10.1152/ajprenal.00419.2003. [DOI] [PubMed] [Google Scholar]
- 123.Nakhoul NL, Hering-Smith KS, Abdulnour-Nakhoul SM, Hamm LL. Transport of NH(3)/NH in oocytes expressing aquaporin-1. Am J Physiol Renal Physiol. 2001;281:F255–F263. doi: 10.1152/ajprenal.2001.281.2.F255. [DOI] [PubMed] [Google Scholar]
- 124.Occleshaw VJ. The distribution of ammonia between chloroform and water at 25°C. J Chem Soc. 1931:1436–1437. [Google Scholar]
- 125.Odgaard E, Jakobsen JK, Frische S, Praetorius J, Nielsen S, Aalkjaer C, Leipziger J. Basolateral Na+-dependent HCO3- transporter NBCn1-mediated HCO3- influx in rat medullary thick ascending limb. J Physiol (Lond) 2004;555:205–218. doi: 10.1113/jphysiol.2003.046474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.OWEN EE, Robinson RR. Amino acid extraction and ammonia metabolism by the human kidney during the prolonged administration of ammonium chloride. J Clin Invest. 1963;42:263–276. doi: 10.1172/JCI104713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pfeiffer R, Rossier G, Spindler B, Meier C, Kuhn L, Verrey F. Amino acid transport of y+L-type by heterodimers of 4F2hc/CD98 and members of the glycoprotein-associated amino acid transporter family. EMBO J. 1999;18:49–57. doi: 10.1093/emboj/18.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Quentin F, Eladari D, Cheval L, Lopez C, Goossens D, Colin Y, Cartron JP, Paillard M, Chambrey R. RhBG and RhCG, the putative ammonia transporters, are expressed in the same cells in the distal nephron. J Am Soc Nephrol. 2003;14:545–554. doi: 10.1097/01.asn.0000050413.43662.55. [DOI] [PubMed] [Google Scholar]
- 129.Rector FC, Jr, Orloff J. The effect of the administration of sodium bicarbonate and ammonium chloride on the excretion and production of ammonia. The absence of alterations in the activity of renal ammonia-producing enzymes in the dog. J Clin Invest. 1959;38:366–372. doi: 10.1172/JCI103810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Richterich RW, Goldstein L. Distribution of glutamine metabolizing enzymes and production of urinary ammonia in the mammalian kidney. Am J Physiol. 1958;195:316–320. doi: 10.1152/ajplegacy.1958.195.2.316. [DOI] [PubMed] [Google Scholar]
- 131.Ripoche P, Bertrand O, Gane P, Birkenmeier C, Colin Y, Cartron JP. Human Rhesus-associated glycoprotein mediates facilitated transport of NH3 into red blood cells. Proc Natl Acad Sci U S A. 2004;101:17222–17227. doi: 10.1073/pnas.0403704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sartorius OW, Roemmelt JC, Pitts RF. The renal regulation of acid-base balance in man. IV. The nature of the renal compensations in ammonium chloride acidosis. J Clin Invest. 1949;28:423–439. doi: 10.1172/JCI102087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sastrasinh S, Sastrasinh M. Glutamine transport in submitochondrial particles. Am J Physiol Renal Physiol. 1989;257:F1050–F1058. doi: 10.1152/ajprenal.1989.257.6.F1050. [DOI] [PubMed] [Google Scholar]
- 134.Sastrasinh S, Tannen RL. Mechanism by which enhanced ammonia production reduces urinary potassium excretion. Kidney Int. 1981;20:326–331. doi: 10.1038/ki.1981.142. [DOI] [PubMed] [Google Scholar]
- 135.Schoolwerth AC, Gesek FA. Intramitochondrial pH and ammonium production in rat and dog kidney cortex. Miner Electrolyte Metab. 1990;16:264–269. [PubMed] [Google Scholar]
- 136.Schoolwerth AC, Strzelecki T, LaNoue KF, Hoover WJ. Effect of pH and alpha-ketoglutarate on mitochondrial ammonia production. Contrib Nephrol. 1982;31:127–133. doi: 10.1159/000406628. [DOI] [PubMed] [Google Scholar]
- 137.Schoolwerth AC. Regulation of renal ammoniagenesis in metabolic acidosis. Kidney Int. 1991;40:961–973. doi: 10.1038/ki.1991.301. [DOI] [PubMed] [Google Scholar]
- 138.Schroeder JM, Liu W, Curthoys NP. pH-responsive stabilization of glutamate dehydrogenase mRNA in LLC-PK1-F+ cells. Am J Physiol Renal Physiol. 2003;285:F258–F265. doi: 10.1152/ajprenal.00422.2002. [DOI] [PubMed] [Google Scholar]
- 139.Schwartz GJ. Physiology and molecular biology of renal carbonic anhydrase. J Nephrol. 2002;15(Suppl 5):S61–S74. [PubMed] [Google Scholar]
- 140.Schwartz GJ, Kittelberger AM, Barnhart DA, Vijayakumar S. Carbonic anhydrase IV is expressed in H+-secreting cells of rabbit kidney. Am J Physiol Renal Physiol. 2000;278:F894–F904. doi: 10.1152/ajprenal.2000.278.6.F894. [DOI] [PubMed] [Google Scholar]
- 141.Seshadri RM, Klein JD, Kozlowski S, Sands JM, Kim YH, Handlogten ME, Verlander JW, Weiner ID. Renal expression of the ammonia transporters, Rhbg and Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol. 2006;290:F397–F408. doi: 10.1152/ajprenal.00162.2005. [DOI] [PubMed] [Google Scholar]
- 142.Seshadri RM, Klein JD, Smith T, Sands JM, Handlogten ME, Verlander JW, Weiner ID. Changes in the subcellular distribution of the ammonia transporter Rhcg, in response to chronic metabolic acidosis. Am J Physiol Renal Physiol. 2006;290:F1443–F1452. doi: 10.1152/ajprenal.00459.2005. [DOI] [PubMed] [Google Scholar]
- 143.Shapiro RA, Morehouse RF, Curthoys NP. Inhibition by glutamate of phosphate-dependent glutaminase of rat kidney. Biochem J. 1982;207:561–566. doi: 10.1042/bj2070561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shashidharan P, Huntley GW, Meyer T, Morrison JH, Plaitakis A. Neuron-specific human glutamate transporter: Molecular cloning, characterization and expression in human brain. Brain Res. 1994;662:245–250. doi: 10.1016/0006-8993(94)90819-2. [DOI] [PubMed] [Google Scholar]
- 145.Silbernagl S. Tubular reabsorption of l-glutamine studied by free-flow micropuncture and microperfusion of rat kidney. Int J Biochem. 1980;12:9–16. doi: 10.1016/0020-711x(80)90034-8. [DOI] [PubMed] [Google Scholar]
- 146.Silbernagl S. Ammoniagenesis catalyzed by hippurate-activated gamma-glutamyltransferase in the lumen of the proximal tubule. A microperfusion study in rat kidney in vivo. Pflugers Arch. 1986;407(Suppl 2):S72–S79. doi: 10.1007/BF00584933. [DOI] [PubMed] [Google Scholar]
- 147.Simon E, Martin D, Buerkert J. Contribution of individual superficial nephron segments to ammonium handling in chronic metabolic acidosis in the rat. Evidence for ammonia disequilibrium in the renal cortex. J Clin Invest. 1985;76:855–864. doi: 10.1172/JCI112043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Simon EE, Hamm LL. Ammonia entry along rat proximal tubule in vivo: Effects of luminal pH and flow rate. Am J Physiol. 1987;253:F760–F766. doi: 10.1152/ajprenal.1987.253.4.F760. [DOI] [PubMed] [Google Scholar]
- 149.Simon EE, Merli C, Herndon J, Cragoe EJ, Jr., Hamm LL. Effects of barium and 5-(N-ethyl-N-isopropyl)-amiloride on proximal tubule ammonia transport. Am J Physiol. 1992;262:F36–F39. doi: 10.1152/ajprenal.1992.262.1.F36. [DOI] [PubMed] [Google Scholar]
- 150.Simon EE, Merli C, Herndon J, Hamm LL. Contribution of luminal ammoniagenesis to proximal tubule ammonia appearance in the rat. Am J Physiol. 1990;259:F402–F407. doi: 10.1152/ajprenal.1990.259.3.F402. [DOI] [PubMed] [Google Scholar]
- 151.Simpson DP, Adam W. Glutamine transport and metabolism by mitochondria from dog renal cortex. General properties and response to acidosis and alkalosis. J Biol Chem. 1975;250:8148–8158. [PubMed] [Google Scholar]
- 152.Singh SK, Binder HJ, Geibel JP, Boron WF. An apical permeability barrier to NH3/NH4+ in isolated, perfused colonic crypts. Proc Natl Acad Sci U S A. 1995;92:11573–11577. doi: 10.1073/pnas.92.25.11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Solbu TT, Boulland JL, Zahid W, Lyamouri Bredahl MK, miry-Moghaddam M, Storm-Mathisen J, Roberg BA, Chaudhry FA. Induction and targeting of the glutamine transporter SN1 to the basolateral membranes of cortical kidney tubule cells during chronic metabolic acidosis suggest a role in pH regulation. J Am Soc Nephrol. 2005;16:869–877. doi: 10.1681/ASN.2004060433. [DOI] [PubMed] [Google Scholar]
- 154.Soria LR, Fanelli E, Altamura N, Svelto M, Marinelli RA, Calamita G. Aquaporin 8-facilitated mitochondrial ammonia transport. Biochem Biophys Res Commun. 2010;393:217–221. doi: 10.1016/j.bbrc.2010.01.104. [DOI] [PubMed] [Google Scholar]
- 155.Star RA, Burg MB, Knepper MA. Luminal disequilibrium pH and ammonia transport in outer medullary collecting duct. Am J Physiol. 1987;252:F1148–F1157. doi: 10.1152/ajprenal.1987.252.6.F1148. [DOI] [PubMed] [Google Scholar]
- 156.Star RA, Kurtz I, Mejia R, Burg MB, Knepper MA. Disequilibrium pH and ammonia transport in isolated perfused cortical collecting ducts. Am J Physiol. 1987;253:F1232–F1242. doi: 10.1152/ajprenal.1987.253.6.F1232. [DOI] [PubMed] [Google Scholar]
- 157.Stumvoll M, Perriello G, Meyer C, Gerich J. Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney. 1999;55:778–792. doi: 10.1046/j.1523-1755.1999.055003778.x. [DOI] [PubMed] [Google Scholar]
- 158.Tannen RL. Ammonia metabolism. Am J Physiol Renal Physiol. 1978;235:F265–F277. doi: 10.1152/ajprenal.1978.235.4.F265. [DOI] [PubMed] [Google Scholar]
- 159.Tannen RL, Sahai A. Biochemical pathways and modulators of renal ammoniagenesis. Miner Electrolyte Metab. 1990;16:249–258. [PubMed] [Google Scholar]
- 160.Tannen RL, Terrien T. Potassium-sparing effect of enhanced renal ammonia production. Am J Physiol. 1975;228:699–705. doi: 10.1152/ajplegacy.1975.228.3.699. [DOI] [PubMed] [Google Scholar]
- 161.Taylor L, Curthoys NP. Glutamine metabolism: Role in acid-base balance. Biochem Mol Biol Educ. 2004;32:291–304. doi: 10.1002/bmb.2004.494032050388. [DOI] [PubMed] [Google Scholar]
- 162.Tong J, Harrison G, Curthoys NP. The effect of metabolic acidosis on the synthesis and turnover of rat renal phosphate-dependent glutaminase. Biochem J. 1986;233:139–144. doi: 10.1042/bj2330139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tong J, Shapiro RA, Curthoys NP. Changes in the levels of translatable glutaminase mRNA during onset and recovery from metabolic acidosis. Biochemistry. 1987;26:2773–2777. doi: 10.1021/bi00384a018. [DOI] [PubMed] [Google Scholar]
- 164.Tsuruoka S, Kittelberger AM, Schwartz GJ. Carbonic anhydrase II and IV mRNA in rabbit nephron segments: Stimulation during metabolic acidosis. Am J Physiol. 1998;274:F259–F267. doi: 10.1152/ajprenal.1998.274.2.F259. [DOI] [PubMed] [Google Scholar]
- 165.Valles P, Lapointe MS, Wysocki J, Batlle D. Kidney vacuolar H+-TPase: Physiology and regulation. Semin Nephrol. 2006;26:361–374. doi: 10.1016/j.semnephrol.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 166.Van Slyke DD, Phillips RA, Hamilton PB, Archibard RM, Futcher PH, Miller A. Glutamine as source material of urinary ammonia. J Biol Chem. 1943;150:481–482. [Google Scholar]
- 167.Verlander JW, Miller RT, Frank AE, Royaux IE, Kim YH, Weiner ID. Localization of the ammonium transporter proteins, Rh B glycoprotein and Rh C glycoprotein, in the mouse kidney. Am J Physiol Renal Physiol. 2003;284:F323–F337. doi: 10.1152/ajprenal.00050.2002. [DOI] [PubMed] [Google Scholar]
- 168.Waisbren SJ, Geibel JP, Modlin IM, Boron WF. Unusual permeability properties of gastric gland cells. Nature. 1994;368:332–335. doi: 10.1038/368332a0. [DOI] [PubMed] [Google Scholar]
- 169.Wall SM. Ouabain reduces net acid secretion and increases pHi by inhibiting NH4+ uptake on rat tIMCD Na(+)-K(+)-ATPase. Am J Physiol. 1997;273:F857–F868. doi: 10.1152/ajprenal.1997.273.6.F857. [DOI] [PubMed] [Google Scholar]
- 170.Wall SM, Fischer MP. Contribution of the Na+-K+-2Cl− cotransporter (NKCC1) to transepithelial transport of H+, NH4+, K+, and Na+ in rat outer medullary collecting duct. J Am Soc Nephrol. 2002;13:827–835. doi: 10.1681/ASN.V134827. [DOI] [PubMed] [Google Scholar]
- 171.Wall SM, Fischer MP, Kim GH, Nguyen BM, Hassell KA. In rat inner medullary collecting duct, NH4 +uptake by the Na,K-ATPase is increased during hypokalemia. Am J Physiol Renal Physiol. 2002;282:F91–F102. doi: 10.1152/ajprenal.0141.2001. [DOI] [PubMed] [Google Scholar]
- 172.Wall SM, Flessner MF, Knepper MA. Distribution of luminal carbonic anhydrase activity along rat inner medullary collecting duct. Am J Physiol. 1991;260:F738–F748. doi: 10.1152/ajprenal.1991.260.5.F738. [DOI] [PubMed] [Google Scholar]
- 173.Wall SM, Koger LM. NH+4 transport mediated by Na(+)-K(+)-ATPase in rat inner medullary collecting duct. Am J Physiol. 1994;267:F660–F670. doi: 10.1152/ajprenal.1994.267.4.F660. [DOI] [PubMed] [Google Scholar]
- 174.Wall SM, Trinh HN, Woodward KE. Heterogeneity of NH4+ transport in mouse inner medullary collecting duct cells. Am J Physiol. 1995;269:F536–F544. doi: 10.1152/ajprenal.1995.269.4.F536. [DOI] [PubMed] [Google Scholar]
- 175.Watts BA, Good DW. Effects of ammonium on intracellular pH in rat medullary thick ascending limb: Mechanisms of apical membrane NH4+ transport. J Gen Physiol. 1994;103:917–936. doi: 10.1085/jgp.103.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Weiner ID. The Rh gene family and renal ammonium transport. Curr Opin Nephrol Hyper. 2004;13:533–540. doi: 10.1097/00041552-200409000-00009. [DOI] [PubMed] [Google Scholar]
- 177.Weiner ID, Hamm LL. Molecular mechanisms of renal ammonia transport. Annu Rev Physiol. 2007;69:317–340. doi: 10.1146/annurev.physiol.69.040705.142215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Weiner ID, Miller RT, Verlander JW. Localization of the ammonium transporters, Rh B glycoprotein and Rh C glycoprotein in the mouse liver. Gastroenterology. 2003;124:1432–1440. doi: 10.1016/s0016-5085(03)00277-4. [DOI] [PubMed] [Google Scholar]
- 179.Weiner ID, Verlander JW. Molecular physiology of the Rh ammonia transport proteins. Curr Opin Nephrol Hypertens. 2010;19:471–477. doi: 10.1097/MNH.0b013e32833bfa4e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Weiner ID, Verlander JW. Role of NH3 and NH4+ transporters in renal acid-base transport. Am J Physiol Renal Physiol. 2011;300:F11–F23. doi: 10.1152/ajprenal.00554.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Weinstein AM, Krahn TA. A mathematical model of rat ascending Henle limb. II. Epithelial function. Am J Physiol Renal Physiol. 2009;298:F525–F542. doi: 10.1152/ajprenal.00231.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Welbourne TC, Dass PD. Gamma glutamyltransferase contribution to renal ammoniagenesis in vivo. Pflugers Arch. 1988;411:573–578. doi: 10.1007/BF00582380. [DOI] [PubMed] [Google Scholar]
- 183.Westhoff CM, Ferreri-Jacobia M, Mak DO, Foskett JK. Identification of the erythrocyte Rh-blood group glycoprotein as a mammalian ammonium transporter. J Biol Chem. 2002;277:12499–12502. doi: 10.1074/jbc.C200060200. [DOI] [PubMed] [Google Scholar]
- 184.Westhoff CM, Siegel DL, Burd CG, Foskett JK. Mechanism of genetic complementation of ammonium transport in yeast by human erythrocyte Rh-associated glycoprotein (RhAG) J Biol Chem. 2004;279:17443–17448. doi: 10.1074/jbc.M311853200. [DOI] [PubMed] [Google Scholar]
- 185.Wilcox CS, Granges F, Kirk G, Gordon D, Giebisch G. Effects of saline infusion on titratable acid generation and ammonia secretion. Am J Physiol. 1984;247:F506–F519. doi: 10.1152/ajprenal.1984.247.3.F506. [DOI] [PubMed] [Google Scholar]
- 186.Windus DW, Cohn DE, Klahr S, Hammerman MR. Glutamine transport in renal basolateral vesicles from dogs with metabolic acidosis. Am J Physiol. 1984;246:F78–F86. doi: 10.1152/ajprenal.1984.246.1.F78. [DOI] [PubMed] [Google Scholar]
- 187.Windus DW, Klahr S, Hammerman MR. Glutamine transport in basolateral vesicles from dogs with acute respiratory acidosis. Am J Physiol. 1984;247:F403–F407. doi: 10.1152/ajprenal.1984.247.3.F403. [DOI] [PubMed] [Google Scholar]
- 188.Winkler CA, Kittelberger AM, Schwartz GJ. Expression of carbonic anhydrase IV mRNA in rabbit kidney: Stimulation by metabolic acidosis. Am J Physiol. 1997;272:F551–F560. doi: 10.1152/ajprenal.1997.272.4.F551. [DOI] [PubMed] [Google Scholar]
- 189.Wright PA, Knepper MA. Glutamate dehydrogenase activities in microdissected rat nephron segments: Effects of acid-base loading. Am Physiol. 1990;259:F53–F59. doi: 10.1152/ajprenal.1990.259.1.F53. [DOI] [PubMed] [Google Scholar]
- 190.Wright PA, Knepper MA. Phosphate-dependent glutaminase activity in rat renal cortical and medullary tubule segments. Am J Physiol. 1990;259:F961–F970. doi: 10.1152/ajprenal.1990.259.6.F961. [DOI] [PubMed] [Google Scholar]
- 191.Yang B, Song Y, Zhao D, Verkman AS. Phenotype analysis of aquaporin-8 null mice. Am J Physiol Cell Physiol. 2005;288:C1161–C1170. doi: 10.1152/ajpcell.00564.2004. [DOI] [PubMed] [Google Scholar]
- 192.Yang B, Zhao D, Solenov E, Verkman AS. Evidence from knockout mice against physiologically significant aquaporin 8-facilitated ammonia transport. Am J Physiol Cell Physiol. 2006;291:C417–C423. doi: 10.1152/ajpcell.00057.2006. [DOI] [PubMed] [Google Scholar]
- 193.Yip KP, Kurtz I. NH3 permeability of principal cells and intercalated cells measured by confocal fluorescence imaging. Am J Physiol. 1995;269:F545–F550. doi: 10.1152/ajprenal.1995.269.4.F545. [DOI] [PubMed] [Google Scholar]
- 194.Zidi-Yahiaoui N, Mouro-Chanteloup I, D’Ambrosio AM, Lopez C, Gane P, Van Kim C, Cartron JP, Colin Y, Ripoche P. Human Rhesus B and Rhesus C glycoproteins: Properties of facilitated ammonium transport in recombinant kidney cells. Biochem J. 2005;391:33–40. doi: 10.1042/BJ20050657. [DOI] [PMC free article] [PubMed] [Google Scholar]