

Abstract
Xeroderma pigmentosum (XP) is a rare autosomal recessive genetic disorder characterized by defective DNA repair leading to clinical and cellular hypersensitivity to ultraviolet (UV) radiation and carcinogenic agents. Important clinical features are: Intense cutaneous photosensitivity, xerosis, poikiloderma, actinic keratosis, acute burning under minimal sun exposure, erythemas, hyperpigmented lentiginous macules, and malignant lesions in sun-exposed areas, including basocellular carcinoma, squamous cell carcinoma, and melanoma. There is a great involvement of many parts of the body, especially head and neck. Oral implications such as severe oral pain and mouth opening limitation were present due to perioral scars. The disorder is associated more commonly in populations where marriage of close blood relatives is common. Treatment of the disorder includes avoidance of UV radiation, topical application of 5-fluorouracil to treat actinic keratoses, and regular evaluation by an ophthalmologist, dermatologist, and neurologist. Genetic counseling is important aspects as an increased incidence of consanguineous marriages have been reported with this disorder. In addition, this paper discuss some important aspects concerning the role of the dental professional management of this entity, since XP patients require constant dental care and follow-up in order to control the occurrence of new lesions on the lips or inside oral cavity.
Keywords: Actinic cheilitis, dental treatment, oral genoderamtosis, xeroderma pigmentosum
INTRODUCTION
Xeroderma pigmentosum (XP) (literally dry pigmented skin) is defined by extreme sensitivity to sunlight, resulting in sunburn, pigment changes in the skin, and a greatly elevated incidence of skin cancers. It is an autosomal recessive disease with the potential of causing more than 1000-fold increase in the frequency of all types of major skin cancers (basal cell cancers, squamous cell cancers, and malignant melanoma) in areas exposed to sunlight compared with normal population along with sub-optimal mental development.[1,2] One enzyme of excision-repair mechanism (DNA repair) is defective, and the patient's skin is abnormally sensitive to the defects of sun-light. In vitro cultures of skin fibroblast from these patients are also abnormally sensitive to ultraviolet (UV) light as well as some chemical carcinogens.[3] Homozygotes with XP show marked tendency to develop skin cancer and have the fully expressed condition, which includes strong disposition to sunlight-induced melanoma, basal cell carcinoma and squamous cell carcinoma of the skin in addition to other nonneoplastic cutaneous and ocular abnormalities such as photophobia, lacrimation, keratitis, opacities and tumors of the eyelid and cornea.[1,4]
The objective of the current paper is to recount the case of XP with oral exposition of the same and to outline the function of the dentist in regulating the disease.
CASE REPORT
A 29-year-old female was referred to the Department of Oral Medicine at the College of Dentistry, King Saud University, Riyadh, Saudi Arabia for diagnosis and treatment of a tongue swelling. Duration of the lesion, according to the patient was 3 months. She also had dark brown pigmentation all over her face. Her history revealed development of pigmentation at the age of 1 year. A positive family history with a similar condition in one of her sibling was given. They were born to normal parents with consanguineous marriage. Both girls showed signs of blisters and erythema after exposure to sunlight. They were diagnosed with XP when they were 1-year-old. Her sister was admitted for treatment of “skin malignancy” at King Faisal Specialist Hospital. No other members of the family had the condition.
On examination, there was a painless, sessile firm tumor mass attached to the tip of the tongue mostly on the dorsum. It measured about 1.5 cm × 1 cm. Oral hygienics was unsatisfactory with presence of moderate to severe periodontitis with substantiate amount of calculus deposits. Spacing between teeth was noticeable especially between upper and lower anterior teeth. Maxillary central incisor showed enamel hypoplasia.
The patient had multiple, small, dark brown colored lentigines appeared as tanned pigmented macules, on the face, varying in size and shape with areas of depigmentation in association with significant visual impairment [Figure 1]. Photophobia was also present. Her sibling also revealed the same abnormalities. The lips were red with areas of bleeding and crusted along with perioral scarring that leads to limited mouth opening along with actinic cheilitis [Figure 2]. Gingiva and alveolar mucosa were red and inflamed with generalized gingival recession. She had protrusion of maxillary anterior teeth and Class II division 2 malocclusion [Figure 3]. Orthopantomogram reveals the severe generalized bone loss along with spacing [Figure 4].
Figure 1.
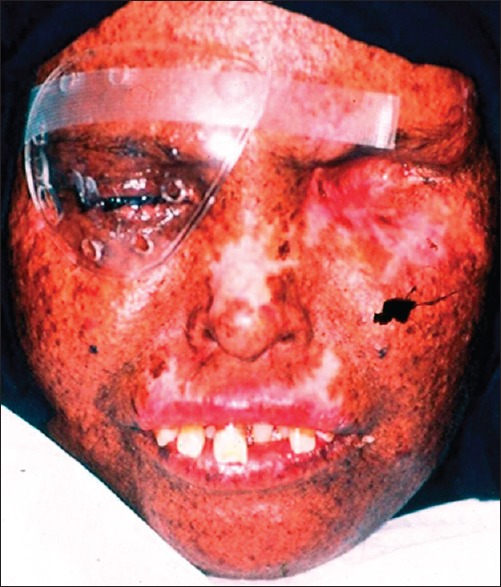
Depicts the macules and areas of hyperpigmentation along with severe ocular abnormalities
Figure 2.
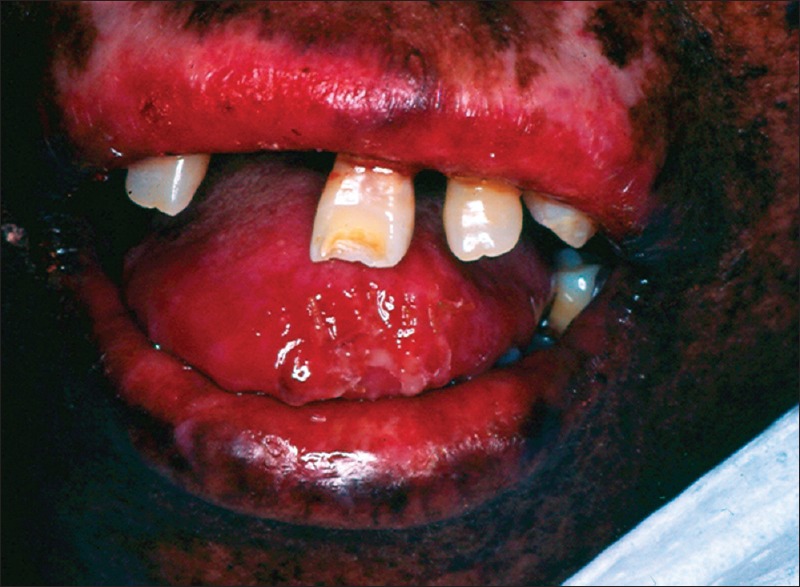
Reveals the perioral scarring due to actinic chilitis
Figure 3.
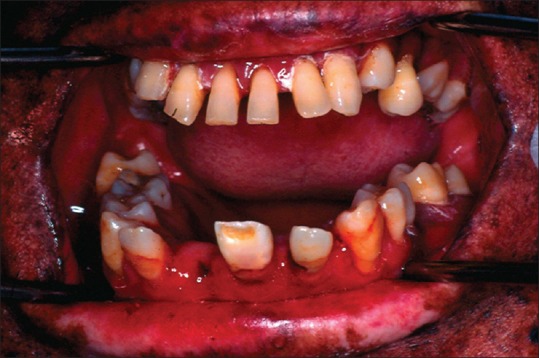
Intraoral view revealed dark brown and gray pigmentation, poor oral hygiene, periodontitis and enamel hypoplasia upper central incisor
Figure 4.
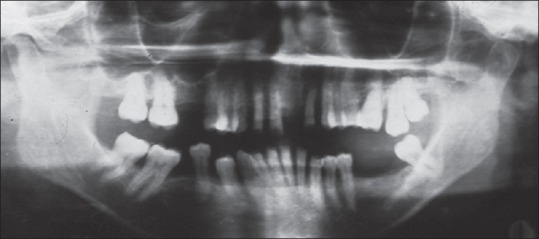
Illustrates the generalized bone loss and spacing
An excisional biopsy of the swelling was obtained with no postoperative complication. Histological diagnosis revealed a diagnosis of pyogenic granuloma with the following description: Tongue tissue with extensive surface tissue necrosis and degeneration. Two epithelial areas showed degeneration and necrosis. Deep tissues showed congested as well as empty blood vessels. The inflammatory cells infiltrate in the necrotic surface and within the tissue were mixed polymorphonuclear leukocytes, plasma cells, lymphocytes, foam cells and macrophages. At higher power, few normal mitosis were evident. There were also some multinucleated giant cells. Large epithelioid macrophages were rather prominent.
Furthermore, patient was sent to dermatological, neurological, and ophthalmological departments for evaluation of the multi-factorial disease.
During the follow-up visit, patient was complaining of dental pain and mobility as a result of trauma she received when she fell down while walking after an eye operation.
DISCUSSION
Xeroderma pigmentosum is known as autosomal recessive disease referred as chromosomal and DNA instability syndrome.[2] XP was first described in 1874 by Hebra and Kaposi. In 1882, Kaposi coined the term XP for the condition, referring to its characteristic dry, pigmented skin.[5] The frequency is approximately 1/250,000 populations. An equal incidence has been reported in males and females. A history of consanguinity has been elicited in our case.[6,7]
Patients diagnosed with XP are at an increased risk of developing skin cancer due to mutation in XP genes, which cause defective DNA repair results in the inability to fix UV-damaged DNA. Normally, the DNA repaired was carried out by nucleotide excision repair or postreplication repair (PRR). One XP gene is linked to PRR, and its complementation group is XP variant. A spectrum of clinical disease exists for XP and its severity depends on the amount of sun exposure combined with the total residual amount of DNA repair and this reflected in our case as the patient has maximum number of lentigines.[8,9,10,14]
Without any protection from sunlight that includes, noncovering up the exposed parts of the body and no use of sunscreens, death may occur within the first decade.[2,5] Individuals with XP develop cancer of the skin caused by exposure to mutational effects of radiant energy. Therefore, the disease is considered both hereditary and environmental.[11] The disease is usually manifested at the age of 1 or 2 years, and the particular girl reported here and her sister was diagnosed within the 1st year of life.
The photosensitivity expressed in XP at variable rate, but the spectrum varies between 280 nm and 330 nm. Ocular problems occur in nearly 75% of individuals with XP that include photophobia, conjunctivitis, eyelid solar lentigines, ectropion, symblepharon, vascular pterygia, and epitheliomas of eyelid.[12] Most of the patients had a history of erythema and blistering on scanty exposure to the sunlight as well as photophobia and corneal opacities. They did not know the side-effect of other lights used in the dental clinic. It is the responsibility of the dermatologist to explain the adverse effect of other sources of light.
Oral signs and symptoms of this disease are rare. Malignancies like squamous cell carcinoma can develop on various parts of the oral mucosa,[13] but in our case patient did not have any signs of malignancy although, she had the trismus along with severe generalized periodontitis. While treating these patients, dentists should know that certain precautions are important for these patients such as avoiding all types of UV light and light-cured filling materials irrespective of all intraoral lesions as the light might induce malignant transformation of the epithelial and connective tissue because DNA repair mechanisms are dysfunctional.[3] Light-cure fillings should be replaced by glass-ionomer or auto-cure filling material.
Mainstay of the treatment is avoidance of all sort of exposure to sunlight. Sunscreens (physical and chemical) should be applied to all exposed parts of the body. Oral retinoids are quite effective in the prevention of skin cancer in the patients with XP. Chemical therapy of 5-fluorouracil might be effective in the treatment of actinic keratoses. A new approach to photoprotection is to repair DNA damage after UV exposure. This can be accomplished by delivery of a DNA repair enzyme into the skin by means of specially engineered liposomes.
Gene therapy for XP is still in the theoretical and experimental stage. Various methods of correcting the defects in XP have been attempted in vitro and in animal studies using viral vectors (adenoviruses and retroviruses) carrying the gene replacement products. Ex vivo skin gene therapy, which refers to grafting skin that has the genetic defect corrected, may be useful in XP in the future. Surgical treatment includes excision of a malignant lesion when encountered.[15]
Dermatologists and dentists have the responsibility of informing their patients about the side effects of light-cure materials and provide these patients with information cards explaining their conditions and certain precautions recommended during dental treatment including dental materials to be avoided. Since there is no known cure, genetic counseling is helpful. Existing lesions may be controlled by cytotoxic drugs. Further studies should be done to know the exact effect of the different types of UV light and light-cure filling materials on these patients.
CONCLUSION
Xeroderma pigmentosum is a rare genetic disorder which is related not only to physical abnormalities but, also associated with psychosocial burden to the patient because of its nonpleasing appearance. The extent of damage depends on the amount of exposure to UV light. Dentists should use the UV light very carefully in these patients. Constant education of the patient is the mainstay treatment. Further studies and research is going on for the treatment of XP and hopes lies in the protein and gene therapy.
Footnotes
Source of Support: Nill.
Conflict of Interest: None declared
REFERENCES
- 1.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–50. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 2.Walter JB, Israel MS. General Pathology. 6th ed. Churchill Livingstone; 1987. Physical carcinogenic agent; pp. 52–56. [Google Scholar]
- 3.Kraemer KH, Lee MM, Scotto J. DNA repair protects against cutaneous and internal neoplasia: Evidence from xeroderma pigmentosum. Carcinogenesis. 1984;5:511–4. doi: 10.1093/carcin/5.4.511. [DOI] [PubMed] [Google Scholar]
- 4.Robbins JH, Kraemer KH, Lutzner MA, Festoff BW, Coon HG. Xeroderma pigmentosum. An inherited diseases with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann Intern Med. 1974;80:221–48. doi: 10.7326/0003-4819-80-2-221. [DOI] [PubMed] [Google Scholar]
- 5.English JS, Swerdlow AJ. The risk of malignant melanoma, internal malignancy and mortality in xeroderma pigmentosum patients. Br J Dermatol. 1987;117:457–61. doi: 10.1111/j.1365-2133.1987.tb04925.x. [DOI] [PubMed] [Google Scholar]
- 6.Hedera P, Fink JK. Xeroderma pigmentosum. eMedicine. [Last accessed on 2006 Apr 20]. Available from: http//www.emedicine.com/neuro/topic399.htm .
- 7.Norgauer J, Idzko M, Panther E, Hellstern O, Herouy Y. Xeroderma pigmentosum. Eur J Dermatol. 2003;13:4–9. [PubMed] [Google Scholar]
- 8.DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–96. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warrick E, Garcia M, Chagnoleau C, Chevallier O, Bergoglio V, Sartori D, et al. Preclinical corrective gene transfer in xeroderma pigmentosum human skin stem cells. Mol Ther. 2012;20:798–807. doi: 10.1038/mt.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lambert WC. Genetic diseases associated with DNA and chromosomal instability. Dermatol Clin. 1987;5:85–108. [PubMed] [Google Scholar]
- 11.Lehmann AR, McGibbon D, Stefanini M. Xeroderma pigmentosum. Orphanet J Rare Dis. 2011;6:70. doi: 10.1186/1750-1172-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol Mar. 2012;132:785–96. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patton LL, Valdez IH. Xeroderma pigmentosum: Review and report of a case. Oral Surg Oral Med Oral Pathol. 1991;71:297–300. doi: 10.1016/0030-4220(91)90303-t. [DOI] [PubMed] [Google Scholar]
- 14.Chavanne F, Broughton BC, Pietra D, Nardo T, Browitt A, Lehmann AR, et al. Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell, protein, and transcript levels. Cancer Res. 2000;60:1974–82. [PubMed] [Google Scholar]
- 15.Kraemer KH, Slor H. Xeroderma pigmentosum. Clin Dermatol. 1985;3:33–69. doi: 10.1016/0738-081x(85)90096-3. [DOI] [PubMed] [Google Scholar]