

Abstract
Nonalcoholic fatty liver disease (NAFLD) encompasses a range of manifestations, including steatosis and cirrhosis. Progressive disease is characterized by hepatic leukocyte accumulation in the form of steatohepatitis. The adhesion molecule vascular adhesion protein-1 (VAP-1) is a membrane-bound amine oxidase that promotes leukocyte recruitment to the liver, and the soluble form (sVAP-1) accounts for most circulating monoamine oxidase activity, has insulin-like effects, and can initiate oxidative stress. Here, we determined that hepatic VAP-1 expression is increased in patients with chronic liver disease and that serum sVAP-1 levels are elevated in patients with NAFLD compared with those in control individuals. In 4 murine hepatic injury models, an absence or blockade of functional VAP-1 reduced inflammatory cell recruitment to the liver and attenuated fibrosis. Moreover, disease was reduced in animals expressing a catalytically inactive form of VAP-1, implicating enzyme activity in the disease pathogenesis. Within the liver, hepatic stromal cells expressed functional VAP-1, and evaluation of cultured cells revealed that sVAP-1 promotes leukocyte migration through catalytic generation of ROS, which depended on VAP-1 enzyme activity. VAP-1 enhanced stromal cell spreading and wound closure and modulated expression of profibrotic genes. Together, these results link the amine oxidase activity of VAP-1 with hepatic inflammation and fibrosis and suggest that targeting VAP-1 has therapeutic potential for NAFLD and other chronic fibrotic liver diseases.
Introduction
Nonalcoholic fatty liver disease (NAFLD) represents a spectrum of liver disease encompassing steatosis, nonalcoholic steatohepatitis (NASH), and cirrhosis and is increasingly recognized as the leading cause of liver dysfunction in Western societies. NAFLD is strongly associated with obesity, dyslipidemia, insulin resistance, and diabetes mellitus (1–4) and is considered to be a hepatic manifestation of the metabolic syndrome (5).
Steatosis sensitizes the liver to the induction of inflammation by a second pathogenic insult that promotes oxidative stress and activation of the inflammasome, resulting in steatohepatitis (6, 7). Oxidative stress may occur as a consequence of dietary or environmental factors on the background of the metabolic syndrome (8). Persistent inflammation in response to liver injury is the critical factor that drives progression to fibrosis, cirrhosis, and hepatocellular carcinoma (9). Inflammation at any site, including the liver, is the result of an accumulation of leukocytes organized into an inflammatory infiltrate. For this to occur, leukocytes must be recruited from the circulation by interactions with endothelium and positioned within the tissue (10).
Vascular adhesion protein-1 (VAP-1) is a 170-kDa homodimeric type 2 transmembrane sialoglycoprotein that has a short cytoplasmic tail with no known signal sequence, a single transmembrane segment, and a large extracellular domain (11). VAP-1 is constitutively expressed on human hepatic endothelium and supports lymphocyte adhesion and transendothelial migration across primary hepatic sinusoidal endothelium in vitro and in several models of liver inflammation in vivo (12–15). Cloning of VAP-1 revealed it to be a copper-dependent semicarbazide-sensitive amine oxidase (SSAO) [E.C.1.4.3.6] known as amine oxidase copper–containing 3 (AOC3), which catalyzes the oxidative deamination of exogenous and endogenous primary amines (such as those generated from sarcosine, creatinine, and adrenaline), resulting in the generation of aldehyde, ammonia, and H2O2 (16). These products activate NFκB-dependent chemokine secretion and adhesion molecule expression in liver endothelium (17, 18) and may also initiate and propagate oxidative stress following the conversion of H2O2 to hydroxyl free radicals. VAP-1 is also found in adipocytes and smooth muscle cells, and provision of SSAO substrates to these cells in vitro has insulin-like effects on their metabolism, which is mediated via PI 3-kinase activation by H2O2 (19–21).
A soluble form of VAP-1 (sVAP-1) accounts for nearly all of the circulating amine oxidase activity in humans (22–24). The level of sVAP-1 in serum is regulated in part by changes in plasma insulin (25), and sVAP-1 levels are elevated in a number of inflammatory conditions (23, 26). Our group has previously shown that circulating levels of sVAP-1 are substantially elevated in the serum of patients with several chronic inflammatory liver diseases (alcoholic liver disease, primary biliary cirrhosis, primary sclerosing cholangitis), but not in rheumatoid arthritis or inflammatory bowel disease. VAP-1 is shed from the hepatic vasculature and adipose tissue, and measurement of VAP-1 levels in hepatic and portal veins suggests that the liver is a major source of sVAP-1 (23, 26). Elevated serum sVAP-1 levels have also been reported in patients with diabetes, obesity, and complications of the metabolic syndrome such as atherosclerosis and congestive heart failure (27–31).
These observations led us to propose that VAP-1 may have an important role in the progression of NAFLD and establishment of chronic liver disease (CLD). Here, we show that hepatic expression of VAP-1 is increased in human NAFLD in association with markedly elevated serum levels of sVAP-1 when compared with those in controls matched for age, sex, and metabolic phenotype. We provide evidence that VAP-1 promotes the progression of steatohepatitis, because VAP-1–deficient mice (Aoc3–/–) and WT animals treated with a neutralizing Ab were protected from the development of severe steatohepatitis and onset of fibrosis in 3 murine models of steatohepatitis and a carbon tetrachloride (CCl4) model of fibrosis. In particular, VAP-1 deficiency protects against the accumulation of intrahepatic T cells, NKT cells, and myeloid subsets. Furthermore, through the use of mice expressing catalytically inactive VAP-1 as a consequence of an Aoc3 point mutation knock-in, we demonstrate that the amine oxidase activity of VAP-1 is crucial for the accumulation of these effector immune cells and the establishment of fibrosis. We demonstrate that hepatic stromal cells express VAP-1, which can promote the expression of fibrotic markers and accelerate wound healing. These findings strongly implicate VAP-1 in the pathogenesis of fibrotic liver disease and provide evidence that VAP-1 is a potential therapeutic target in NAFLD and other chronic inflammatory liver diseases.
Results
Circulating serum [sVAP-1] is significantly elevated in human NAFLD and is independently associated with the presence of liver disease.
Because circulating levels of sVAP-1 are significantly elevated in the serum of patients with chronic inflammatory liver diseases and in those with complications of the metabolic syndrome (23, 26–30), we investigated whether sVAP-1 levels in patients with the metabolic syndrome are associated with the presence and severity of NAFLD. We compared serum levels of sVAP-1 in 144 patients with biopsy-proven NAFLD and 74 control patients matched for age and metabolic phenotype but without biochemical or radiological evidence of liver disease (Table 1). The cohorts were well matched for age and BMI. A larger proportion of NAFLD patients were male than in the control cohort, but sVAP-1 levels were higher in females when compared with those in males in both cohorts. An increased number of patients in the NAFLD cohort had hypertension and diabetes, which is unsurprising, given the close association between these conditions and NAFLD. A large proportion of both cohorts had impaired glucose tolerance. Cholesterol levels were similar in each cohort, but those with NAFLD had higher triglyceride levels.
We found that soluble VAP-1 levels were significantly increased in the NAFLD group when compared with those in controls matched for age and metabolic phenotype (median, 837 ng/ml [interquartile range {IQR}, 655–1,051 ng/ml] versus 256 ng/ml [IQR, 212–308 ng/ml], P < 0.0001; Figure 1A and Table 1). sVAP-1 levels in the NAFLD cohort correlated with the amine oxidase activity of the patients’ serum (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI73722DS1). Univariate analysis showed that the homeostatic model of insulin resistance (HOMA-IR) (rs = 0.3, P = 0.03) and diabetic status (rs = 0.32, P = 0.0002) were correlated with sVAP-1 in the NAFLD cohort (Supplemental Table 1), whereas fibrosis stage was the only factor independently associated with sVAP-1 after multiple logistic regression modeling (Table 2). To date, 17 patients from the NAFLD cohort have died (8 from cancer: 4 hepatocellular carcinoma, 1 esophageal cancer, 1 colon cancer, 1 breast cancer, and the eighth patient died from a combination of breast cancer and Ewing’s sarcoma), 13 of whom were cirrhotic. The concentration of sVAP-1 in those who subsequently died (median, 1,287 ng/ml; IQR, 1,243–2,116 ng/ml) was significantly higher (P = 0.000107 by Mann-Whitney U test) than that in patients who are still alive (median, 814 ng/ml; IQR, 653–1,001 ng/ml).
Figure 1. Serum sVAP-1 is elevated in patients with NAFLD and correlates with histological markers of liver injury.

(A) sVAP-1 levels in patients with NAFLD (n = 133) were compared with those in a control cohort of patients with metabolic syndrome but no evidence of liver injury (n = 74). (B–E) Elevated sVAP-1 levels in patients with NAFLD were associated with lobular inflammation (B), hepatocyte ballooning (C), fibrosis (D), and NASH (E). (F) sVAP-1 concentrations in patient serum increased with the fibrosis stage as determined from biopsy samples (Kleiner classification). (A–E) P values were calculated by Mann-Whitney U test; (F) Kruskal-Wallis test (P = 0.0003) and Cuzick trend test (P ≤ 0.0001); P’ was calculated by Mann-Whitney U test with Bonferroni’s correction.
Table 1.
Comparison of physical and biochemical parameters associated with the NAFLD and control cohorts
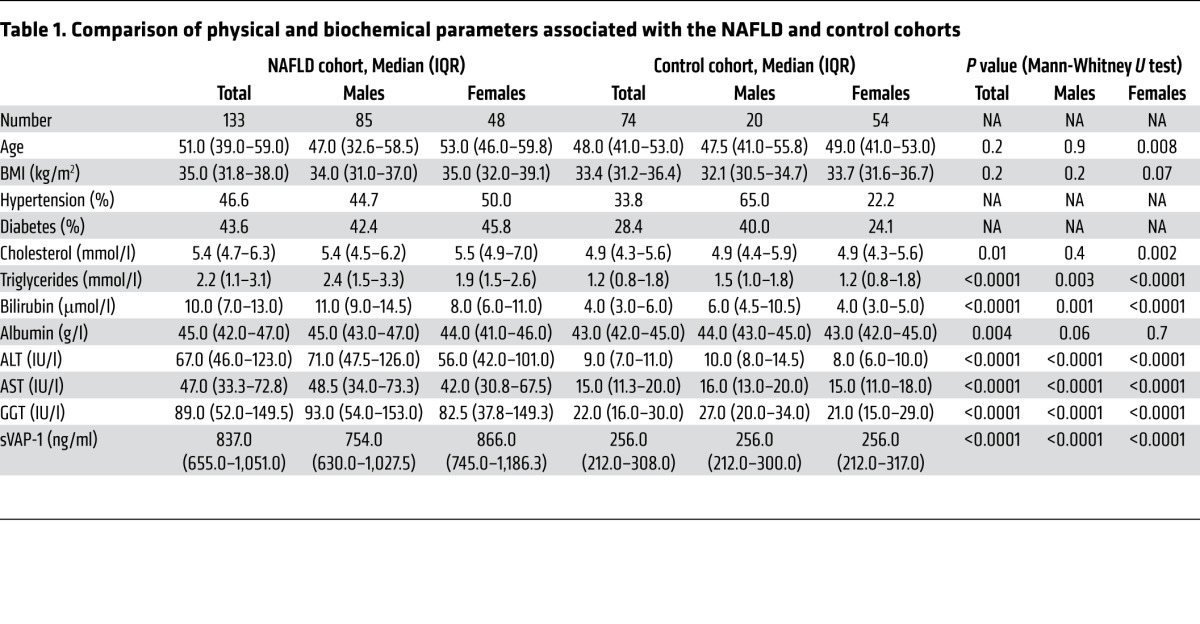
Table 2.
Parameters that independently correlated with sVAP-1 concentrations following multivariate analysis

Circulating sVAP-1 correlates with disease severity and is predictive of NASH and the presence of significant fibrosis on biopsy in NAFLD.
The histological spectrum of NAFLD includes simple steatosis, steatohepatitis with different levels of liver injury, and fibrosis leading to cirrhosis. We assessed sVAP-1 levels in relation to the presence or absence of hepatocyte ballooning, lobular inflammation, and fibrosis and found significantly higher levels when each of these histological features was present (Figure 1, B–D). Furthermore, we found that sVAP-1 levels were significantly increased in patients with NASH (defined as steatosis plus lobular inflammation, hepatocyte ballooning, and/or fibrosis) compared with those in patients with simple steatosis (Figure 1E, P < 0.0001). The area under the receiver operating curve (AUROC) to predict histological NASH by serum sVAP-1 in our cohort was 0.82 (95% CI, 0.74–0.90), and a cutoff level of greater than 793.5 ng/ml estimated NASH with a sensitivity of 67.7% and specificity of 89.7% (data not shown).
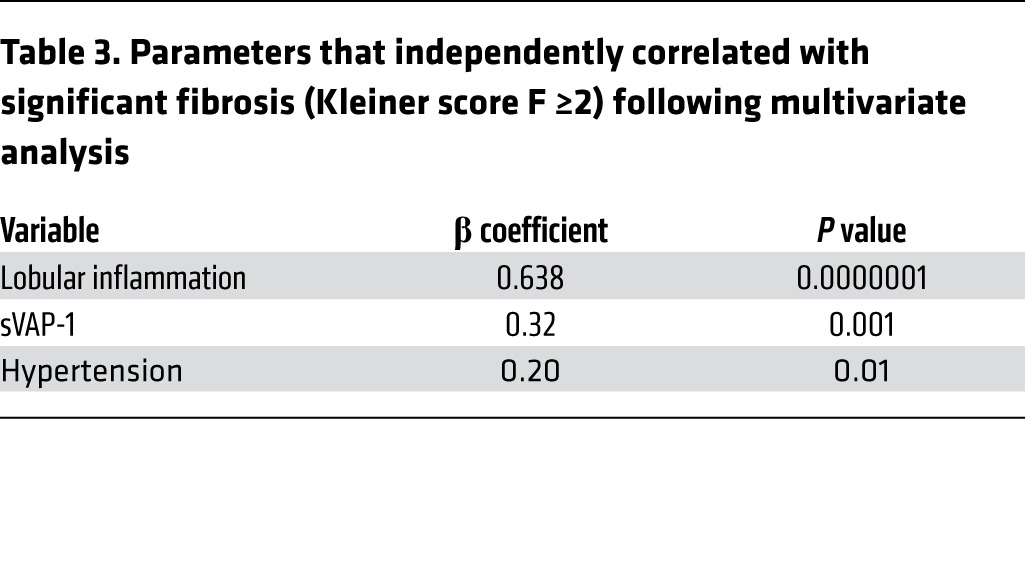
We determined the clinical and biochemical characteristics of our NAFLD cohort that were independently associated with the presence of significant fibrosis (Kleiner classification F ≥2; ref. 32) on liver biopsy (Figure 1F) using univariate descriptive statistics followed by multiple logistic regression analysis with backward elimination (Table 3 and Supplemental Table 2). This revealed sVAP-1 to be a good predictor of the presence of significant fibrosis (F ≥2) (β coefficient, 0.32, P = 0.001). The AUROC to predict significant fibrosis (F ≥2) by serum sVAP-1 was 0.71 (95% CI, 0.62–0.79), and a cutoff level of greater than 1,018 ng/ml estimated F ≥2 with a sensitivity of 45.2% and specificity of 91.5% (data not shown).
Table 3.
Parameters that independently correlated with significant fibrosis (Kleiner score F ≥2) following multivariate analysis
Hepatic VAP-1 expression is increased in human CLD.
We investigated hepatic expression of VAP-1 in the spectrum of NAFLD by immunohistochemistry (Figure 2A). In normal liver and in patients with simple steatosis, we observed that VAP-1 expression was confined to vascular structures, portal vessels, central veins, and sinusoids. In patients with steatohepatitis, we detected more pronounced VAP-1 staining in septal areas when compared with staining in normal liver tissue or simple steatosis, which was partly associated with neovessels in the hepatic scar (Figure 2A). We extended our study to other end-stage CLDs, namely primary biliary cirrhosis, primary sclerosing cholangitis, and alcoholic liver disease. In all 3 diseases, immunohistochemistry demonstrated increased staining of VAP-1 in fibrotic septa (Figure 2B). Multicolor confocal microscopy showed that rather than having a predominantly endothelial (CD31+) distribution, VAP-1 stained α-SMA+ and CD90+ stromal cells within the NASH liver with markedly increased VAP-1 expression throughout the expanded space of Disse (Figure 2D). In contrast, we did not detect VAP-1 in biliary epithelial cells (EpCAM+) or hepatocytes (CK18+) (Figure 2C).
Figure 2. Expression of VAP-1 is increased in liver tissue from patients with evidence of liver disease and localizes to hepatic stromal cells.

(A) Immunohistochemical detection of VAP-1 (brown) in liver sections taken from patients across the spectrum of NAFLD (mild-to-moderate-to-severe steatosis and cirrhosis). Sections were counterstained with hematoxylin (blue). Scale bars: 50 μm. (B) VAP-1 staining (red) was detected in sinusoids and fibrotic septa in CLD (primary biliary cirrhosis, top panel; primary sclerosing cholangitis, middle panel; and alcoholic liver disease, lower panel). Scale bars: 200 μm. (C) Confocal fluorescence microscopy with dual fluorescent staining for VAP-1 (green) and cell-specific markers in cirrhotic liver (red). VAP-1 was present on some CD31+ endothelial cells, but was predominantly associated with the stromal cell markers α-SMA and CD90. VAP-1 did not colocalize with markers of cholangiocytes (EpCAM) or hepatocytes (CK18). Scale bars: 50 μm. (D) Multicolor confocal microscopy showed increased VAP-1 expression levels (green) in the expanded space of Disse in the sinusoids of NASH livers (lower panel) when compared with levels in normal livers (upper panel). The endothelial marker CD31 is shown in red (coexpression is yellow). Scale bars: 20 μm.
Stromal cells express VAP-1, which promotes amine oxidase–dependent lymphocyte migration in vitro.
To determine whether the VAP-1 protein could be synthesized by stromal cells, we isolated activated hepatic stellate cells (HSCs) from nonfibrotic human tissue and in vivo–activated liver myofibroblasts (aLMFs) from end-stage chronically diseased human liver tissue. Both cell types had detectable VAP-1 protein localized to cytoplasmic vesicles with little membranous expression (Figure 3A), and these cells were capable of de novo synthesis of AOC3 mRNA at levels that were comparable to those in hepatic sinusoidal endothelial cells (HSECs). We observed similar levels in the HSC cell line LX2, allowing us to use this cell line to model HSC responses (Figure 3B). Expression of AOC3 mRNA in LX2 cells was upregulated by PDGF-BB and TGF-β1 (Figure 3C), consistent with increased expression of VAP-1 in fibrotic tissues. The addition of recombinant VAP-1 (rVAP-1) had no effect on AOC3 expression in LX2, indicating that transcription of VAP-1 is not modulated by extracellular VAP-1 in this system.
Figure 3. Isolated human hepatic stromal cells express functional VAP-1 capable of driving lymphocyte migration in vitro.

(A) VAP-1 was detected in isolated human aHSCs (left) and aLMFs (right) (DAPI blue). Scale bars: 50 μm. (B) AOC3 mRNA was expressed by HSECs, aLMFs, aHSCs (4 independent patient isolates each) and by the HSC cell line LX2 (3 repeats) at similar levels (dotted line represents negative controls). (C) Expression of AOC3 could be induced following a 24-hour incubation with PDGF-BB or TGF-β1 (both at 10 ng/ml) but not with rVAP-1 (500 ng/ml). Data represent 2 independent experiments measured in triplicate. (D) Amine oxidase activity of aHSCs (n = 4 isolates) in the absence and presence of VAP-1/LOX inhibitor BEA or the LOX-specific inhibitor 2-APN. (E) CD3+ lymphocytes (left) and CD68+ macrophages (right) were closely associated with VAP-1+ vessels and stroma in cirrhotic livers. Scale bars: 50 μm. (F) Conditioned media from primary cell cultures of aHSCs (n = 3 isolates) promoted VAP-1–dependent migration of peripheral blood lymphocytes in modified Boyden chamber assays. (G) Highly purified sVAP-1 induced the migration of peripheral blood lymphocytes in modified Boyden chamber assays in vitro (n = 3 lymphocyte preparations, 12 random fields/sample). (H) Lymphocyte migration was induced by 100 ng/ml sVAP-1 and is shown compared with the baseline response to media alone (n = 3 lymphocyte preparations; 12 random fields per sample). Data were calculated by 1-way ANOVA with Tukey’s post-hoc test (C, G, and H), paired Student’s t test (D), and Mann-Whitney U test (F). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We detected sVAP-1 in conditioned media from in vitro–activated HSC (aHSC) and aLMF cultures and confirmed enzyme activity by detecting H2O2 generation in response to the SSAO substrate benzylamine (Figure 3D). This activity could be blocked by the SSAO inhibitor 2-bromoethylamine (BEA), but not by the lysyl oxidase–specific (LOX-specific) inhibitor β-aminopropionitrile (β-APN). The release of VAP-1 from the stromal compartment in vitro and the increased circulating levels of sVAP-1 in patients with steatohepatitis prompted us to investigate whether sVAP-1 could provide a promigratory signal for leukocytes. Multicolor confocal microscopy of tissue sections taken from the liver of patients with NASH cirrhosis demonstrated a close association between VAP-1+ stromal cells and infiltrating leukocytes (Figure 3E). Conditioned media from in vitro–cultured aHSCs promoted the migration of lymphocytes in Boyden chamber assays, and the addition of a function-blocking anti–VAP-1 Ab (TK8-14) reduced their migratory capacity by more than 50% (Figure 3F). In parallel studies, highly purified human sVAP-1 induced dose-dependent migration of peripheral blood lymphocytes in Boyden chamber assays with a maximal response between 10 and 100 ng/ml (Figure 3G) that was comparable to that seen with 10 ng/ml CXCL12, a potent lymphocyte chemotactic factor. The related amine oxidase LOX has been shown to promote the migration of smooth muscle cells and monocytes through the action of H2O2 (33, 34), and a recent study in zebrafish reported that H2O2 produced by an unspecified amine oxidase promotes leukocyte recruitment during wound healing (35). This led us to investigate whether the promigratory effects of sVAP-1 were attenuated by the VAP-1 enzyme inhibitor BEA or by the removal of H2O2 by catalase (Figure 3H). The inhibition of catalysis or removal of H2O2 reduced the number of cells migrating to sVAP-1 by more than 50%, suggesting that the promigratory action of sVAP-1 is, in part, a consequence of the catalytic generation of ROS by VAP-1 enzyme activity. The addition of the GPCR inhibitor pertussis toxin (PTx) reduced the number of migrating cells to a similar degree, suggesting that VAP-1–induced migration is dependent on GPCR signaling, consistent with studies showing that ROS activate the GPCR (36). Exposure to VAP-1 did not lead to activation of leukocyte β1 or β2 integrins or changes in the surface expression of the chemokine receptors CCR1-10, CXCR1-7, or CX3CR1 (data not shown).
These human data demonstrated that enzymatically active VAP-1 expressed by aHSCs is upregulated in the diseased liver and is capable of modulating leukocyte recruitment. This led us to study the role of VAP-1 in liver fibrogenesis in vivo.
VAP-1 deficiency attenuates CCl4-induced hepatic fibrosis.
We used a CCl4 model to induce hepatic injury in WT (C57BL/6) mice, VAP-1–KO (Aoc3–/–) mice, and WT mice that received weekly doses of a monoclonal anti–VAP-1 Ab (BTT-1029). Control animals received mineral oil (MO) alone. One group of animals was sacrificed after 8 weeks of CCl4 treatment and a second group following an additional 4-week period of resolution during which CCl4 treatment was stopped. We found that untreated Aoc3–/– animals had an overtly normal phenotype with macroscopically and microscopically normal livers, consistent with previous reports (37).
In line with previous studies, we detected bridging fibrosis by picosirius red staining at 8 weeks in WT animals, with significant fibrosis resolution 4 weeks after stopping CCl4 treatment (Figure 4A). Alanine aminotransferase (ALT) levels were comparable between WT, Aoc3–/–, and Ab-treated animals given CCl4, suggesting that the absence of VAP-1 did not diminish the degree of initial liver injury (Figure 4B). This finding was similar to that seen for blockade of a related amine oxidase, lysyl oxidase–like protein-2 (LOXL2), in a CCl4-induced injury model (38).
Figure 4. VAP-1 contributes to scar formation in CCl4-induced murine fibrosis.

(A) Picrosirius red staining of liver from WT mice receiving an 8-week treatment of MO, CCl4, or CCl4 plus a function–blocking anti–VAP-1 Ab (WT CCl4 + Ab) or liver from Aoc3–/– mice that received CCl4 (Aoc3–/– CCl4). Lower panels depict the same experimental groups following an additional 4 weeks of resolution after stopping CCl4 at 8 weeks. Scale bars: 200 μm. (B) Serum ALT levels and (C and D) sirius red (C) and α-SMA (D) staining following 8 weeks of vehicle or CCl4 administration (white bars) and 8 weeks of treatment followed by 4-week resolution (black bars). Fibrosis was also determined by qRT-PCR for Col1a1 and Acta2 (E and F). 3–4 animals per group. 1-way ANOVA with Tukey’s post-hoc test: *P < 0.05, **P < 0.01 and ***P < 0.001 versus WT MO at 8 weeks; †P < 0.05 and #P < 0.001 versus WT MO at 12 weeks.
We found that Aoc3–/– mice and WT animals treated with anti–VAP-1 Ab had significantly less fibrosis at 8 weeks, as demonstrated by picosirius red staining and reduced staining for α-SMA (Figure 4, C and D). Four weeks after stopping CCl4, we observed that fibrosis was greatly reduced in all animals, although WT animals had significantly more extensive picosirius red staining compared with that seen in MO-treated control mice (Figure 4C). We confirmed an increase in fibrosis in the CCl4-treated WT mice relative to that in Aoc3–/– mice at 8 weeks by real-time quantitative reverse transcription PCR (qRT-PCR) of whole liver for collagen 1 (Col1a1) and α-SMA (Acta2), and there was a trend toward a decrease in Col1a1 and Acta2 in WT mice that received Ab (Figure 4, E and F). In all groups, mRNA levels for these markers of fibrosis returned to those seen in mice receiving MO following 4 weeks of resolution. The extent of picosirius red and α-SMA staining was not significantly different between the 8- and 12-week time points in the animals receiving BTT-1029, suggesting that the Ab offers protection against CCl4-induced fibrosis (Figure 4, C and D).
VAP-1 deficiency attenuates leukocyte infiltration into the liver during fibrogenesis.
VAP-1 promotes leukocyte recruitment to the liver (39), suggesting a mechanism to explain the attenuated fibrosis observed in the absence of functional VAP-1. In order to determine whether leukocyte infiltration preceded fibrosis, we quantified lymphocyte and monocyte subsets in the livers of WT and Aoc3–/– animals prior to CCl4 administration (base), following 2, 4, or 8 weeks of CCl4, and at 12 weeks (resolution). Lymphocyte recruitment occurred within the first 2 weeks, whereas myeloid cells peaked at 8 weeks following injury (Figure 5, A–C). There was a significant reduction in the absolute number of all subsets in the livers of Aoc3–/– mice compared with that seen in WT animals receiving the same dose of CCl4. The numbers of cells detected in the WT livers increased progressively up to the 8-week treatment time point and returned to the levels observed for the Aoc3–/– group 4 weeks after cessation of CCl4. At the early (2-week) time point, we observed a marked reduction in the numbers of CD8+ T cells and CD19+ B cells in Aoc3–/– animals and significantly fewer CD4+ T cells, and we detected NK1.1+ and CD3+NK1.1+ NKT cells at 4 weeks, 8 weeks, and 4 weeks, respectively (Figure 5, A and B). Infiltration with macrophages and myeloid cells peaked later at 8 weeks and was significantly reduced in Aoc3–/– animals at both the 4-week and 8-week time points (Figure 5, A and C). We observed no significant differences in the numbers of cells within the WT and Aoc3–/– livers prior to the administration of CCl4 (Figure 5A, Base). Our finding that leukocytes were recruited during the first 2 weeks following CCl4 administration prior to the development of fibrosis (Figure 5B) is consistent with reports implicating them in fibrogenesis (40).
Figure 5. Reduced leukocyte infiltration of the liver after CCl4 treatment in Aoc3–/– mice.

The number of liver-infiltrating cells in WT or Aoc3–/– mice after CCl4 for 2, 4, or 8 weeks or following 8 weeks of CCl4 treatment followed by 4 weeks of resolution (Res) was quantified by flow cytometry. (A) There were significantly more T cells, B cells, NKT cells, and NK cells in WT animals (black bars) treated with CCl4 compared with those in Aoc3–/– mice (white bars). Data are expressed as the number of infiltrating cells per gram of fresh liver tissue (A) or fold change over baseline (Base) values (B and C: upper panels: WT; lower panels: Aoc3–/–) for 3 to 5 animals per group. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by 2-way ANOVA with Bonferroni’s correction.
VAP-1 promotes the development of severe steatohepatitis in a murine methionine choline–deficient diet.
Mice fed a methionine choline–deficient (MCD) diet develop liver injury with histology similar to that of human NASH including steatosis, inflammatory cell infiltration, and early fibrosis (41). To further investigate the role of VAP-1 in the development of liver disease in vivo, we administered an MCD diet for 6 weeks to 3 groups of 6 mice: (a) WT (C57BL/6), (b) Aoc3–/–, and (c) WT mice treated with weekly monoclonal anti–VAP-1 Ab started 1 week prior to the diet (WT plus BTT-1029). At sacrifice, we found that serum ALT levels were elevated in all mice, with no significant differences between the 3 groups, similar to that seen for the CCl4 model (Figure 6A). Bilirubin levels were raised in the Ab-treated group compared with those in the WT and KO animals, but we observed no differences in weight loss or blood glucose levels in these groups (Supplemental Figure 2). Low-power microscopy of H&E-stained liver sections demonstrated macrovesicular steatosis in association with inflammatory foci in WT mice (Figure 6B). We found significantly fewer inflammatory foci in the livers of mice treated with anti–VAP-1 Ab and in Aoc3–/– mice compared with those found in WT mice (Figure 6C). Steatosis determined by Oil red O staining was present in all mice, but was less extensive in anti–VAP-1–treated mice and particularly reduced in Aoc3–/– mice (Figure 6, D and E). This finding was unexpected, because SSAO activity modulates glucose uptake, whereas the MCD diet model does not induce insulin resistance.
Figure 6. WT mice receiving anti–VAP-1 Ab (BTT-1029) and Aoc3–/– animals are protected against MCD diet–induced steatohepatitis.

(A) Serum ALT levels of animals fed an MCD diet. (B and C) Inflammatory score based on sections stained with H&E, determined as: 0 = no foci per field, 1 = 1–2 foci, 2 = 3–4 foci, 3 = >4 foci. 5 fields of view per animal. Scale bars: 50 μm. (D) Accumulation of lipid droplets in murine tissue sections was assessed by Oil red O staining (red) with hematoxylin counterstaining (blue) and expressed as a percentage of surface area occupied by the lipid (E, 5 fields of view per animal). 6 animals per group. Scale bars: 50 μm. ****P < 0.0001 by 1-way ANOVA with Tukey’s post-hoc test (A and E) and ***P < 0.001 by Kruskal-Wallis test with Dunn’s multiple comparisons (C).
Aoc3–/– mice show a reduction in intrahepatic CD4+ and NKT cell populations in response to MCD diet–induced steatohepatitis.
Having demonstrated by histology that there was a reduction in inflammatory infiltrates in Aoc3–/– or anti–VAP-1–treated animals on an MCD diet, we characterized changes in the infiltrate in detail at 6 weeks by quantifying and phenotyping liver-infiltrating leukocytes (Figure 7). WT mice fed an MCD diet showed a 2.2-fold increase in intrahepatic CD3+ T cells compared with that seen in control mice that received normal chow (NC) (Figure 7A). The number of intrahepatic CD3+ T cells was significantly reduced in Aoc3–/– mice compared with that in WT animals due to a reduction in the numbers of intrahepatic CD4+ T cell and CD3+NK1.1+ NKT cell populations (Figure 7, B and E). In contrast to the CCl4 model, we found no significant differences in the expansion of intrahepatic CD8+ T cells (~4.5-fold increase vs. NC-fed mice) and CD19+ B cells (~1.7-fold increase vs. NC-fed mice) between WT and Aoc3–/– mice (Figure 7, C and F). MCD diet–fed WT mice demonstrated a 4.4-fold increase in CD11b+F4/80+ macrophages and a 6.8-fold increase in CD11b+GR1+ monocytes compared with NC-fed mice. MCD diet–fed Aoc3–/– mice had similar numbers of CD11b+F4/80+ macrophages but fewer intrahepatic CD11b+GR1+ cells compared with what was observed in WT animals fed an MCD diet (Figure 7, G and H).
Figure 7. Aoc3–/– mice have fewer intrahepatic CD4+ and NK1.1+ T cells and CD11b+GR1+ monocytes compared with WT animals fed an MCD diet for 6 weeks.

Numbers and phenotype of liver-infiltrating cells per gram of fresh liver tissue from WT (black) and Aoc3–/– (blue) mice following 6 weeks of an NC or MCD diet were evaluated by flow cytometry. All cells were gated from a live CD45+ population isolated from livers perfused with PBS to deplete peripheral blood. 3–5 animals per group. *P < 0.05 and ***P < 0.001 by Mann-Whitney U test.
Aoc3–/– and Ab-treated mice show reduced fibrogenesis in response to an MCD diet.
Parenchymal infiltration by leukocytes drives the development of fibrosis in NAFLD (5, 42), leading us to investigate whether Aoc3–/– and Ab-treated mice are protected against fibrosis following 6 weeks of an MCD diet. Both VAP-1 deficiency and prophylactic anti–VAP-1 Ab blockade resulted in significant and similar reductions in α-SMA expression (Figure 8, A and B) and collagen deposition (Figure 8, C and D) compared with that seen in WT mice when assessed by histological morphometric analysis and by qRT-PCR of total Acta2 and Col1a1 mRNA from whole mouse liver (Figure 8E). We observed a similar pattern in WT mice that received therapeutic doses of anti–VAP-1 Ab for the last 4 weeks of the diet: the recruitment of CD11b+F4/80+ macrophages was significantly reduced in WT mice receiving anti–VAP-1 Ab, and this was associated with an attenuated fibrotic response (Figure 9, A and B).
Figure 8. Administration of anti–VAP-1 Ab (BTT-1029) or deficiency of Aoc3 limits fibrosis in animals fed a 6-week MCD diet.

Immunohistochemical staining for α-SMA (A and B) and Van Gieson’s collagen staining (C and D) were expressed as a percentage of the surface area of the field of view, calculated using ImageJ software (B, α-SMA; D, Van Gieson’s/collagen). 3–5 animals per group with 3 to 5 fields of view per animal. Scale bars: 50 μm. (E) Changes in Acta2 and Col1a1 mRNA expression in the liver tissue of these animals were quantified by qRT-PCR (4–5 animals per group). **P < 0.01, ***P < 0.001, and ****P < 0.0001 by 1-way ANOVA with Tukey’s post-hoc test.
Figure 9. Therapeutic administration of anti–VAP-1 Ab BTT-1029 reduces hepatic leukocyte recruitment and fibrosis in WT mice fed an MCD diet for 6 weeks.

(A) The proportion of leukocyte subsets relative to the live CD45+ cell population was assessed by flow cytometry. DX-5 is a pan-NK marker. (B) Changes in the expression of Acta2 and Col1a1 in the liver tissue of these animals were quantified by qRT-PCR. 5–6 animals per group. **P < 0.01 by Mann-Whitney U test.
Although the MCD diet model of steatohepatitis recapitulates many of the histological features of human NASH, it is an acute model resulting in weight loss and is not associated with systemic insulin resistance and as such does not mirror the physiological changes that occur during metabolic syndrome–induced human NAFLD. Thus, we felt it important to investigate the effects of VAP-1 deficiency in chronic models of steatohepatitis that are associated with obesity and insulin resistance.
C57BL/6 mice fed a high-fat diet (HFD) for 18 weeks develop obesity, glucose intolerance, hyperinsulinemia, hyperleptinemia, and hypercholesterolemia (43). The main hepatic pathology seen following an HFD is macrovesicular steatosis, with the development of steatohepatitis and modest fibrosis after a prolonged HFD (44). We observed that WT mice fed an HFD for 18 weeks developed severe macrovesicular steatosis, inflammatory foci, and thin, fibrous septa, all of which were greatly reduced in Aoc3–/– animals, together with reduced Acta2 and Col1a1 expression (Supplemental Figures 3–6). Although the HFD model more closely resembles the pathophysiology of human NASH, the extent of steatohepatitis is not as severe as that seen with the MCD diet model (45). Tetri et al. described a murine model of steatohepatitis that develops in response to a diet high in trans fats and fructose and interventions to promote sedentary behavior (46). This Western lifestyle model (WLM) is characterized by obesity with impaired glucose tolerance and increased plasma insulin, resistin, and leptin comparable to the pathological changes seen in NAFLD and at 16 weeks, animals develop severe steatosis with significant necroinflammatory activity. We exposed WT and Aoc3–/– mice to this regimen and studied the pathological changes at 6, 9, and 12 months during evolution of NAFLD and compared these outcomes with mice fed NC and standard drinking water and allowed unrestricted activity.
Serum ALT levels were elevated in mice exposed to the WLM by 6 months, but we observed no significant differences in ALT levels between WT or Aoc3–/– mice at either 6 or 9 months (Figure 10A). Aoc3–/– mice gained more weight during the first month of the diet compared with that seen in the WT group (Supplemental Figure 7).
Figure 10. Liver tissue from Aoc3–/– animals exhibits reduced steatosis and fibrosis when fed a WLM diet.

(A) Serum ALT levels of WT or Aoc3–/– animals fed either a NC or WLM diet for 6 or 9 months and Aoc3 expression in WT animals at 9 months. 3–5 animals per group. (B) Collagen deposition in the WLM animals was visualized by Van Gieson’s staining at 9 and 12 months, and the surface area covered by the stain at the 12-month time point was evaluated by ImageJ software (C). 5–6 animals per group; 5 fields per animal. Scale bars: 50 μm. (D) Changes in Acta2 and Col1a1 expression in the liver tissue of these animals at 12 months were quantified by qRT-PCR. 4–6 animals per group. (E and F) The extent of steatosis in WT and Aoc3–/– animals fed a WLM diet at 9 months was evaluated by Oil red O staining and ImageJ software. 4 animal per group; 3–5 fields per animal. Scale bars: 50 μm. ***P < 0.001 and ****P < 0.0001 by Mann-Whitney U test.
Aoc3 expression was increased in WT mice following 9 months of the diet when compared with that detected in animals on a NC diet (Figure 10A), and the extent of fibrosis at both the 9- and 12-month time points was reduced in the Aoc3–/– animals, as evaluated by collagen deposition levels (Figure 10, B and C) and Acta2 and Col1a1 expression (Figure 10D).
Although steatosis was present in all the WLM diet–fed animals, we found that the extent of steatosis was reduced in Aoc3–/– mice compared with that in WT mice at the 9-month time point (Figure 10, E and F). This was similar to what we observed with the MCD diet–fed mice (compare Figure 6).
We quantified immune cells isolated from the livers of WT and Aoc3–/– mice fed NC or those on the WLM diet at 6 and 9 months. We observed only a modest increase in CD8+ T cells, NK1.1+ cells, CD11b+F4/80+ cells, and Cd11b+GR1+ myeloid cells at 6 months, and there were no significant differences between WT and Aoc3–/– mice in the numbers of any cell type (Figure 11). However, by 9 months, the WLM mice developed an inflammatory infiltrate comparable in profile and absolute cell numbers to what we observed in mice after 6 weeks on an MCD diet (compare Figures 7 and 11). Moreover, VAP-1 deficiency was associated with reduced numbers of intrahepatic CD4+ and NK1.1+ T cell populations at 9 months. Thus, VAP-1 promotes a cellular infiltrate that precedes the development of fibrosis.
Figure 11. Intrahepatic leukocyte populations are increased in animals fed a WLM diet for 9 months but not 6 months, with fewer CD4+ and NK1.1+ T cells present in Aoc3–/– mice fed a WLM diet for 9 months.

The numbers and phenotype of liver-infiltrating cells per gram of fresh liver tissue from WT and Aoc3–/– mice following 6 and 9 months of a NC or WLM diet were evaluated by flow cytometry. All cells were gated from a live CD45+ cell population isolated from livers perfused with PBS to deplete peripheral blood. 2–4 animals per group. For clarity, only the Mann-Whitney U tests between WT and Aoc3–/– at 9 months are shown; *P < 0.05.
The amine oxidase activity of VAP-1 promotes progression of steatohepatitis.
Because we had demonstrated that VAP-1 SSAO activity is involved in lymphocyte chemotaxis and transendothelial migration (12), we investigated the requirement for SSAO activity in the development of steatohepatitis. We generated mice in which WT Aoc3 was deleted and replaced by an Aoc3 point–mutation knock-in (SSAO KO), in which the change of a single amino acid in the active site from Tyr to Phe renders VAP-1 catalytically inactive. These mice show a normal tissue distribution of VAP-1 with undetectable SSAO enzyme activity (Supplemental Figure 8).
Since the inflammatory changes in the MCD diet model were similar to those seen in the more relevant WLM, we used the shorter MCD diet model to study the development of steatohepatitis in SSAO-KO mice. Following 4.5 weeks of an MCD diet, the presence of catalytically inactive VAP-1 resulted in the same reductions in hepatic CD4+ and NK1.1+ T cells as those seen in the Aoc3–/– mice (Figure 12), confirming that the amine oxidase activity of VAP-1 is involved in the generation of hepatitis in vivo. While mice with catalytically inactive VAP-1 had levels of injury similar to those seen in the WT mice at 4.5 weeks (Supplemental Figures 9–12), catalytic inactivation of VAP-1 resulted in a significant loss of Acta2 expression at 6 weeks (Figure 12B), suggesting that the reduced infiltrate results in an attenuated fibrotic response.
Figure 12. Mice harboring an enzyme-dead mutant of Aoc3 (SSAO KO) show reduced hepatic infiltration of leukocytes and reduced fibrosis compared with WT animals fed an MCD diet.

(A) The numbers and phenotype of liver-infiltrating cells per gram of fresh liver tissue from WT and SSAO-KO mice following 4.5 weeks of an MCD diet were evaluated by flow cytometry. All cells were gated from a live CD45+ cell population isolated from livers perfused with PBS to deplete peripheral blood. 4–5 animals per group. (B) Changes in the expression of Acta2 and Col1a1 in the liver tissue of these animals were quantified at 6 weeks by qRT-PCR. 5–6 animals per group. *P < 0.05 and **P < 0.01 by Mann-Whitney U test.
VAP-1 enhances wound healing by hepatic stromal cells in vitro.
The presence of VAP-1 protein within fibrotic septa of chronically diseased human liver tissue suggested that it might contribute to remodeling of the scar via direct effects on the hepatic stroma. Short-term (24-hour) treatment with sVAP-1 at 500 ng/ml had no effect on aHSC activation, proliferation, or apoptosis in vitro based on detachment assays, cell proliferation measurements, and qRT-PCR for the prosurvival factor BCL2 and the associated proapoptotic factor BAX (Supplemental Figure 13). However, we observed increased cell spreading in impedance-based assays upon treatment with either sVAP-1 or a recombinant form of the protein produced in CHO cells (rVAP-1) (Figure 13A). The potent mitogen PDGF-BB had no effect on cell spreading over the same time course. This prompted us to investigate the effect of de novo synthesized VAP-1 on stromal cell function. Overexpression of an N-terminal GFP-tagged form of VAP-1 (GFP-WT-VAP-1, described in ref. 47) in LX2 cells accelerated wound healing in a scratch wound assay when compared with that in the control transfectant plasmid Lifeact-GFP (pLifeact-GFP), which is known to have little-to-no effect on cell motility (ref. 48 and Figure 13B). This effect was not dependent on enzyme activity, as expression of the enzyme-dead mutant of VAP-1 (GFP-Y471F-VAP-1) had a similar effect (Figure 13B). In complementary experiments, siRNA-mediated knockdown of AOC3 in LX2 cells retarded wound closure when compared with that seen in scrambled controls (Figure 13C).
Figure 13. VAP-1 induces spreading of aHSCs and promotes wound healing in LX2 cells independently of enzyme activity.

(A) Treatment of aHSCs with soluble rVAP-1 or highly purified serum-derived VAP-1 (sVAP-1) induced cell spreading as assessed by xCELLigence impedance assay. Responses to sVAP-1, rVAP-1 (both 500 ng/ml), and PDGF-BB (10 ng/ml) were measured (n = 2, each measured in quadruplicate). (B) Transfection of LX2 with plasmids encoding WT or enzyme-dead (Y471F) VAP-1 enhanced wound healing when compared with that observed in control transfectants (Lifeact). Representative image from 1 experiment is shown, with data taken from 3 independent experiments. (C) Conversely, knockdown of AOC3 gene expression with siRNA retarded wound closure when compared with that detected in scrambled control siRNA sequences. Representative image from 1 experiment is shown, with data taken from 3 independent experiments. *P < 0.05, and **P < 0.01 by 1-way ANOVA with Tukey’s post-hoc test (A) and 2-way ANOVA with Bonferroni’s correction (B and C) .
Expression of VAP-1 regulates the transcription of profibrotic factors.
To investigate the mechanism by which VAP-1 drives the fibrotic response, we measured the expression levels of genes associated with fibrosis in transfected LX2 cells by qRT-PCR. Overexpression of AOC3 in cells transfected with GFP-tagged VAP-1 plasmid constructs was demonstrated by a 2,000-fold increase in message when compared with that in control cells transfected with Lifeact-GFP (Figure 14A). We found that expression of PDGFRB, which is a characteristic of HSC activation, was increased in GFP-WT-VAP-1–transfected cells, and this was dependent on the enzymatic activity of VAP-1, as the enzyme-dead mutant (GFP-Y471F-VAP-1) did not elicit a similar response, suggesting that expression of catalytically active VAP-1 provides a profibrogenic stimulus. This hypothesis was supported by the observation that GFP-WT-VAP-1 induced upregulation of both COL1A1 and LOXL2 (Figure 14A), the latter being a related amine oxidase that has been implicated in the development of fibrosis in a number of disease models (38). We confirmed the close association between VAP-1 and the expression of profibrotic genes in siRNA-mediated knockdown experiments, in which reduced AOC3 expression in LX2 cells downregulated the expression of both COL1A1 and LOXL2 (Figure 14B).
Figure 14. VAP-1 is associated with the expression of fibrosis-associated genes in transfected LX2 cells.

(A) Overexpression of WT but not enzyme-dead (Y471F) VAP-1 in LX2 cells led to a significant increase in COL1A1, PDGFRB, and LOXL2 expression at 72 hours when compared with that in control transfectants (Lifeact). (B) Knockdown of AOC3 in LX2 cells significantly reduced the expression of COL1A1 and LOXL2 at 72 hours following nucleofection. Data are representative of 2 experiments, each conducted in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by 1-way ANOVA with Tukey’s post-hoc test (A) and unpaired Student’s t test (B).
Discussion
In NAFLD, the progression of steatosis to cirrhosis is driven by steatohepatitis, which develops as a consequence of activation of the inflammasome in response to liver injury (49–52). Steatohepatitis is characterized by infiltration of the hepatic parenchyma with T cells, macrophages, and neutrophils (53), and previous findings that VAP-1 plays an important role in the recruitment of lymphocytes and monocytes to the liver prompted us to study its role in NAFLD (12, 15, 39). We have reported that VAP-1 is shed into the circulation as a soluble enzyme that accounts for most of the circulating monoamine oxidase activity in human blood and that levels are increased in chronic inflammatory liver diseases, but not in acute liver injury (23, 26). We now report elevated sVAP-1 levels in a well-defined cohort of NAFLD patients that is associated with increased VAP-1 expression in the liver. Because sVAP-1 levels are elevated in patients with obesity and type 2 diabetes, we compared these levels in NAFLD with those in a group of patients matched for BMI and metabolic phenotype who had no evidence of liver involvement. We found that sVAP-1 levels were elevated in the metabolic syndrome cohort compared with those in healthy controls, but were significantly lower compared with levels in NAFLD patients. Circulating sVAP-1 levels also correlated with histological evidence of steatohepatitis and fibrosis on liver biopsy, and multivariate analysis showed that VAP-1 was independently associated with fibrosis stage. Reliable methods to assess steatohepatitis, fibrosis, and the risk of progression to cirrhosis in NAFLD are limited. The current findings suggest that sVAP-1 levels, particularly if combined with other biomarkers of fibrosis, could have clinical utility as a noninvasive biomarker to differentiate between simple steatosis and NASH.
The association between sVAP-1 and fibrosis led us to investigate the expression of VAP-1 protein in patients with other types of CLD. Detailed studies revealed that VAP-1 was expressed by the hepatic stroma as well as by endothelial cells, and we observed no detectable staining of hepatocytes or cholangiocytes. Stellate cells are closely associated with sinusoidal endothelium and act as pericytes, contributing to the regulation of endothelial differentiation and function (54). In response to injury, quiescent HSCs proliferate and transdifferentiate into activated myofibroblasts, which deposit matrix during hepatic fibrogenesis. We found that both in vivo–activated aLMFs and in vitro–activated HSCs expressed and secreted enzymatically active VAP-1, which was a potent leukocyte chemotactic factor at levels comparable to those found in vivo. Thus, VAP-1 can modulate leukocyte migration in both its transmembranous and soluble forms. This property was dependent in part on SSAO activity associated with the generation of H2O2 and, to our knowledge, is the first report of SSAO directly stimulating leukocyte motility. It is likely that VAP-1 production by endothelial cells and myofibroblasts generates a local gradient of sVAP-1/H2O2, which stimulates leukocyte migration and recruitment. Our finding that the chemotactic effect of sVAP-1 was reduced by treatment with pertussis toxin is consistent with a contribution from autocrine activation of chemotactic factors that signal through GPCRs, as has been described for neutrophils migrating in response to H2O2 (55). Autocrine chemoattractant production and direct modification of redox-sensitive intracellular receptors has been shown in monocyte migration in response to lysyl oxidase, another enzyme that generates H2O2 (56, 57). This model is consistent with a study in zebrafish, in which a tissue gradient of H2O2 produced by an unidentified amine oxidase recruits leukocytes to the scar during wound healing (35). Thus VAP-1–generated H2O2 could operate in a similar way, resulting in a local tissue gradient that promotes and sustains the recruitment of leukocytes to the hepatic parenchyma. We suggest that endothelial VAP-1 promotes the recruitment of effector cells to the inflamed liver and that VAP-1 produced by HSCs positions them within the hepatic scar. These findings are consistent with, and provide a potential mechanism to explain, previous observations that VAP-1 expressed by specialized splenic fibroblasts guides lymphocyte migration within the spleen (58).
In order to test the hypothesis that VAP-1 links inflammation and fibrosis in response to liver injury, we compared the response to CCl4-induced liver injury in WT mice, Aoc3–/– animals, and WT mice treated with a neutralizing anti–VAP-1 Ab. In response to CCl4, WT mice developed septal fibrosis and increased leukocyte infiltration. The absence of VAP-1 in Aoc3–/– mice or its blockade by Ab in WT animals markedly reduced fibrosis and accelerated recovery in response to CCl4, implicating VAP-1 in fibrogenesis and fibrosis resolution. In the absence of VAP-1, the absolute numbers of both lymphoid and myeloid cells were substantially reduced, consistent with a model in which VAP-1 promotes the recruitment of an infiltrate that drives the development of fibrosis.
Several properties of VAP-1 SSAO suggest that it could have roles in NAFLD progression in addition to the ability to recruit inflammatory cells. These include the local generation of ROS by SSAO activity and the ability of SSAO to sensitize insulin signaling. Our current data generated in 3 different models of NAFLD suggest that VAP-1 contributes to the establishment of steatohepatitis and progression to fibrosis. This was evidenced by reduced inflammation, steatosis, and fibrosis in the absence of VAP-1 in an HFD, an MCD diet, and in the WLM. Using the WLM of NAFLD, in which mice given a high-fructose and trans-fat diet develop NASH over a 9- to 12-month period, we were able to study the kinetics of hepatitis and fibrosis development in a model that recapitulates human NAFLD. Both WT and Aoc3–/– mice developed steatosis and liver injury, as measured by ALT levels after 6 months, before the development of steatohepatitis that developed between 6 and 9 months, and they developed fibrosis that was apparent at 12 months. The severity of steatosis was reduced, though not abolished, in Aoc3–/– animals (similar to our findings with MCD diet– and HFD-fed mice), suggesting that VAP-1 promotes steatosis as well as steatohepatitis. We expected this would be an SSAO-dependent effect of VAP-1, but surprisingly, steatosis was reduced by Ab treatment, which does not inhibit SSAO activity toward small conventional amine substrates, and was unaffected in mice expressing enzyme-dead VAP-1. The reasons for this are currently unclear. VAP-1 substrates have been shown to activate glucose transport (59), and feeding mice the SSAO inhibitor semicarbazide limits weight gain and inhibits fat deposition in adipose tissue (60). However, Bour et al. demonstrated that Aoc3–/– animals have reduced leukocyte infiltration into white adipose tissue associated with increased fat deposition, which could not be rescued by expression of VAP-1 from the human adipocyte–specific aP2 promoter (61). These data suggest that the role of VAP-1 in the development of hepatic steatosis is complex and that the effects of adipocyte-specific VAP-1 on the release of adipokines must also be taken into consideration.
Steatohepatitis in response to the WLM was characterized by a mixed inflammatory cell infiltrate that was markedly reduced in Aoc3–/– animals. The composition of the infiltrate showed a specific reduction in CD4 and NKT cells in the Aoc3–/– animals, and these mice failed to develop significant fibrosis at 12 months. The changes in infiltrate were very similar to those seen in the MCD diet–induced model of steatohepatitis, in which we also observed a reduction in CD4 and NKT cell infiltration and a subsequent reduction in fibrosis in Aoc3–/– animals. The shorter time frame of the MCD diet model allowed us to show that we could achieve similar effects in WT animals using an anti–VAP-1–blocking Ab with both prophylactic and therapeutic dosing strategies.
As VAP-1 has both enzyme and adhesive properties, we determined whether the effect of VAP-1 on leukocyte recruitment was dependent on SSAO activity. Because there are limited highly specific enzyme inhibitors for murine SSAO, we generated a mouse in which native Aoc3 was replaced by a mutated Aoc3, in which a single conservative (Tyr to Phe) amino acid change renders the molecule enzymatically inactive. We showed that the mutant protein was expressed with the same tissue distribution as that of WT VAP-1, consistent with a recent report (62), and then subjected the mice to an MCD diet. Analysis of the infiltrate and expression of fibrotic genes showed a pattern of changes similar to that observed in Aoc3–/– animals, suggesting that SSAO activity contributes to VAP-1–mediated steatohepatitis and fibrosis in NAFLD. This is consistent with the finding that caffeine, an inhibitor of amine oxidase activity (63), has been associated with a reduction in the risk of fibrosis in NASH patients (64). However, the fact that we were able to effectively prevent fibrosis using a VAP-1–blocking Ab that does not bind to the enzyme active site or inhibit deamination of small soluble substrates in vitro suggests a more complex mechanism. It is possible that the Ab prevents access of endogenous, cell-associated macromolecular substrates to the enzyme in vivo, although until the endogenous ligands of murine VAP-1 are defined, it is difficult to test this hypothesis. An alternative explanation is that recruitment requires sequential interactions involving enzyme-dependent and -independent steps; if either of these is blocked, recruitment fails. A similar mechanism was proposed for the recruitment of granulocytes across endothelial monolayers (65). Both hypotheses are consistent with our murine data and suggest that both VAP-1–blocking Abs and enzyme inhibitors have therapeutic potential.
We used wound-healing and transfection studies to examine the direct effects of VAP-1 expression on the stromal cell population. VAP-1 promoted the wound-healing response, induced spreading of aHSCs, and enhanced expression of profibrotic genes (COL1A1, LOXL2), indicating that VAP-1 has direct effects on stellate cells that could promote scarring. There is a precedent for the involvement of VAP-1 in matrix remodeling, because its overexpression in arterial smooth muscle cells increases elastin deposition in the arterial wall (66). The contribution of SSAO activity to these functions remains unclear, however, since accelerated wound healing was independent of enzyme activity, whereas expression of profibrotic genes was unaffected by the transfection of LX2 cells with a catalytically inactive form of the protein (Figures 13 and 14). A similar effect has been observed for LOXL2, in which enzyme-dead forms of the protein retained function (67, 68).
We suggest that VAP-1 plays a complex role in the pathogenesis of CLD. Its ability to promote the recruitment of leukocytes to the liver in response to initial liver injury and its contribution to the development of fibrosis suggest that targeting VAP-1 therapeutically, through Ab blockade or enzyme inhibitors, could prevent the progression of liver disease.
Methods
Study populations.
Serum from NAFLD patients (n = 133) was obtained at the time of the patient’s admission for liver biopsy. All patients included in the study had histologically confirmed NAFLD, with biopsies graded and staged according to the Kleiner classification; extensive clinical and biochemical characteristics were recorded at the time of biopsy (Table 1). A control cohort matched for age, BMI, and metabolic phenotype was established and fasting serum samples and anthropometric data collected (n = 74, Table 1). Exclusion criteria for all patients were: age of less than 18 years, consumption of greater than 20 g ethanol per day if female and greater than 30 g per day if male, infection with viral hepatitis or HIV, treatment with corticosteroids or immunosuppressive therapy within the past 12 weeks, and treatment with monoamine oxidase inhibitors. Further exclusion criteria for the control cohort were biochemical or radiological evidence of liver disease. Serum was stored at –80°C until later use.
Human tissue.
Human tissue and blood samples were collected from patients admitted to the University Hospitals Birmingham NHS Foundation Trust and the Newcastle-Upon-Tyne Hospitals NHS Foundation Trust. Liver tissue from explanted livers was taken from normal organ donor liver tissue that was surplus for surgical requirements or from uninvolved liver removed at hepatic resection for secondary liver tumors; diseased tissue was obtained from patients undergoing liver transplantation for CLD.
Isolation of human HSCs and aLMFs and maintenance of the LX2 cell line.
HSCs were isolated from nonfibrotic human liver tissue, and aLMFs were purified from fibrotic tissue as described previously (69). Cell viability was confirmed using trypan blue exclusion, and autofluorescence of HSCs suggested a greater than 90% purity. Cells were plated on plastic in DMEM containing 16% FCS (Invitrogen) and used within 4 passages. HSCs that adhered to plastic after isolation were termed aHSCs to differentiate them from their quiescent phenotype.
The HSC line LX2 (a gift of S. Friedman, Mount Sinai School of Medicine, New York, New York, USA) was maintained in DMEM containing 2% FCS (Invitrogen).
Soluble VAP-1 measurements.
Soluble VAP-1 levels in human serum were measured by time-resolved immunofluorometric assay at Biotie Therapies Corp. Briefly, a biotin-conjugated monoclonal anti–VAP-1 Ab (clone TK8-14; eBioscience) was immobilized onto a streptavidin-coated microtiter plate, to which the serum samples were added. sVAP-1 that had bound to the plate was then detected with a europium-conjugated anti–VAP-1 Ab (clone TK8-18; eBioscience). VAP-1 levels were measured by time-resolved fluorescence at 615 nm (VICTOR2 Multilabel Counter; PerkinElmer). Quantification of VAP-1 concentration in serum was based on a reference sample of highly purified human serum VAP-1 (Biovian Ltd.). The calibration range of the method was from 1.5 to 1,500 ng/ml. The linearity (R2) of the calibration (standard) curves was 0.997–1.000.
Cell migration assay.
Chemotaxis assays were performed as previously described (70). Briefly, 30 μl aHSC-conditioned medium, highly purified sVAP-1 (Biotie Therapies Corp.), or recombinant CXCL12 (10 ng/ml; Peprotech) was placed in the lower wells of a custom-made 48-well chemotaxis chamber (Neuro Probe) separated from an upper chamber containing cell suspension (peripheral blood leukocytes, 1.5 × 106 cells/ml) by a polycarbonate membrane (5-μm pores, Nucleopore Filtration; Neuro Probe). Negative controls containing media alone or media containing inhibitor alone were included in each assay. The assembled chamber was incubated for 2 hours at 37°C in a humidified CO2–enriched atmosphere. Following incubation, the filter was removed from the chamber and nonmigrated cells on the upper surface removed by wiping before being air dried, fixed in methanol, and stained with hematoxylin (Diff-Quik; Sigma-Aldrich). The number of stained cells in 4 high-power fields (×40) for each of 6 wells per sample was counted, giving 24 fields of view per condition, and migration was calculated as the median of these 24 counts expressed as the number of migrating cells per high-power field. To assess the contribution of GPCR, leukocytes were pretreated with 100 ng/ml pertussis toxin (PTx; Sigma-Aldrich) for 30 minutes at 37°C, washed, and resuspended in RPMI containing 0.1% BSA prior to the assay. The viability of the leukocytes was not affected by this treatment. In some assays, 100 U/ml catalase (Sigma-Aldrich) was included in the test wells to assess the contribution of H2O2; in others, the enzyme activity of VAP-1 was inhibited by preincubation with 2.5 mM BEA (Sigma-Aldrich) for 30 minutes prior to the assay.
Cellular proliferation assays.
A CyQUANT NF Cell Proliferation Assay Kit (Invitrogen) was used to measure the total nucleic acid content of aHSCs after 8 days in culture in the absence and presence of 10 ng/ml PDGF-BB (Peprotech) or 500 ng/ml highly purified serum–derived sVAP-1 (Biotie Therapies Corp.) according to the manufacturer’s instructions.
Cellular attachment assay.
Activated HSCs were seeded onto plates and allowed to adhere for 24 hours in DMEM containing 16% FCS in the presence of 1.5 μM gliotoxin (Sigma-Aldrich) or sVAP-1 (500 ng/ml) at 37°C and 5% CO2. Adherent cells were counted in 12 high-power fields and normalized to the cells receiving media alone. The data represent 3 separate aHSC isolates measured in triplicate.
Cell-spreading assay and xCELLigence impedance measurements.
The xCELLigence system was used according to the manufacturer’s instructions (Roche Applied Science) to measure the impedance every minute for 4 hours of a monolayer of 10,000 aHSCs per well in a pre-equilibrated E-Plate using integrated software. Data were normalized at the 40-minute time point to produce a dimensionless normalized cell index (NCI), and cell spreading was calculated using real-time cell analyzer (RTCA) software (Roche Applied Science). Two independent isolates of aHSCs were studied, with each treatment measured in quadruplicate.
Murine models of liver injury.
All mice were maintained and housed under conventional conditions in the Biomedical Services Unit at the University of Birmingham or at the University of Turku. Up to 5 mice were housed per cage under alternating 12-hour light/12-hour dark cycles at 23°C. WT C57BL/6 (WT), Aoc3–/–, and Aoc3 point–mutation knock-in mice (SSAO KO, harboring a mutation in the sequence for an active site tyrosine to render the protein catalytically inactive) aged 6 to 8 weeks were used in the studies. C57BL/6 mice were obtained from Charles River Laboratories. Aoc3–/– mice were re-derived from the previously described Aoc3–/– line (37) by blastocyst transfer at Taconic, the contract breeder. Aoc3 point–mutation knock-in mice were backcrossed onto a C57BL/6 genetic background by genOway and supplied by the contract breeder Charles River Laboratories. Mutant allele mice were identified by PCR screening of purified genomic DNA with specific primers.
CCl4 model.
Female mice were injected twice weekly for 2, 4, 6, or 8 weeks with either CCl4 (1.0 ml/kg CCl4 diluted 1:4 in MO; Sigma-Aldrich) or MO alone. Animals were sacrificed 96 hours after the final dose of CCl4. In parallel experiments, animals received vehicle or CCl4 for 8 weeks, as above, at which point vehicle or CCl4 treatment was stopped, and the animals were sacrificed following 4 weeks of resolution.
Animal diets.
MCD diet: Male mice were fed an MCD diet (MP Biomedicals) ad libitum for 4.5 to 6 weeks. HFD: Male mice were fed an HFD diet that provided 55% of calories from fat (Special Diets Services) ad libitum for 18 weeks. WLM diet: Male mice were fed a diet with 45% of calories obtained from fat and 30% of the fat in the form of partially hydrogenated vegetable oil (Custom Research Diet TD.06303; Harlan Laboratories) in conjunction with fructose-supplemented drinking water (55% fructose, 45% glucose by weight at a concentration of 42 g/l) ad libitum for 6, 9, or 12 months. Control animals received NC and nonsupplemented drinking water.
Animals treated with the VAP-1 function–blocking Ab (mouse anti-mouse BTT-1029, 9 mg/kg; Biotie Therapies Corp.) received a) prophylactic weekly i.v. injections 1 week prior to and weekly throughout the experiment, or b) therapeutic twice-weekly i.v. injections for the final 4 weeks of the 6-week experiment. BTT1029 is a mouse IgG2a mAb generated in Aoc3–/– animals by immunizing with human recombinant VAP-1. The Ab was selected from several hybridomas recognizing recombinant human VAP-1. A secondary screening was made to select antibodies that bound to recombinant mouse VAP-1 (Biotie Therapies Corp.).
Blood samples were withdrawn by cardiac puncture during isofluorane anesthesia, after which the mice were sacrificed by cervical dislocation. Livers were dissected and processed for paraffin embedding, cryosectioning, and RNA extraction (stored in RNAlater; QIAGEN). Bilirubin and ALT were measured at the clinical biochemistry laboratory of the Women’s Hospital Birmingham (Birmingham, United Kingdom), and serum glucose concentrations were obtained using Accu-Chek (Roche).
qRT-PCR.
Total cellular RNA was isolated using an RNeasy kit (QIAGEN). RNA concentration and purity were measured using a Nanophotometer (Geneflow Ltd.) or a Nanodrop 1000 (Thermo Scientific), and 1 μg of total RNA was transcribed to cDNA using the First Strand Synthesis Kit (Roche) or SuperScript III System (Invitrogen). Real-time PCR was carried out on each sample in triplicate using a Roche Lightcycler 480 machine and Probes Master Mix (200 nM probe, 500 nM primers; Roche Diagnostics) or TaqMan Assay Mix (Invitrogen), as detailed in Supplemental Table 3. Cycling conditions were as follows: 95°C for 10 seconds, 55–60°C (target dependent) for 50 seconds, and 72°C for 1 second. For relative quantification analysis, data were normalized to the appropriate housekeeping gene using “E-analysis” (Roche Diagnostics) and the device software (version 1.5.0.39). Relative expression levels are shown as fold change compared with the appropriate unstimulated control, tissue, or animal. For absolute quantification studies, the number of copies of AOC3 mRNA in matched amounts of starting total RNA from each cell type was determined from a calibration curve of known dilutions of a plasmid encoding AOC3.
Histopathology and immunohistochemistry.
For morphological analysis, murine liver specimens were fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 4-μm sections. Slides were deparaffinized according to standard procedures and stained with freshly made H&E. To evaluate fibrosis, the sections were stained with Van Gieson’s solution (Sigma-Aldrich) or Picosirius Red (Sigma-Aldrich) staining according to standard protocols. For semiquantitative determination of hepatic infiltrate, 5 random fields of H&E-stained sections at 20× objective magnification were assessed per animal. Inflammatory scores were determined as follows: 0 = no foci per field; 1 = 1–2 foci; 2 = 3–4 foci; 3 = >4 foci. For VAP-1 staining in murine tissue, acetone-fixed sections were sequentially incubated with an anti–VAP-1 mAb (TK7-88; ref. 71) and HRP-conjugated anti–rat Ig (Dako). Immunoreactivity was visualized using DAB as a chromogen. The sections were counterstained with hematoxylin, and micrographs were acquired using an Olympus BX60 microscope and the Cell D program.
Oil red O staining of freshly cut sections of murine liver tissue (7-μm thickness) was carried out to evaluate the extent of steatosis. Sections were incubated in 60% propan-2-ol for 5 minutes, followed by a 15-minute incubation with Oil red O reagent (0.3% Oil red O in propan-2-ol/water; Sigma-Aldrich). Next, the samples were washed with propan-2-ol, then water, and mounted in Immunomount (Fisher Scientific). A Mouse-on-Mouse kit (Vector Laboratories) was used in conjunction with an anti–α-SMA mAb (clone 1A4, murine IgG2a, 5 μg/ml; Sigma-Aldrich) to detect α-SMA in 7-μm-thick tissue slices. The staining was visualized with a VECTASTAIN Elite ABC kit and NovaRed substrate (Vector Laboratories). Determination of the surface area covered by the stain was performed by analysis of 3 to 5 random fields per animal (depending on the size of the section) at ×20 magnification and threshold analysis with ImageJ software version 1.42q (NIH).
Human tissue samples (10 × 10 × 5 mm) were taken from livers exhibiting a normal phenotype, mild steatosis, moderate steatosis, severe steatosis, or cirrhosis (4 individuals per group, based on histology of hematoxylin-stained tissue sections as assessed by a trained pathologist [SGH]). These samples were fixed in formalin and processed through to paraffin wax. Tissues were deparaffinized and rehydrated to water and, after a low temperature retrieval technique (ALTER), were immunostained on a Dako Autostainer using a Prestige rabbit anti–human VAP-1 polyclonal Ab (HPA000980, 1:200; Sigma-Aldrich), a Vector ImmPRESS secondary reagent kit, and NovaRED as a chromogen (both from Vector Laboratories). Immunohistochemistry on frozen sections taken from patients who underwent transplantation for end-stage CLD (alcoholic liver disease, primary biliary cirrhosis, primary sclerosing cholangitis) was performed in a similar manner using TK8-14 as the anti–VAP-1 Ab (BMS1033; eBioscience).
Immunofluorescence.
Fresh-frozen, acetone-fixed tissue or 4% paraformaldehyde-fixed cells were incubated for 1 hour at room temperature with PBS containing 0.1% Triton X-100 and 10% goat serum (serum-PBST). Following overnight incubation with primary Abs against the antigens α-SMA (clone 1A4, 5 μg/ml; Sigma-Aldrich); CD31 (clone 9G11, 20 μg/ml; R&D Systems); VAP-1 (clone TK8-14, 2.5 μg/ml; Biotie Therapies Corp.); VAP-1 (clone 174-5, 2 μg/ml; Novus Biologics); EpCAM (clone HEA125, 100 μg/ml; Progen); CK18 (clone DC10, 1 μg/ml; Dako); CD90 (clone 5E10, 1 μg/ml; eBioscience); CD3 (clone UCHT1, 1 μg/ml; AbDSerotec); and CD68 (clone KP1, 1 μg/ml; Abcam), samples were washed 3 times in PBST and incubated with Alexa Fluor–conjugated secondary Abs (1:500; Invitrogen) for 1 hour at room temperature. Nuclei were visualized using DAPI nucleic acid stain (Invitrogen). Fluorescence images were acquired using a Zeiss LSM 510 confocal fluorescence microscope (ZEISS).
Quantification of liver-infiltrating immune cells.
Mouse livers were harvested and perfused with PBS via cardiac puncture following disconnection of the hepatic portal vein. The organs were then weighed, dissociated in a gentleMACS C Tube (Miltenyi Biotec), and the resulting immune cells were then purified and analyzed by flow cytometry. Inflammatory cells were gated as a CD45+ cell population (anti–CD45-PerCP-Cy5.5, clone 30-F11; BD Biosciences), and nonviable cells were excluded using a LIVE/DEAD fixable stain (Invitrogen). Lymphocytes were characterized based on staining using a cocktail of anti–CD3-Pacific blue (clone 500A2); anti–CD4-PE (clone RM4-5); anti–CD8a-APC (clone 53-6.7); anti–CD19-APC-Cy7 or anti–CD19-BV510 (both clone 1D3); and anti–NK1.1-FITC (clone PK136) or DX5-FITC (clone DX5) Abs (all from BD Biosciences), while the monocyte subsets were identified by staining with anti–CD11b-PE (clone M1/70; BD Biosciences); anti–GR1-APC (clone RB6-8C5; BD Biosciences); and anti–F4/80-FITC (clone BM8; eBioscience) Abs. Absolute cell counts were determined with AccuCheck Counting Beads (Invitrogen), and the number of cells was normalized to the total liver weight. Data were analyzed using a CyAn ADP flow cytometer (Beckman Coulter) or a BD LSRII using Summit version 4.3 or FlowJo version 10.0.7 software where appropriate.
Determination of amine oxidase activity in murine tissues and human serum samples.
SSAO activity from human sera was measured as previously described (31). Briefly, 2.4 μl serum was incubated in the absence or presence of the SSAO inhibitor hydroxylamine (5 μM) for 30 minutes prior to the addition of SSAO substrate (benzylamine, 0.5 mM), HRP, and Amplex Red reagent (Molecular Probes). The SSAO-dependent production of hydrogen peroxide was followed for 60 minutes using a Tecan Infinite M200 spectrofluorometer. A calibration curve of exogenously added hydrogen peroxide was used to normalize the data. The SSAO activities in sera are reported as nmol hydrogen peroxide produced by 1 ml serum in 1 hour. SSAO activity from mouse fat lysates was measured as previously described (37). Briefly, fat tissue was lysed in PBS containing 0.2% NP-40. Protein concentrations were determined from clarified lysates, and 30 μg lysate was subjected to Amplex Red assays as described above.
Transfection of cells with siRNA or plasmid DNA encoding GFP fusion proteins.
Cells were transiently transfected with plasmid DNA or siRNA using a Nucleofector II (Lonza) according to the manufacturer’s instructions. Briefly, 5 μg DNA or 500 nM siRNA (AOC3 silencer from Invitrogen; s16435, sense: 5′-AGACUAGUUUAUGAGAUAATT-3′, antisense: 5′-UUAUCUCAUAAACUAGUCUTT-3′; silencer controls 1 and 2 from the same company were used as nontargeting siRNA) was nucleofected into 1.0 × 106 cells in 100 μl basic fibroblast Nucleofection solution (Lonza) using program A-023. The plasmids coding for GFP-WT-VAP-1 and the enzyme-dead mutant GFP-Y471F-VAP-1 have been previously described (47). Lifeact-GFP was obtained from Thistle Scientific.
Monitoring wound closure with the Cell-IQ system.
Real-time cell images of scratch wound assays were captured by a Cell-IQ system (CM Technologies) running Imagen software version 2.8.12.0 and analyzer version 3.3.0. For scratch wound assays, cells were transfected with the appropriate plasmid or siRNA and plated at a cell density of 100,000 cells per well (diluted in DMEM containing 2 % FCS, 1 ml per well) in a 24-well plate. Following overnight incubation, the media were replaced, and the cells were incubated for an additional 24 hours to reach confluence. A scratch wound was then effected using a 200-μl capacity pipette tip, and the media were replaced. The well plate was then inserted into the Cell-IQ, and the closure of each wound was monitored over a 24-hour period in a 5% CO2 and air environment maintained at 37°C.
Statistics.
Statistical analysis was performed using GraphPad Prism software version 6.0b (GraphPad Software) or SPSS version 22 (SPSS Inc.). Univariate data were calculated using Spearman’s Rho. All data in the figures are shown as the mean ± SEM. Data presented in Table 1 are shown as the median (IQR) where appropriate. Two-tailed Student’s t tests were used when appropriate. A P value of less than 0.05 was considered significant.
Study approval.
Serum and tissue samples from patients were obtained with written informed consent and with local ethics committee approval (LREC references 10/H0906/41, Newcastle & North Tyneside 1, UK; 04/Q2707/278, South Birmingham, Birmingham, UK; 06/Q2702/61, South Birmingham, Birmingham, UK; 98/CA5192, South Birmingham, Birmingham, UK; 04/Q2708/41, South Birmingham, Birmingham, UK). Animal studies were performed in accordance with the UK Animals (Scientific Procedures) Act of 1986, with approval granted by the UK Home Office (project license PPL 40/3201, Shrewsbury office) and with approval of the IACUC of the State Provincial Office of Southern Finland and in compliance with Finnish laws relating to the conduct of animal experimentation.
Supplementary Material
Acknowledgments
We are grateful to our clinical colleagues and patient donors for the provision of blood and tissue samples and would like to thank Janine Youster, Miroslava Blahova, and Bridget Gunson for their technical and statistical assistance. The authors would like to dedicate this work to the memory of our friend and colleague Nina Westerlund, who contributed so much to this study, but passed away before its completion. These studies were funded by the Wellcome Trust (083684/Z/07/Z and 091019/Z/09/Z) and the Finnish Academy.
References
- 1.Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006;40(suppl 1):S5–S10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- 2.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2):S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 3.Kojima H, et al. Difference and similarity between non-alcoholic steatohepatitis and alcoholic liver disease. Alcohol Clin Exp Res. 2005;29(12):259S–263S. doi: 10.1097/01.alc.0000191776.37626.30. [DOI] [PubMed] [Google Scholar]
- 4.Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. 2013;10(6):330–344. doi: 10.1038/nrgastro.2013.41. [DOI] [PubMed] [Google Scholar]
- 5.Day CP. From fat to inflammation. Gastroenterology. 2006;130(1):207–210. doi: 10.1053/j.gastro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 6.Henao-Mejia J, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Day CP. Non-alcoholic fatty liver disease: current concepts and management strategies. Clin Med. 2006;6(1):19–25. doi: 10.7861/clinmedicine.6-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lieber CS. Metabolic effects of ethanol and its interaction with other drugs, hepatotoxic agents, vitamins, and carcinogens: a 1988 update. Semin Liver Dis. 1988;8(1):47–68. doi: 10.1055/s-2008-1040528. [DOI] [PubMed] [Google Scholar]
- 9.Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123(5):1887–1901. doi: 10.1172/JCI66028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lalor PF, Shields P, Grant A, Adams DH. Recruitment of lymphocytes to the human liver. Immunol Cell Biol. 2002;80(1):52–64. doi: 10.1046/j.1440-1711.2002.01062.x. [DOI] [PubMed] [Google Scholar]
- 11.Jalkanen S, Salmi M. Cell surface monoamine oxidases: enzymes in search of a function. EMBO J. 2001;20(15):3893–3901. doi: 10.1093/emboj/20.15.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lalor PF, Edwards S, McNab G, Salmi M, Jalkanen S, Adams DH. Vascular adhesion protein-1 mediates adhesion and transmigration of lymphocytes on human hepatic endothelial cells. J Immunol. 2002;169(2):983–992. doi: 10.4049/jimmunol.169.2.983. [DOI] [PubMed] [Google Scholar]
- 13.McNab G, Reeves JL, Salmi M, Hubscher S, Jalkanen S, Adams DH. Vascular adhesion protein 1 mediates binding of T cells to human hepatic endothelium. Gastroenterology. 1996;110(2):522–528. doi: 10.1053/gast.1996.v110.pm8566600. [DOI] [PubMed] [Google Scholar]
- 14.Yoong KF, McNab G, Hubscher SG, Adams DH. Vascular adhesion protein-1 and ICAM-1 support the adhesion of tumor-infiltrating lymphocytes to tumor endothelium in human hepatocellular carcinoma. J Immunol. 1998;160(8):3978–3988. [PubMed] [Google Scholar]
- 15.Bonder CS, et al. Rules of recruitment for Th1 and Th2 lymphocytes in inflamed liver: a role for alpha-4 integrin and vascular adhesion protein-1. Immunity. 2005;23(2):153–163. doi: 10.1016/j.immuni.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 16.Lyles GA. Mammalian plasma and tissue-bound semicarbazide-sensitive amine oxidases: biochemical, pharmacological and toxicological aspects. Int J Biochem Cell Biol. 1996;28(3):259–274. doi: 10.1016/1357-2725(95)00130-1. [DOI] [PubMed] [Google Scholar]
- 17.Lalor PF, Sun PJ, Weston CJ, Martin-Santos A, Wakelam MJ, Adams DH. Activation of vascular adhesion protein-1 on liver endothelium results in an NF-κB-dependent increase in lymphocyte adhesion. Hepatology. 2007;45(2):465–474. doi: 10.1002/hep.21497. [DOI] [PubMed] [Google Scholar]
- 18.Jalkanen S, et al. The oxidase activity of vascular adhesion protein-1 (VAP-1) induces endothelial E- and P-selectins and leukocyte binding. Blood. 2007;110(6):1864–1870. doi: 10.1182/blood-2007-01-069674. [DOI] [PubMed] [Google Scholar]
- 19.Enrique-Tarancon G, et al. Substrates of semicarbazide-sensitive amine oxidase co-operate with vanadate to stimulate tyrosine phosphorylation of insulin-receptor-substrate proteins, phosphoinositide 3-kinase activity and GLUT4 translocation in adipose cells. Biochem J. 2000;350(pt 1):171–180. [PMC free article] [PubMed] [Google Scholar]
- 20.Enrique-Tarancon G, et al. Role of semicarbazide-sensitive amine oxidase on glucose transport and GLUT4 recruitment to the cell surface in adipose cells. J Biol Chem. 1998;273(14):8025–8032. doi: 10.1074/jbc.273.14.8025. [DOI] [PubMed] [Google Scholar]
- 21.Marti L, et al. Tyramine and vanadate synergistically stimulate glucose transport in rat adipocytes by amine oxidase-dependent generation of hydrogen peroxide. J Pharmacol Exp Ther. 1998;285(1):342–349. [PubMed] [Google Scholar]
- 22.Abella A, et al. Adipocytes release a soluble form of VAP-1/SSAO by a metalloprotease-dependent process and in a regulated manner. Diabetologia. 2004;47(3):429–438. doi: 10.1007/s00125-004-1346-2. [DOI] [PubMed] [Google Scholar]
- 23.Kurkijarvi R, Yegutkin GG, Gunson BK, Jalkanen S, Salmi M, Adams DH. Circulating soluble vascular adhesion protein 1 accounts for the increased serum monoamine oxidase activity in chronic liver disease. Gastroenterology. 2000;119(4):1096–1103. doi: 10.1053/gast.2000.18163. [DOI] [PubMed] [Google Scholar]
- 24.Stolen CM, Yegutkin GG, Kurkijarvi R, Bono P, Alitalo K, Jalkanen S. Origins of serum semicarbazide-sensitive amine oxidase. Circ Res. 2004;95(1):50–57. doi: 10.1161/01.RES.0000134630.68877.2F. [DOI] [PubMed] [Google Scholar]
- 25.Salmi M, et al. Insulin-regulated increase of soluble vascular adhesion protein-1 in diabetes. Am J Pathol. 2002;161(6):2255–2262. doi: 10.1016/S0002-9440(10)64501-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurkijarvi R, Adams DH, Leino R, Mottonen T, Jalkanen S, Salmi M. Circulating form of human vascular adhesion protein-1 (VAP-1): increased serum levels in inflammatory liver diseases. J Immunol. 1998;161(3):1549–1557. [PubMed] [Google Scholar]
- 27.Meszaros Z, Szombathy T, Raimondi L, Karadi I, Romics L, Magyar K. Elevated serum semicarbazide-sensitive amine oxidase activity in non-insulin-dependent diabetes mellitus: correlation with body mass index and serum triglyceride. Metabolism. 1999;48(1):113–117. doi: 10.1016/S0026-0495(99)90019-7. [DOI] [PubMed] [Google Scholar]
- 28.Karadi I, et al. Serum semicarbazide-sensitive amine oxidase (SSAO) activity is an independent marker of carotid atherosclerosis. Clin Chim Acta. 2002;323(1):139–146. doi: 10.1016/s0009-8981(02)00189-4. [DOI] [PubMed] [Google Scholar]
- 29.Boomsma F, et al. Plasma semicarbazide-sensitive amine oxidase is elevated in patients with congestive heart failure. Cardiovasc Res. 1997;33(2):387–391. doi: 10.1016/S0008-6363(96)00209-X. [DOI] [PubMed] [Google Scholar]
- 30.Li HY, et al. Serum vascular adhesion protein-1 is increased in acute and chronic hyperglycemia. Clin Chim Acta. 2009;404(2):149–153. doi: 10.1016/j.cca.2009.03.041. [DOI] [PubMed] [Google Scholar]
- 31.Aalto K, et al. Soluble vascular adhesion protein-1 correlates with cardiovascular risk factors and early atherosclerotic manifestations. Arterioscler Thromb Vasc Biol. 2012;32(2):523–532. doi: 10.1161/ATVBAHA.111.238030. [DOI] [PubMed] [Google Scholar]
- 32.Kleiner DE, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 33.Lazarus HM, Cruikshank WW, Narasimhan N, Kagan HM, Center DM. Induction of human monocyte motility by lysyl oxidase. Matrix Biol. 1995;14(9):727–731. doi: 10.1016/S0945-053X(05)80015-0. [DOI] [PubMed] [Google Scholar]
- 34.Lucero HA, et al. Lysyl oxidase oxidizes cell membrane proteins and enhances the chemotactic response of vascular smooth muscle cells. J Biol Chem. 2008;283(35):24103–24117. doi: 10.1074/jbc.M709897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459(7249):996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ushio-Fukai M. Vascular signaling through G protein-coupled receptors: new concepts. Curr Opin Nephrol Hypertens. 2009;18(2):153–159. doi: 10.1097/MNH.0b013e3283252efe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stolen CM, et al. Absence of the endothelial oxidase AOC3 leads to abnormal leukocyte traffic in vivo. Immunity. 2005;22(1):105–115. doi: 10.1016/j.immuni.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 38.Barry-Hamilton V, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16(9):1009–1017. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]
- 39.Lee WY, Salmi M, Kelly MM, Jalkanen S, Kubes P. Therapeutic advantage of anti-vascular adhesion protein-1 over anti-α4 integrin antibody in concanavalin a-induced hepatitis. Hepatology. 2013;58(4):1413–1423. doi: 10.1002/hep.26469. [DOI] [PubMed] [Google Scholar]
- 40.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117(3):539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Varela-Rey M, Embade N, Ariz U, Lu SC, Mato JM, Martinez-Chantar ML. Non-alcoholic steatohepatitis and animal models: understanding the human disease. Int J Biochem Cell Biol. 2009;41(5):969–976. doi: 10.1016/j.biocel.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 42.Diehl AM. Alcoholic liver disease: natural history. Liver Transpl Surg. 1997;3(3):206–211. doi: 10.1002/lt.500030303. [DOI] [PubMed] [Google Scholar]
- 43.Biddinger SB, Almind K, Miyazaki M, Kokkotou E, Ntambi JM, Kahn CR. Effects of diet and genetic background on sterol regulatory element-binding protein-1c, stearoyl-CoA desaturase 1, and the development of the metabolic syndrome. Diabetes. 2005;54(5):1314–1323. doi: 10.2337/diabetes.54.5.1314. [DOI] [PubMed] [Google Scholar]
- 44.DeLeve LD, Wang X, Kanel GC, Atkinson RD, McCuskey RS. Prevention of hepatic fibrosis in a murine model of metabolic syndrome with nonalcoholic steatohepatitis. Am J Pathol. 2008;173(4):993–1001. doi: 10.2353/ajpath.2008.070720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8(1):35–44. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 46.Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol. 2008;295(5):G987–G995. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weston CJ, Shepherd EL, Adams DH. Cellular localization and trafficking of vascular adhesion protein-1 as revealed by an N-terminal GFP fusion protein. J Neural Transm. 2013;120(6):951–961. doi: 10.1007/s00702-013-1003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riedl J, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5(7):605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henao-Mejia J, Elinav E, Strowig T, Flavell RA. Inflammasomes: far beyond inflammation. Nat Immunol. 2012;13(4):321–324. doi: 10.1038/ni.2257. [DOI] [PubMed] [Google Scholar]
- 50.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143(5):1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 51.Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54(1):133–144. doi: 10.1002/hep.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bohinc BN, Diehl AM. Mechanisms of disease progression in NASH: new paradigms. Clin Liver Dis. 2012;16(3):549–565. doi: 10.1016/j.cld.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 53.Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander-Tetri BA. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology. 2011;53(3):810–820. doi: 10.1002/hep.24127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klyubin, Kirpichnikova KM, Gamaley IA. Hydrogen peroxide-induced chemotaxis of mouse peritoneal neutrophils. Eur J Cell Biol. 1996;70(4):347–351. [PubMed] [Google Scholar]
- 56.Lazarus HM, Cruikshank WW, Narasimhan N, Kagan HM, Center DM. Induction of human monocyte motility by lysyl oxidase. Matrix Biol. 1995;14(9):727–731. doi: 10.1016/S0945-053X(05)80015-0. [DOI] [PubMed] [Google Scholar]
- 57.Lucero HA, et al. Lysyl oxidase oxidizes cell membrane proteins and enhances the chemotactic response of vascular smooth muscle cells. J Biol Chem. 2008;283(35):24103–24117. doi: 10.1074/jbc.M709897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Steiniger B, Barth P, Hellinger A. The perifollicular and marginal zones of the human splenic white pulp: do fibroblasts guide lymphocyte immigration? Am J Pathol. 2001;159(2):501–512. doi: 10.1016/S0002-9440(10)61722-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iffiu-Soltesz Z, et al. Chronic benzylamine administration in the drinking water improves glucose tolerance, reduces body weight gain and circulating cholesterol in high-fat diet-fed mice. Pharmacol Res. 2010;61(4):355–363. doi: 10.1016/j.phrs.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 60.Mercader J, Iffiu-Soltesz Z, Bour S, Carpene C. Oral administration of semicarbazide limits weight gain together with inhibition of fat deposition and of primary amine oxidase activity in adipose tissue. J Obes. 2011;2011:475786. doi: 10.1155/2011/475786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bour S, et al. Semicarbazide-sensitive amine oxidase/vascular adhesion protein-1 deficiency reduces leukocyte infiltration into adipose tissue and favors fat deposition. Am J Pathol. 2009;174(3):1075–1083. doi: 10.2353/ajpath.2009.080612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Noonan T, et al. The oxidase activity of vascular adhesion protein-1 (VAP-1) is essential for function. Am J Clin Exp Immunol. 2013;2(2):172–185. [PMC free article] [PubMed] [Google Scholar]
- 63.Olivieri A, Tipton K. Inhibition of bovine plasma semicarbazide-sensitive amine oxidase by caffeine. J Biochem Mol Toxicol. 2011;25(1):26–27. doi: 10.1002/jbt.20356. [DOI] [PubMed] [Google Scholar]
- 64.Molloy JW, Calcagno CJ, Williams CD, Jones FJ, Torres DM, Harrison SA. Association of coffee and caffeine consumption with fatty liver disease, nonalcoholic steatohepatitis, and degree of hepatic fibrosis. Hepatology. 2012;55(2):429–436. doi: 10.1002/hep.24731. [DOI] [PubMed] [Google Scholar]
- 65.Koskinen K, et al. Granulocyte transmigration through the endothelium is regulated by the oxidase activity of vascular adhesion protein-1 (VAP-1). Blood. 2004;103(9):3388–3395. doi: 10.1182/blood-2003-09-3275. [DOI] [PubMed] [Google Scholar]
- 66.Gokturk C, et al. Overexpression of semicarbazide-sensitive amine oxidase in smooth muscle cells leads to an abnormal structure of the aortic elastic laminas. Am J Pathol. 2003;163(5):1921–1928. doi: 10.1016/S0002-9440(10)63550-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cuevas EP, Moreno-Bueno G, Canesin G, Santos V, Portillo F, Cano A. LOXL2 catalytically inactive mutants mediate epithelial-to-mesenchymal transition. Biol Open. 2014;3(2):129–137. doi: 10.1242/bio.20146841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lugassy J, et al. The enzymatic activity of lysyl oxidas-like-2 (LOXL2) is not required for LOXL2-induced inhibition of keratinocyte differentiation. J Biol Chem. 2012;287(5):3541–3549. doi: 10.1074/jbc.M111.261016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Holt AP, Haughton EL, Lalor PF, Filer A, Buckley CD, Adams DH. Liver myofibroblasts regulate infiltration and positioning of lymphocytes in human liver. Gastroenterology. 2009;136(2):705–714. doi: 10.1053/j.gastro.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 70.Hathaway M, Burnett D, Elias E, Adams DH. Secretion of soluble chemotactic factors, including interleukin-6: a mechanism for the recruitment of CD8-positive T lymphocytes to human liver allografts during rejection. Hepatology. 1993;18(3):511–518. [PubMed] [Google Scholar]
- 71.Merinen M, Irjala H, Salmi M, Jaakkola I, Hanninen A, Jalkanen S. Vascular adhesion protein-1 is involved in both acute and chronic inflammation in the mouse. Am J Pathol. 2005;166(3):793–800. doi: 10.1016/S0002-9440(10)62300-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.