

Abstract
While 30%–70% of RSV-infected infants develop bronchiolitis, 2% require hospitalization. It is not clear why disease severity differs among healthy, full-term infants; however, virus titers, inflammation, and Th2 bias are proposed explanations. While TLR4 is associated with these disease phenotypes, the role of this receptor in respiratory syncytial virus (RSV) pathogenesis is controversial. Here, we evaluated the interaction between TLR4 and environmental factors in RSV disease and defined the immune mediators associated with severe illness. Two independent populations of infants with RSV bronchiolitis revealed that the severity of RSV infection is determined by the TLR4 genotype of the individual and by environmental exposure to LPS. RSV-infected infants with severe disease exhibited a high GATA3/T-bet ratio, which manifested as a high IL-4/IFN-γ ratio in respiratory secretions. The IL-4/IFN-γ ratio present in infants with severe RSV is indicative of Th2 polarization. Murine models of RSV infection confirmed that LPS exposure, Tlr4 genotype, and Th2 polarization influence disease phenotypes. Together, the results of this study identify environmental and genetic factors that influence RSV pathogenesis and reveal that a high IL-4/IFN-γ ratio is associated with severe disease. Moreover, these molecules should be explored as potential targets for therapeutic intervention.
Introduction
Respiratory syncytial virus (RSV) is the most frequent cause of hospitalization in infants worldwide (1). An estimated 30%–70% of infants develop bronchiolitis upon primary RSV infection, 1%–3% of whom are hospitalized (1–3). Even though risk factors for hospitalization have been identified (2, 4), the mechanism of severe RSV bronchiolitis is not well understood.
Three hypotheses have been widely accepted as probable explanations for disease severity (4). The first attributes severe bronchiolitis to RSV damage to the respiratory tract and is supported by studies in which higher doses of candidate intranasal vaccine viruses increased symptoms in recipients (5). A second hypothesis postulates that severity is a consequence of inflammation (6). The final explanation ascribes severe RSV bronchiolitis to a CD4+ Th2 polarization of the immune response in the respiratory tract (7, 8) and is supported by the resemblance of symptoms between bronchiolitis and asthma, high Th2 cytokine levels in severely ill infants, and genetic association studies with Th2 genes (8–11).
TLR4 is associated with all 3 hypotheses, and its activation has been reported to affect RSV titers in the lungs, inflammation, and Th bias (12, 13). However, studies of the role of TLR4 in RSV pathogenesis have yielded conflicting results in mice (12–14) and in children living in different environments (refs. 15–19 and Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI75183DS1). To our knowledge, environmental factors have not been considered in evaluations of RSV pathogenesis. Yet, gene-environment interactions involving TLRs modulate other wheezing illnesses (20, 21).
Two SNPs in TLR4 encode substitutions (Asp299Gly and Thr399Ile) in the ectodomain that are in high linkage disequilibrium and have been associated with an endotoxin (LPS) hyporesponsive phenotype (22). LPS interacts with molecules that allow activation of TLR4, altering responses to different stimuli (23).
We prospectively studied 2 independent populations of infants with bronchiolitis to test the hypothesis that the interaction of TLR4 SNPs with the environment modulates RSV disease severity. We confirmed the role of the candidate molecules associated with disease severity in this study in mechanistic experiments using a mouse model of RSV bronchiolitis that reproduced the phenotype of human disease.
Results
Infants with RSV bronchiolitis are exposed to different levels of environmental LPS.
In our first study, 768 infants with bronchiolitis (426 with severe disease and 342 with mild disease) were prospectively recruited in the emergency rooms (ERs) and outpatient clinics (OPCs) of participating institutions during the winter seasons of 2003 to 2006. Fifty-four (7%) parents declined their infant’s participation; these infants had characteristics similar to those of infants enrolled in the study (not shown).
RSV was detected in nasal secretions of 418 (54%) participating patients. Severe RSV bronchiolitis was found in 246 cases (59%), while 172 (41%) infants had mild RSV bronchiolitis and served as controls.
Infants from 3 geographical regions of Buenos Aires participated in the first study. Those from the western and southern rural regions of the Buenos Aires district lived in families of low socioeconomic status (SES) (Supplemental Table 2). Variables reported to affect the risk of severe RSV bronchiolitis (1, 2) were similar in infants from the western and southern low-income regions (Supplemental Table 3).
The third group of infants lived in urban middle-class families (central region; Supplemental Table 2). In a comparison of the frequency of risk factors in the low- and middle-income groups, exposure to cigarette smoke (24) was observed more often in low-income families (Supplemental Table 3). Only age and number of siblings under the age of 14 years living at home influenced the severity of RSV bronchiolitis (Supplemental Table 4). We detected no differences in symptom duration, oxygen requirement, or need for intensive care between RSV-infected populations in the rural and urban groups (Supplemental Table 5).
Interestingly, we found that socioeconomic status was associated with differences in LPS levels in infants’ bedrooms (Figure 1A). Homes in low-SES, rural regions had similar levels of bedroom LPS (P = 0.89), which were significantly higher than those in urban neighborhoods of middle SES (P < 0.01). Yet, other microbial signals in addition to LPS modulate immune responses through TLR4. Therefore, we expanded our characterization of these signals by exploring nasopharyngeal (NP) colonization with 3 bacteria encoding TLR4 agonists: Streptococcus pneumoniae (S. pneumoniae) (25), nontypable Haemophilus influenzae (H. influenzae) (26), and Moraxella catharrhalis (M. catharrhalis) (27). NP carriage of each bacterium was more frequent and carriage more diverse in infants of low SES from rural homes (Supplemental Figure 1 and Figure 1B). Therefore, infants of low SES are exposed to higher levels of TLR4 bacterial agonists, evidenced directly by higher bedroom levels of LPS (Figure 1A) and more frequent NP carriage of each and all 3 bacteria (Figure 1B and Supplemental Figure 1) and indirectly by demographic indicators like lack of sewage (62% vs. 0% in high SES) and crowding (45% vs. 3% in high SES) at home (Supplemental Table 2 and refs. 28, 29). For simplicity hereafter in the text and figures, we refer to infants from rural homes with NP bacterial carriage and indicators of low SES as having exposure to high levels of LPS.
Figure 1. Pattern recognition receptors and environmental interactions.

(A) LPS concentrations categorized as low (<48 EU/ml), medium (48–480 EU/ml), and high (>480 EU/ml), as determined using an LAL assay in cradles and bed sheets of infants from low- versus middle-SES groups. P < 0.01 by Fisher’s exact test. (B) Diversity of NP colonization, defined as carriage of all 3 S. pneumoniae, H. influenzae, and M. catarrhalis bacteria in a convenience sample of 100 infants of low SES versus those of high SES. Percentage of infants with detectable bacterial DNA. P < 0.001 by χ2 test. (C and D) Association of the TLR4 Asp299Gly heterozygous genotype in RSV-infected infants with mild (white bars) or severe (black bars) bronchiolitis in groups of infants from rural homes with high NP bacterial carriage, indicators of low SES, exposure to high environmental levels of LPS (High LPS), and groups of infants from urban homes with low NP bacterial carriage, middle-SES status, and low LPS exposure levels (Low LPS) in 2003–2006 (C; P = 0.003 for interaction) and 2010–2013 (D; P = 0.002 for interaction). (E) ORs and 95% CIs for disease severity in infants from high and low LPS level environments in population 1 (2003–2006), population 2 (2010–2013), and both populations combined (P < 0.001 for interaction in combination). Fisher’s exact and Zelen’s exact tests for C–E (see Table 1). (F) CD14 C-159T and (G) CD14 C-550T alleles in RSV-infected infants with severe (black bars) or mild (white bars) bronchiolitis in groups exposed to low or high environmental levels of LPS. Fisher’s exact; P = NS for both alleles.
The interaction between TLR4 genotype and the environment is associated with severity of RSV bronchiolitis.
The cohort was genotyped for the Asp299Gly and Thr399Ile alleles (Supplemental Table 6). The overall distributions of the 2 alleles were in Hardy-Weinberg equilibrium, and the frequencies were similar to those reported for other populations (30). Thirty-three ethnicity-specific genomic markers revealed neither evidence of admixture nor significant ethnic differences between the groups (Supplemental Table 7).
The TLR4 D299G allele was present in 27 of 397 (6.8%) RSV-infected infants and 21 of 331 (6.3%) infants infected with other agents (P = 0.9; Supplemental Table 6). The number of individuals homozygous for the SNP was too small for statistical analyses. We performed identical analyses with the Thr399Ile allele and the Asp299Gly/Thr399Ile haplotype. The results were similar to those observed with the Asp299Gly allele alone. Therefore, hereafter only analyses with the Asp299Gly SNP are presented.
In infants from the rural western and southern regions, characterized by low SES and environments with high LPS levels, Asp299Gly was more frequently found in children with mild RSV bronchiolitis (Supplemental Figure 2). In fact, both low-income groups had similar risk factors (Supplemental Table 3), additional indicators of disease severity (Supplemental Table 5), and distribution of TLR4 genotypes (P = 0.3) and were therefore subsequently analyzed as 1 group (Figure 1C). Conversely, in infants from urban homes who had low-level LPS exposure and high SES, Asp299Gly was more frequently found in the infants with severe disease (Figure 1C). The impact on severity of illness of the interaction between TLR4 and environments with different LPS levels was significant, even after adjusting for risk factors affecting the severity of RSV bronchiolitis (Table 1 and refs. 1, 2). TLR4 genotypes did not affect the severity of illness in patients with non–RSV bronchiolitis, suggesting that the gene-environment interaction is virus specific (Table 1).
Table 1.
OR for severe bronchiolitis: association with TLR4 mutation according to the environment
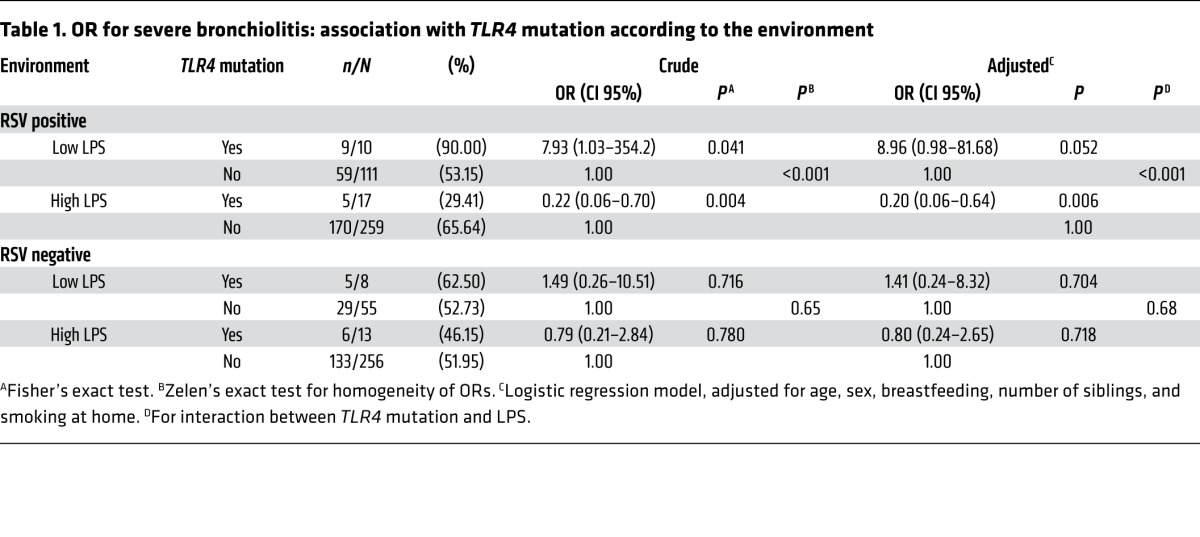
CD14 is a component of the CD14/TLR4 signaling that enhances TLR4 responses (31, 32). To determine whether CD14 affected RSV disease severity, we asked whether the CD14 C-159T and CD14 C-550T SNPs were also associated with RSV bronchiolitis (Figure 1, F and G, and Supplemental Table 8). Allelic frequencies for both SNPs were in Hardy-Weinberg equilibrium and did not differ from those reported previously (16, 17, 31). Unlike TLR4 Asp299Gly, CD14 C-159T and CD14 C-550T did not affect RSV disease severity.
Role of RSV titers in disease severity.
To explore candidate variables that may affect the severity of RSV bronchiolitis, we first compared RSV titers in respiratory secretions. A comparison between cases and controls revealed no differences (Figure 2A). Time from initiation of symptoms to sampling was similar between groups (Figure 2B). Neither differential LPS exposure levels (Figure 2C) nor different genotypes (Figure 2D) affected RSV titers. These observations do not support an association between the severity of RSV disease and virus titers in respiratory secretions. Our comparisons of RSV subgroups A and B also failed to find differences between severe and mild cases (Supplemental Figure 4).
Figure 2. RSV lung titers are not associated with severity.

(A) RSV titers in respiratory secretions from infants with severe (black box) or mild (white box) bronchiolitis; P = 0.659. Data represent the mean ± SEM. (B) Time from initiation of symptoms to collection of respiratory secretions in RSV-infected infants with severe (black bars) or mild (white bars) bronchiolitis. P = 0.990. (C) RSV titer in respiratory secretions from infants from rural homes with high NP bacterial carriage, indicators of low SES, and exposure to high environmental levels of LPS (High LPS; white box) and infants from urban homes with low NP bacterial carriage, middle SES, and low LPS exposure levels (Low LPS; black box). P = 0.608. Data represent the mean ± SEM. (D) RSV titers in respiratory secretions from RSV-infected infants with 2 major alleles (black box) and infants with 1 TLR4 D299G allele (white box). P = 0.399. All statistical analyses were performed using 2-tailed Student’s t tests.
RSV-mediated inflammation in the lungs is determined by environmental exposure.
We then examined whether severity of RSV bronchiolitis was associated with inflammation in the respiratory tract. For this purpose, we measured levels of IL-6, IL-8, TNF-α, and IL-1β in respiratory secretions from RSV-infected infants and found no differences in levels between mildly and severely ill patients (Figure 3, A–D).
Figure 3. Inflammation in the lungs is determined by environmental exposure.

(A) IL-6, (B) IL-8, (C) TNF-α, and (D) IL-1β levels in respiratory secretions from infants with severe (black bars) or mild (white bars) bronchiolitis. P = NS for all comparisons. Data represent the mean ± SEM. (E) IL-6 and (F) IL-8 levels in respiratory secretions from infants from urban homes with low NP bacterial carriage, high SES, and low LPS exposure levels (Low LPS) and infants from rural homes with high NP bacterial carriage, indicators of low SES with exposure to high environmental levels of LPS (High LPS). P = 0.04 for Il-6; P = 0.003 for IL-8. Data represent the mean ± SEM. (G–I) TLR4, CD14, and TLR2 mRNA expression in respiratory secretions from healthy infants exposed to low (black bar) or high (white bar) environmental levels of LPS. P < 0.01 for all comparisons by Mann-Whitney U test.
Interestingly, we found that inflammatory cytokines were more abundant in secretions from patients living in urban areas who had low levels of LPS exposure versus those in rural areas with chronically high levels of LPS exposure (Figure 3, E and F). These findings suggest that rural environments rich in LPS influence cytokine production levels during RSV infection. To explore whether different inflammatory responses in these populations were associated with differential expression of pattern recognition receptors (PRRs) in the respiratory tract before infection, we compared TLR4 expression in asymptomatic infants residing in the same neighborhoods as infants infected with RSV during the study. We found that expression of TLR4 in the respiratory tract of asymptomatic infants from rural neighborhoods with high environmental LPS levels was suppressed compared with that detected in infants from urban neighborhoods with low LPS levels (Figure 3G). We reasoned that other PRRs might also be modulated by environmental exposure. Indeed, CD14, TLR2, and MD2 mRNA levels were downregulated in infants chronically exposed to LPS (Figure 3, H and I, not shown, and ref. 33).
The TLR4-environment interaction modulates GATA3/T-bet ratios affecting RSV disease severity.
In mice, exposure to high levels of pathogen-associated molecular patterns (PAMPs) promotes Th1 bias, and low-level exposure promotes Th2 responses through PRRs (34). We therefore reasoned that the population of poor infants from rural environments with low PRR expression would have weak PRR-RSV interactions (equivalent to a low inoculum of a PAMP) and bias the immune response to Th2, while middle-class infants from urban environments with high PRR expression would bias the immune response to Th1 in RSV infection.
To explore this hypothesis, we examined in both populations expression of the Th2 and Th1 transcription factors GATA3 and T-bet (35, 36). We hypothesized that the ratio between these master regulators during RSV infection would be affected by the interaction between TLR4 and the environment and play a role in the severity of illness.
Interestingly, in the subgroup of infants with available measurements (n = 120), we found that Th2 GATA3/T-bet ratios greater than 1 were associated with RSV disease severity (Figure 4A; odds ratio [OR] = 2.82 [CI, 1.14–6.96]; P = 0.02). Furthermore, infants at high risk for severe disease (low-level LPS exposure and a TLR4 D299G allele plus high-level LPS exposure and 2 major alleles) had a higher frequency of GATA3/T-bet ratios greater than 1 than did low-risk infants (Figure 4B; OR = 7.61 [CI, 2.83–20.4]; P < 0.0001). Finally, we found that the effect of the TLR4-environment interaction on severe RSV bronchiolitis was weakened by adjusting for GATA3/T-bet (OR = 2.03 [CI, 0.88–4.69]; P = 0.1), even after adjusting for RSV risk factors (P = 0.1; Figure 4C). These findings suggest that the GATA3/T-bet ratio is involved in the LPS/TLR4 pathway to severe RSV bronchiolitis.
Figure 4. Th2 bias and RSV bronchiolitis.

(A) Percentage of RSV-infected infants with severe or mild RSV bronchiolitis with a GATA3/Tbet mRNA ratio greater than 1 in respiratory secretions. P = 0.02 by χ2 test. (B) Percentage of high-risk (WT and high-level LPS exposure plus TLR4 D299G and low-level LPS exposure; black bar) and low-risk (WT and low-level LPS exposure plus TLR4 D299G and high-level LPS exposure; white bar) RSV-infected infants with a GATA3/Tbet mRNA ratio greater than 1 in respiratory secretions. P < 0.0001 by χ2 test. (C) Effect of TLR4-environment interaction on severity of RSV bronchiolitis in unadjusted analysis and after adjusting for GATA3/T-bet ratios greater than 1. (D) IFN-γ, (E) IL-4, and (F) IL-4/IFN-γ levels in respiratory secretions from infants with severe (black bar) or mild (white bar) RSV bronchiolitis (range = 0.33–9.2 for severe in IL-4/IFN-γ and 0.01–2.3 for mild). Data represent the mean ± SEM. (G) IL-4/ IFN-γ levels in respiratory secretions from high- (black bar) or low- (white bar) risk infants. Data represent the mean ± SEM. (H) IL-9, (I) IL-13, and (J) IL-5 levels in respiratory secretions from infants with severe (black bar) or mild (white bar) RSV bronchiolitis. Data represent the mean ± SEM. Mann-Whitney U test for D–J.
Low IFN-γ and high IL-4 levels in respiratory secretions from infants with severe RSV bronchiolitis.
We therefore investigated the role of Th1 (IFN-γ) and Th2 (IL-4, IL-5, IL-9, and IL-13) cytokines in bronchiolitis severity. IFN-γ levels were lower in infants with severe illness compared with those in infants with mild illness (Figure 4D). Conversely, IL-4 levels were higher in infants with severe RSV infection (Figure 4E). In fact, IL-4/ IFN-γ ratios were also significantly higher in severely ill and at-risk infants, confirming our observations (Figure 4, F and G). We detected no differences in IL-9 or IL-13 levels in respiratory secretions from mildly or severely ill patients (Figure 4, H and I). Interestingly, IL-5 levels were associated with protection (Figure 4J). IL-17 was undetectable in all infants.
Finally, we investigated the distribution of a gain-of-function IL4 SNP (C-590T) in infants with RSV bronchiolitis. Consistent with studies in industrialized countries (9, 10), C-590T was associated with severity in environments with low LPS levels. We observed no effect in the rural population exposed to high levels of LPS (Supplemental Figure 3 and Supplemental Table 9). Conversely, IL13 SNP (A-445G) analysis was not associated with differences in severity (Supplemental Figure 3 and Supplemental Table 9).
TLR4 genotype and environment in a second infant population.
To confirm our observations, we studied 433 infants with RSV lower respiratory infection (LRI) between 2010 and 2013. These infants were admitted to hospitals in the same central and southern regions of Buenos Aires (Figure 1, D and E). Two hundred and thirty-five infants lived in rural, low-income homes in the southern region, which was characterized by high levels of environmental LPS, while 198 infants lived in an urban, middle-class, low-level LPS environment in the central region (Supplemental Table 10). In this case, we detected the Asp299Gly allele in 12 infants in the southern region versus 14 in the central region, for an overall rate of 6.0% (P = 0.64 vs. the first population). Again, the Asp299Gly allele rates were higher in severe cases among infants from urban, low-level LPS exposure environments (OR = 4.72 [CI, 1.00–44.31]) and in mild cases among those in rural, low-income, high-level LPS exposure environments (OR = 0.20 [CI, 0.03–0.83]; Figure 1E). Moreover, the impact of the TLR4-environmental interaction on disease severity was significant when we analyzed this second population (P = 0.001) and both populations together (P < 0.001; Figure 1E).
A mechanistic role for Th2 bias in a mouse model of RSV bronchiolitis.
Finally, we investigated whether a Th bias played a mechanistic role in RSV disease severity in mice. For this purpose, we evaluated airway hyperreactivity (AHR) in rodents infected with RSV.
First, we replicated the gene-environment interaction observed in infants with RSV bronchiolitis (Figure 5, A and B). Preexposure of C57BL/10 and Tlr4+/– mice to a chronically high dose of LPS (mimicking infants of low SES) followed by RSV i.n. inoculation led to enhanced AHR in WT mice compared with that observed in Tlr4+/– animals (Figure 5A). Conversely, no LPS (high-SES mice) followed by RSV resulted in increased AHR in Tlr4+/– mice compared with that seen in WT mice (Figure 5B). As expected, we observed no differences in viral pulmonary titers or inflammation, as determined by IL-6 and IL-1β immunoassays in BAL fluids, between groups with different disease severity (not shown). As in infants, we found that TLR4 expression was suppressed in mice exposed to LPS (Figure 5C).
Figure 5. Th2 bias promotes RSV disease in mice.

AHR in mice exposed to 50 mg aerosolized mCh 5 days after i.n. infection with 106 PFU RSV line 19 after high (100 μg qod) (A) or no (B) exposure to bacterial LPS. Student’s t test; P = 0.029 (A) and P = 0.006 (B) for WT versus Tlr4+/–. No statistically significant differences were observed between uninfected WT and Tlr4+/– controls (not shown). Results are representative of duplicate experiments (6–8 mice/group). (C) TLR4 mRNA expression levels in WT mice exposed to high LPS levels versus no LPS. P = 0.012. (D) IFN-γ, (E) IL-4, and (F) IL-4/IFN-γ ratios in lung homogenates of high-risk (no LPS exposure for Tlr4+/– plus high LPS exposure for WT) compared with low-risk (no LPS exposure for WT plus high-level LPS exposure for Tlr4+/–) mice 5 days after inoculation with RSV line 19. P = 0.002 for IFN-γ, P = 0.018 for IL-4, and P = 0.011 for IL-4/IFN-γ ratios; Mann-Whitney U test for D–F. 6–9 mice/group. (G and H) AHR in BALB/c (WT), Stat1–/– (deficient T-bet activation), and Stat6–/– (deficient GATA3 activation) mice exposed to aerosolized mCh 5 days after infection with 106 PFU RSV line 19. P <0.05 for Stat1–/– versus WT and Stat6–/– and P < 0.05 for Stat6–/– versus WT in E; ANOVA with Tukey’s post test. Data represent the mean ± SEM. No significant differences were observed between uninfected controls. Results are representative of duplicate experiments (5 mice/group). (I) AHR in RSV-infected, tamoxifen-treated C57BL/6 WT and in mice conditionally deficient for GATA3 (GATA3fl/fl). P < 0.05 by Mann-Whitney U test. Data represent the mean ± SEM (5 mice/group).
Following these results and our observations in infants, we compared IFN-γ and IL-4 levels in respiratory secretions from mice at high versus low risk for severe disease. As described in infants, at-risk mice had lower IFN-γ levels and higher IL-4 levels and IL-4/IFN-γ ratios in the respiratory tract (Figure 5, D–F). We then explored the cells responsible for IL-4 production in the lungs of mice during RSV infection. Interestingly, we detected IL-4 in CD4+ T lymphocytes obtained from lung homogenates of mice infected with RSV 5 days after infection (Supplemental Figure 5). In addition, IL-4 was produced by invariant NKT (iNKT) cells isolated from the lungs of mice infected with RSV (Supplemental Figure 6).
Finally, we tested the role of Th2 bias in disease severity using Stat1–/– and Stat6–/– mice and mice conditionally deficient for GATA3 (GATA3fl/fl). T-bet activation is directly associated with prior STAT1 activation (37). Induction of GATA3 in naive CD4+ T cells requires STAT6 activation (38). Five days after RSV inoculation, we observed that Stat6–/– mice had lower AHR than did WT controls. Stat1–/– mice had higher AHR than did both of the other groups (Figure 5, G and H). The role of GATA3 was confirmed in GATA3fl/fl mice (Figure 5I). Five days after RSV infection, these mice had lower AHR than did WT mice, supporting a role for Th2 bias in the pathogenesis of RSV disease.
Discussion
Our study identifies an important and specific pathogenic mechanism for severe RSV bronchiolitis. TLR4-environment interactions modulated GATA3/T-bet ratios during RSV infection, and a Th2 bias with high levels of IL-4 and low levels of IFN-γ increased disease severity in infants. We also observed the role of these transcription factors and cytokines in mice.
Our study does not find evidence supporting a causative role for virus titers or RSV strains in disease severity (39). In addition, while our 2003–2006 population exhibited a greater exposure to RSV A in high-level versus low-level LPS exposure environments, RSV B prevailed in high-level LPS exposure homes between 2010 and 2013 (data not shown). Therefore, our data suggest that RSV strains do not affect the TLR4-environment interaction either. The poor record of corticosteroids against RSV (40), studies reporting low levels of inflammatory cytokines during severe illness (41), absent inflammation in fatal RSV cases (42), and discrepant inflammatory responses in severe bronchiolitis caused by different viruses (43) suggest that an augmented production of inflammatory cytokines may not explain severe bronchiolitis. In our study, severe RSV bronchiolitis correlated with a predominance of GATA3 over T-bet and high IL-4/IFN-γ ratios in the respiratory tract (11). Previous studies associated IL-9 and IL-13 with severe disease (8, 10), but ours did not replicate those observations. It is, however, clear that these forms of sampling represent the mixture of conventional Th2 contributions (in ours and other studies, ref. 44) with those of a number of other cell types. With respect to CD4+ T cells, for instance, a major source of IL-9 would be the Th9 cell population (45). More broadly, a hallmark of the innate lymphoid cell type 2 (ILC2) subset is scant IL-4 production, yet detectable IL-5, IL-9, and IL-13 production (46). As such, our findings are consistent with Th2 (sensu stricto) being one influence tipping the balance between severe and mild RSV disease and that hallmarks shared with Th9 and ILC2 cells (IL-5, IL-9, and IL-13) do not provide the positive correlation observed with IL-4 and IL-4/IFN-γ ratios. Additional studies will be required to fully characterize the cell types, in addition to CD4+ T lymphocytes and iNKT cells, contributing to disease enhancement.
The role of TLR4 in RSV disease has been a source of controversy. Environmental exposure (15, 19), lack of power (17–19), choice of controls (15–19), or specific genetic or demographic characteristics of the population (16) may explain the differences between human studies. In fact, our observations and those of others (47) suggest that the effect of TLR4 in disease severity does not occur through interaction of this PRR with the virus but rather through prior environmental TLR4 activation conditioning the immune response to infection. Moreover, not all individuals with this TLR4 haplotype display hyporesponsiveness to LPS (22), and this discrepancy may contribute to the lack of complete correlation in our data.
Importantly, our study identifies a group of full-term infants who may be highly susceptible to RSV: TLR4+/– infants from urban environments with low levels of LPS exposure. Over 80% of these infants in our studies were hospitalized when visiting the ER or OPC with respiratory symptoms due to RSV (15). Even if only 7%–10% of all TLR4+/– infants visit the ER with RSV infections every year (48), hospitalization rates for them would approach those of extremely premature babies (49). But while extremely premature babies constitute approximately 1.5% of the population in western industrialized societies, TLR4+/– full-term infants represent 6%–10% (30). Moreover, 89.5% of North American premature infants hospitalized with RSV LRI in an earlier study were found to be TLR4+/–, while the rate of heterozygosity in the general population was 10.5% (50). Should additional studies in TLR4+/– infants confirm our observations, preventive interventions aimed at virus neutralization and/or novel anti–Th2 approaches (51) may be needed to protect these children.
On the other hand, our findings that approximately 94% of infants in the rural region had 2 major alleles for TLR4 and high LPS exposure levels — which are typical of low-income environments and may result in increased severity of RSV bronchiolitis among these children (P = 0.03) — help to explain why poverty is associated with severe RSV LRI (52, 53). After all, treatment for RSV disease is supportive. Hence, sophisticated medical interventions are unlikely to explain all outcome inequalities between socioeconomic groups. In fact, risk factors such as crowding, day-care attendance, exposure to cigarette smoke, and numerous siblings may further contribute to disease severity by enhancing the LPS environmental effect on TLR4.
Our observations suggest that environmental factors — interacting with the TLR4 genotype — modulate PRR expression in the respiratory tract and can result in an exaggerated Th2 response during RSV infection associated with severe bronchiolitis. While TLR4 studies provide an interesting window into RSV pathogenesis, other genes and risk factors have also been noted to influence disease severity (6–8, 10, 53). In fact, GATA3/T-bet ratios and Th2 bias may be part of a downstream pathogenic pathway modified at various levels by different genes affecting pulmonary function (54, 55) and/or immune responses (8–10, 34) in the context of the environment and its epigenetic modifications.
Our results have caveats. First, in studying human populations we recognize that other environmental factors or unmeasured confounders may affect the results. Second, even though we examined highly representative inflammatory cytokines, other unmeasured inflammatory molecules or even RSV load at other time points could still influence disease severity. In addition, studying healthy human subjects requires extrapolation of immune manifestations in the lungs through the analysis of upper respiratory tract secretions. The direct correlation of cytokine responses and viral titers during LRI has been described in other reports (41). Finally, although the limulus amebocyte lysate (LAL) assay is the industry standard for assessment of endotoxicity, certain endotoxins may have different reactivity to the test, and variability is common (56, 57). Therefore, based on the difficulty of drawing absolute environmental conclusions on single–time point, single-molecule measurements (56, 57), we based our environmental assessments on a combined evaluation of LPS levels, NP bacterial carriage, and living conditions (28, 29, 56, 57).
However, this study has important strengths (58). First, it presents hundreds of infants from different environments with mild and severe RSV bronchiolitis and relevant RSV-negative controls in various populations. Second, phenotypes in infants and mice were defined by clinically relevant criteria, even in the context of the limitations of murine models of human respiratory viruses. Third, this study identifies a novel population at high risk for severe disease: urban, middle-class TLR4+/– infants. Finally, this study proposes a plausible mechanistic paradigm for environmental, genetic, epidemiological, and immune factors affecting the pathogenesis of RSV bronchiolitis in infants and, simultaneously, offers mechanistic support for its observations in mice.
In summary, we describe an association between the TLR4 genotype and environmental conditions that modulate RSV severity through a Th bias. High IL-4/IFN-γ ratios are associated with severe RSV bronchiolitis. Therapeutic interventions should explore modulation of these molecules.
Methods
Patients.
This prospective, case-control study was conducted in Buenos Aires, Argentina, between 2003 and 2006 and between 2010 and 2013. Participating hospitals in 2003–2006 cared for infants in the central (at Hospital Francés), western (Hospital Nacional Dr. Alejandro Posadas), and southern (Hospital Evita Pueblo de Berazategui and Hospital Mi Pueblo de Florencio Varela) regions of the city. These last 2 regions are separated by 40 highway miles and by other areas of the city. Therefore, the possibility of even small groups of people from either population (who typically lack private transportation) moving to the other district is extremely low.
Previously healthy full-term infants under 1 year of age and born after September 15 of the previous year, 15 days after the end of the RSV season in Buenos Aires (3, 43), with signs and symptoms of bronchiolitis for the first time in their lives, were invited to participate. Bronchiolitis was defined as a constellation of clinical signs including wheezing with or without cough, rales, dyspnea, and increased respiratory rate and retractions of the respiratory muscles. The diagnosis was made by trained pediatricians.
The selection criterion for RSV disease severity was based on a clinically relevant endpoint: previously healthy full-term infants with bronchiolitis were recruited to the study if their oxygen saturation upon enrollment was lower than 93% when breathing room air. Control subjects included infants with oxygen saturation at or above 93% while breathing room air. In addition, these criteria allow comparison with numerous previous studies (15–19).
Exclusion criteria included known or suspected impairment of immunological function, major congenital oral malformations, chronic lung disease, cardiac disease, prematurity (gestational age of less than 37 weeks), neuromuscular disorders affecting swallowing, and known or suspected coagulation disorders or bleeding tendency. Epidemiological data reported by parents or guardians and clinical data by physical examination were collected by a pediatric investigator. Epidemiological information included age, gender, breastfeeding at the time of enrollment, smoking by others in the home, day-care attendance, parental history of asthma, and presence of siblings younger than 14 years of age in the household. Clinical information included need for and duration of oxygen supplementation, need for and duration of hospitalization, need for intensive care, and death. Investigators monitored the clinical evolution of participating infants through phone calls and/or hospital visits up to 7 days after enrollment. Therefore, any change in the clinical status of the infants was accounted for in the study (i.e., worsening clinical condition converting a control into a case).
Collection and testing of nasal aspirates.
Nasal secretions were obtained from all infants at the time of enrollment by nasal aspiration using 1 ml sterile saline solution (Analyticals; ChemIT). Nasal aspirates (2 ml) were immediately aliquoted, snap frozen in dry ice, and transported to the laboratory. The initial diagnosis of RSV infection was made by direct immunofluorescence assay (Light Biodiagnostic). Results were immediately reported to the participating pediatricians. Infants with positive RSV tests were asked to return for a second visit 5–9 days after enrollment. Aliquots of nasal aspirates were stored at –80°C for cytokine, mRNA, and viral titer determinations. Viral titers were quantified in nasal aspirates using real-time PCR (RT-PCR). Briefly, RNA was extracted using an RNeasy Mini Kit (QIAGEN). RT-PCR was performed using an RSV TaqMan probe with the sequence 6FAMCAATGATCATGATTTACCTATTGMGBNFQ. Viral titers were estimated using a standard curve with known concentrations of RSV.
Collection and processing of blood samples (2003–2006).
Blood was collected from all participating infants at the time of enrollment by venipuncture in the first population (nasal aspirates were used for genotyping in the 2010–2013 population). Blood samples were transferred to the central laboratory and 500 μl stored at –80°C for genotyping.
Assessment of LPS environmental levels.
Samples from cradle and bed sheets from the bedroom of participating infants (2003–2006) were collected in fiberglass filters. LPS was assessed by an LAL Test Endotoxin Assay (Laboratorio Dr. Rapela) in sample dilutions. Results are reported as EU/ml (endotoxin units). A similar procedure was used to assess LPS levels in a convenience sample from 120 homes from the 2010–2013 cohort.
Identification of bacteria in NP aspirates.
DNA was isolated with the QIAmp DNA Kit (QIAGEN) in NP aspirates, and DNA concentrations were determined using a NanoDrop (Thermo Scientific). S. pneumoniae, H. influenzae, and M. catarrhalis were analyzed by RT-PCR using the StepOnePlus Real-Time PCR system (Invitrogen) with the following sequences: S. pneumoniae forward primer, GCTGTTTTAGCAGATAGTGAGATCGA; S. pneumoniae reverse primer, TCCCAGTCGGTGCTGTCA; S. pneumoniae probe, 5′-FAMAATGTTACGCAACTGACGAGMGBNFQ1-3′; M. catarrhalis forward primer, GTGAGTGCCGCTTTACAACC; M. catarrhalis reverse primer, TGTATCGCCTGCCAAGACAA; M. catarrhalis probe, 5′-FAMTGCTTTCAGCTGTTAGCCAGCCTAAMGBNFQ1-3′; H. influenzae forward primer, CCAGCTGCTAAAGTATTAGTAGAAG; H. influenzae reverse primer, TTCACCGTAAGATACTGTGCC; and H. influenzae probe, 5′-FAMCAGATGCAGTTGAAGGTTATTTAGMGBNFQ1-3′ (all from Applied Biosystems).
Cytokine determinations.
IL-6, IL-8, IL-1β, and TNF-α levels were measured in nasal aspirates obtained upon enrollment of the patients using a microbead array (BD). IL-4, IFN-γ, IL-5, IL-9, IL-13, and IL-17 levels in nasal aspirates were assessed in aliquots of available samples using BioSource ELISA kits following the manufacturer’s instructions.
GATA3, Tbet, and TLR4 mRNA expression in nasal secretions.
GATA3, Tbet, TLR4, CD14, MD2, and TLR2 mRNA expression was assessed by RT-PCR (Applied Biosystems) in nasal aspirates from 120 randomly selected subjects from all study groups. RNA was extracted using the RNeasy Mini Kit (QIAGEN). cDNA synthesis was performed by SuperScript First-Strand cDNA Synthesis Assay (Invitrogen) and expression assayed by TaqMan (Invitrogen). β-Actin mRNA expression was used as a housekeeping gene (Applied Biosystems).
Genotyping.
DNA was isolated from whole-blood samples using the Gentra Puregene kit (Gentra Systems) and characterized for purity and concentration on a Beckman Coulter DU640 spectrophotometer. Low-yield samples were whole-genome amplified with the REPLI-g kit (QIAGEN). TLR4 Asp299Gly and Ile399Thr mutations were assessed by allelic discrimination with preoptimized assays from Applied Biosystems.
Briefly, the TLR4 polymorphic sites were amplified from 10 ng genomic DNA, 12.5 μl TaqMan Genotyping Master Mix, and 1.25 μl of a ×20 preoptimized assay mix containing C_11722238 (Asp299Gly, rs4986790) or C_11722237 (Ile399Thr, rs4986791). Each ×20 mix consisted of 18 μM forward and reverse primers and 8 μM of each allele-specific fluorescence-labeled probe. Standard PCR cycling conditions were used, with initial denaturation at 95°C for 10 minutes, followed by 35 cycles at 92°C for 15 seconds and 60°C for 1 minute. Allele-specific PCR products were detected on an ABI 7000 (Applied Biosystems) and clustered by genotype using ABI 7000 sequence detection software. Ambiguous samples were clustered manually and verified by sequencing. Control samples with a known genotype, as verified by DNA sequencing, were run with each plate. Comparison of IL-6 levels in respiratory secretions from RSV-infected infants with 2 major TLR4 alleles with infants with 1 TLR4 D299G allele showed significantly lower cytokine levels in secretions from infants with a minor allele. No difference in TNF-α or IL-1β levels was observed between WT TLR4 and heterozygous infants (not shown). No difference in the time from initiation of symptoms to sampling was observed (not shown).
CD14 C-159T, CD14 C-550T, and IL4 C-590T mutations were analyzed by RFLP.
Polymorphic sites were amplified from 50 ng genomic DNA, 12.5 I FailSafe buffer (Epicentre Biotechnologies), 1 μM forward and reverse primers, and 0.25 μl Taq polymerase enzyme mix (Epicentre Biotechnologies). The resulting PCR products were digested with a specific restriction enzyme (New England Biolabs) at 37°C for 4 hours and run on 3% agarose gels for fragment detection. Control samples with a known genotype, as verified by DNA sequencing, were run with each plate. Supplemental Table 11 lists the FailSafe buffers, primer sequences, and restriction enzymes used for the RFLP assays.
For quality control, assays were repeated on 5% of the samples, and results were 100% concordant. In addition, because of the potential ethnic diversity in our study population, 33 ethnicity-specific genomic markers were evaluated on the Sequenom iPLEX platform by BioServe to rule out admixture.
Second population (2010–2013).
The same recruitment criteria were used as in 2003–2006 for infants at hospitals caring for middle-income, insured families (Swiss Medical Center, CEMIC, Hospital Español) in the central region and at public institutions caring for low-income families (Hospital Pedro de Elizalde) in the southern region. Genotyping was performed in nasal aspirates.
Mice.
Seven-day-old C57BL/10 mice and C57BL/10 mice heterozygous for Tlr4lps–del (Tlr4+/– mice) (The Jackson Laboratory) were inoculated with 100 μg LPS or placebo every 2 days for 14 days and challenged i.n. with 106 PFU RSV line 19 at 28 days of age. Unlike RSV A2, line 19 induces dose-dependent AHR in mice (59). Four-week-old BALB/c (WT, Stat 6–/–, and Stat 1–/–) mice and tamoxifen-treated C57BL/6 WT and C57BL/6 mice conditionally deficient for GATA3 (GATA3fl/fl) were infected with RSV line 19 nine days later to evaluate the role of transcription factors in RSV bronchiolitis.
AHR to increasing concentrations of methacholine (mCh) was determined as previously described (60). Briefly, 5 days after challenge, we anesthetized the mice with a mix of ketamine (100 mg/kg) and xilazine (10 mg/kg) i.p., intubated them, and ventilated them at a rate of 120 breaths per minute with a constant tidal volume of air (0.2 ml). We then paralyzed the mice with decamethonium bromide (25 μg/kg) and, after establishing a stable airway pressure, determined airway resistance to aerosolized mCh (0.01–30 mg/kg). Immunoassays for inflammatory cytokines in BAL fluids were determined using kits from Biosource, following the manufacturer’s instructions.
Statistics.
Data were analyzed using the χ2 test or Fisher’s exact test for proportions. Continuous variables were analyzed using 2-tailed Student’s t, Mann-Whitney U, or ANOVA tests, where appropriate. For categorical outcomes, logistic regression was performed using models selected a priori. The Hardy-Weinberg equilibrium was calculated for every SNP examined. Statistical analyses were performed using a Stata 11.2 package for IBM-PC (StataCorp) and SAS 9.3 (SAS Institute Inc.). A P value of less than 0.05 was considered significant.
Study approval.
The study protocol was approved by the IRBs of Hospital Francés; Evita Pueblo de Berazategui; Hospital Posadas; Hospital Mi Pueblo de Florencio Varela; Maternidad Suizo-Argentina in Argentina; and Vanderbilt University. Written informed consent was obtained from the parents or guardians of all patients. All murine studies were approved by the IACUC of Vanderbilt University.
Supplementary Material
Acknowledgments
This work was supported by grants from the Bill & Melinda Gates Foundation (to F.P. Polack); the Intramural Research Program of the NIH (to U.J. Buchholz); the NIEHS, Department of Health and Human Services (to S.R. Kleeberger, J. Marzec, and M. Shi); a Director’s Challenge Award from the NIEHS (to F.P. Polack and S.R. Kleeberger); the Intramural Research Program of the NIAID, NIH (to U.J. Buchholz); and by grants AI-054952 (to F.P. Polack) and AI-104853 (to F.P. Polack) from the NIAID, NIH.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exits.
Reference information:J Clin Invest. 2015;125(2):571–582. doi:10.1172/JCI75183.
References
- 1. Collins PL, Chanock RM, Murphy BR. Respiratory syncytial virus disease. In: Knipe D, et al., eds. Fields Virology. 4th ed. Vol.1. Philadelphia, Pennsylvania, USA: Lippincott Williams & Wilkins; 2001:1443–1486. [Google Scholar]
- 2.Simoes EA. Environmental and demographic risk factors for respiratory syncytial virus lower respiratory tract disease. J Pediatr. 2003;143(5 suppl):S118–S126. doi: 10.1067/s0022-3476(03)00511-0. [DOI] [PubMed] [Google Scholar]
- 3.Ferolla FM, et al. Macronutrients during pregnancy and life-threatening respiratory syncytial virus infections in children. Am J Respir Crit Care Med. 2013;187(9):983–990. doi: 10.1164/rccm.201301-0016OC. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman SJ, Laham FR, Polack FP. Mechanisms of illness during respiratory syncytial virus infection: the lungs, the virus and the immune response. Microbes Infect. 2004;6(8):767–772. doi: 10.1016/j.micinf.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 5.Karron RA, et al. Identification of a recombinant live attenuated respiratory syncytial virus vaccine candidate that is highly attenuated in infants. J Infect Dis. 2005;191(7):1093–1104. doi: 10.1086/427813. [DOI] [PubMed] [Google Scholar]
- 6.Hull J, Thomson A, Kwiatkowski D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax. 2000;55(12):1023–1027. doi: 10.1136/thorax.55.12.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Openshaw PJ. Immunity and immunopathology to respiratory syncytial virus. The mouse model. Am J Respir Crit Care Med. 1995;152(4 pt 2):S59–62. doi: 10.1164/ajrccm/152.4_Pt_2.S59. [DOI] [PubMed] [Google Scholar]
- 8.McNamara PS, Flanagan BF, Baldwin LM, Newland P, Hart CA, Smyth RL. Interleukin 9 production in the lungs of infants with severe respiratory syncytial virus bronchiolitis. Lancet. 2004;363(9414):1031–1037. doi: 10.1016/S0140-6736(04)15838-8. [DOI] [PubMed] [Google Scholar]
- 9.Hoebee B, et al. Association of severe respiratory syncytial virus bronchiolitis with interleukin-4 and interleukin-4 receptor α polymorphisms. J Infect Dis. 2003;187(1):2–11. doi: 10.1086/345859. [DOI] [PubMed] [Google Scholar]
- 10.Webb DC, Mahalingam S, Cai Y, Matthaei KI, Donaldson DD, Foster PS. Antigen-specific production of interleukin (IL)-13 and IL-5 cooperate to mediate IL-4Rα-independent airway hyperreactivity. Eur J Immunol. 2003;33(12):3377–3385. doi: 10.1002/eji.200324178. [DOI] [PubMed] [Google Scholar]
- 11.Legg JP, Hussain IR, Warner JA, Johnston SL, Warner JO. Type 1 and type 2 cytokine imbalance in acute respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med. 2003;168(6):633–639. doi: 10.1164/rccm.200210-1148OC. [DOI] [PubMed] [Google Scholar]
- 12.Kurt-Jones EA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1(5):398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 13.Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol. 2001;75(22):10730–10737. doi: 10.1128/JVI.75.22.10730-10737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehl S, et al. The role of Toll-like receptor 4 versus interleukin-12 in immunity to respiratory syncytial virus. Eur J Immunol. 2004;34(4):1146–1153. doi: 10.1002/eji.200324449. [DOI] [PubMed] [Google Scholar]
- 15.Tal G, et al. Association between common Toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J Infect Dis. 2004;189(11):2057–2063. doi: 10.1086/420830. [DOI] [PubMed] [Google Scholar]
- 16.Inoue Y, et al. CD14 -550 C/T, which is related to the serum level of soluble CD14, is associated with the development of respiratory syncytial virus bronchiolitis in the Japanese population. J Infect Dis. 2007;195(11):1618–1624. doi: 10.1086/516790. [DOI] [PubMed] [Google Scholar]
- 17.Puthothu B, Forster J, Heinzmann A, Krueger M. TLR-4 and CD14 polymorphisms in respiratory syncytial virus associated disease. Dis Markers. 2006;22(5-6):303–308. doi: 10.1155/2006/865890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paulus SC, Hirschfeld AF, Victor RE, Brunstein J, Thomas E, Turvey SE. Common human Toll-like receptor 4 polymorphisms — role in susceptibility to respiratory syncytial virus infection and functional immunological relevance. Clin Immunol. 2007;123(3):252–257. doi: 10.1016/j.clim.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 19.Kresfelder TL, Janssen R, Bont L, Pretorius M, Venter M. Confirmation of an association between single nucleotide polymorphisms in the VDR gene with respiratory syncytial virus related disease in South African children. J Med Virol. 2011;83(10):1834–1840. doi: 10.1002/jmv.22179. [DOI] [PubMed] [Google Scholar]
- 20.Smit LA, et al. CD14 and toll-like receptor gene polymorphisms, country living, and asthma in adults. Am J Respir Crit Care Med. 2009;179(5):363–368. doi: 10.1164/rccm.200810-1533OC. [DOI] [PubMed] [Google Scholar]
- 21.Martinez FD. Gene-environment interactions in asthma: with apologies to William of Ockham. Proc Am Thorac Soc. 2007;4(1):26–31. doi: 10.1513/pats.200607-144JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arbour NC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25(2):187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 23.Braun-Fahrlander C, et al. Environmental exposure to endotoxin and its relation to asthma in school-age children. N Engl J Med. 2002;347(12):869–877. doi: 10.1056/NEJMoa020057. [DOI] [PubMed] [Google Scholar]
- 24.Larsson L, et al. Identification of bacterial and fungal components in tobacco and tobacco smoke. Tob Induc Dis. 2008;4:4. doi: 10.1186/1617-9625-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malley R, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A. 2003;100(4):1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, et al. Toll-like receptor 4 mediates innate immune responses to Haemophilus influenzae infection in mouse lung. J Immunol. 2002;168(2):810–815. doi: 10.4049/jimmunol.168.2.810. [DOI] [PubMed] [Google Scholar]
- 27.Hassan F, Ren D, Zhang W, Merkel TJ, Gu XX. Moraxella catarrhalis activates murine macrophages through multiple toll like receptors and has reduced clearance in lungs from TLR4 mutant mice. PLoS One. 2012;7(5):e37610. doi: 10.1371/journal.pone.0037610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waser M, et al. Determinants of endotoxin levels in living environments of farmers’ children and their peers from rural areas. Clin Exp Allergy. 2004;34(3):389–397. doi: 10.1111/j.1365-2222.2004.01873.x. [DOI] [PubMed] [Google Scholar]
- 29.Thorne PS, Cohn RD, Mav D, Arbes SJ, Zeldin DC. Predictors of endotoxin levels in U.S. housing. Environ Health Perspect. 2009;117(5):763–771. doi: 10.1289/ehp.11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferwerda B, et al. TLR4 polymorphisms, infectious diseases, and evolutionary pressure during migration of modern humans. Proc Natl Acad Sci U S A. 2007;104(42):16645–16650. doi: 10.1073/pnas.0704828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simpson A, et al. Endotoxin exposure, CD14, and allergic disease: an interaction between genes and the environment. Am J Respir Crit Care Med. 2006;174(4):386–392. doi: 10.1164/rccm.200509-1380OC. [DOI] [PubMed] [Google Scholar]
- 32.Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3(1):36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 33.Perros F, Lambrecht BN, Hammad H. TLR4 signalling in pulmonary stromal cells is critical for inflammation and immunity in the airways. Respir Res. 2011;12:125. doi: 10.1186/1465-9921-12-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196(12):1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glimcher LH. Trawling for treasure: tales of T-bet. Nat Immunol. 2007;8(5):448–450. doi: 10.1038/ni0507-448. [DOI] [PubMed] [Google Scholar]
- 36.Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE. GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 2006;16(1):3–10. doi: 10.1038/sj.cr.7310002. [DOI] [PubMed] [Google Scholar]
- 37.Robinson DS, O’Garra A. Further checkpoints in Th1 development. Immunity. 2002;16(6):755–758. doi: 10.1016/S1074-7613(02)00331-X. [DOI] [PubMed] [Google Scholar]
- 38.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4(3):313–319. doi: 10.1016/S1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 39.Wright PF, et al. Illness severity, viral shedding, and antibody responses in infants hospitalized with bronchiolitis caused by respiratory syncytial virus. J Infect Dis. 2002;185(8):1011–1018. doi: 10.1086/339822. [DOI] [PubMed] [Google Scholar]
- 40.Corneli HM, et al. A multicenter, randomized, controlled trial of dexamethasone for bronchiolitis. N Engl J Med. 2007;357(4):331–339. doi: 10.1056/NEJMoa071255. [DOI] [PubMed] [Google Scholar]
- 41.Sheeran P, et al. Elevated cytokine concentrations in the nasopharyngeal and tracheal secretions of children with respiratory syncytial virus disease. Pediatr Infect Dis J. 1999;18(2):115–122. doi: 10.1097/00006454-199902000-00007. [DOI] [PubMed] [Google Scholar]
- 42.Welliver TP, et al. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J Infect Dis. 2007;195(8):1126–1136. doi: 10.1086/512615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laham FR, et al. Differential production of inflammatory cytokines in primary infection with human metapneumovirus and with other common respiratory viruses of infancy. J Infect Dis. 2004;189(11):2047–2056. doi: 10.1086/383350. [DOI] [PubMed] [Google Scholar]
- 44.Ripple MJ, et al. Immunomodulation with IL-4R α antisense oligonucleotide prevents respiratory syncytial virus-mediated pulmonary disease. J Immunol. 2010;185(8):4804–4811. doi: 10.4049/jimmunol.1000484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perumal NB, Kaplan MH. Regulating Il9 transcription in T helper cells. Trends Immunol. 2011;32(4):146–150. doi: 10.1016/j.it.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells--how did we miss them? Nat Rev Immunol. 2013;13(2):75–87. doi: 10.1038/nri3349. [DOI] [PubMed] [Google Scholar]
- 47.Marr N, Turvey SE. Role of human TLR4 in respiratory syncytial virus-induced NF-κB activation, viral entry and replication. Innate Immun. 2012;18(6):856–865. doi: 10.1177/1753425912444479. [DOI] [PubMed] [Google Scholar]
- 48.Hall CB, et al. The burden of respiratory syncytial virus infection in young children. N Engl J Med. 2009;360(6):588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.[No authors listed]. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. The IMpact-RSV Study Group. Pediatrics. 1998;102(3 pt 1):531–537. [PubMed] [Google Scholar]
- 50.Awomoyi AA, et al. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J Immunol. 2007;179(5):3171–3177. doi: 10.4049/jimmunol.179.5.3171. [DOI] [PubMed] [Google Scholar]
- 51.Wenzel S, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368(26):2455–2466. doi: 10.1056/NEJMoa1304048. [DOI] [PubMed] [Google Scholar]
- 52.Hall CB. Respiratory syncytial virus in young children. Lancet. 2010;375(9725):1500–1502. doi: 10.1016/S0140-6736(10)60401-1. [DOI] [PubMed] [Google Scholar]
- 53.Nair H, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet. 2010;375(9725):1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prince LS, Dieperink HI, Okoh VO, Fierro-Perez GA, Lallone RL. Toll-like receptor signaling inhibits structural development of the distal fetal mouse lung. Dev Dyn. 2005;233(2):553–561. doi: 10.1002/dvdy.20362. [DOI] [PubMed] [Google Scholar]
- 55.Soutiere SE, Tankersley CG, Mitzner W. Differences in alveolar size in inbred mouse strains. Respir Physiol Neurobiol. 2004;140(3):283–291. doi: 10.1016/j.resp.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 56.Gereda JE, et al. Relation between house-dust endotoxin exposure, type 1 T-cell development, and allergen sensitisation in infants at high risk of asthma. Lancet. 2000;355(9216):1680–1683. doi: 10.1016/S0140-6736(00)02239-X. [DOI] [PubMed] [Google Scholar]
- 57.Gereda JE, Klinnert MD, Price MR, Leung DY, Liu AH. Metropolitan home living conditions associated with indoor endotoxin levels. J Allergy Clin Immunol. 2001;107(5):790–796. doi: 10.1067/mai.2001.115245. [DOI] [PubMed] [Google Scholar]
- 58.Rosenthal N, Schwartz RS. In search of perverse polymorphisms. N Engl J Med. 1998;338(2):122–124. doi: 10.1056/NEJM199801083380210. [DOI] [PubMed] [Google Scholar]
- 59.Lukacs NW, et al. Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am J Pathol. 2006;169(3):977–986. doi: 10.2353/ajpath.2006.051055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delgado MF, et al. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat Med. 2009;15(1):34–41. doi: 10.1038/nm.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.