

Abstract
For a targeted cancer vaccine to be effective, the antigen of interest needs to be naturally processed and presented on MHC by the target cell or an antigen-presenting cell (APC) in the tumor stroma. The presence of these characteristics is often assumed based on animal models, evaluation of antigen-overexpressing APCs in vitro, or assays of material-consuming immune precipitation from fresh solid tissue. Here, we evaluated the use of an alternative approach that uses the proximity ligation assay (PLA) to identify the presentation of an MHC class II–restricted antigen in paraffin-embedded tissue sections from patients with brain tumors. This approach required a specific antibody directed against the epitope that was presented. We used an antibody that specifically binds an epitope of mutated isocitrate dehydrogenase type 1 (IDH1R132H), which is frequently expressed in gliomas and other types of tumors. In situ PLA showed that the IDH1R132H epitope colocalizes with MHC class II in IDH1R132H-mutated glioma tissue. Moreover, PLA demonstrated colocalization between the class II epitope-containing melanoma antigen New York esophageal 1 and MHC class II. Collectively, our data suggest that PLA may be a useful tool to acquire information on whether an antigen is presented in situ, and this technique has potential to guide clinical studies that use antigen-specific cancer immunotherapy.
Introduction
Active cancer immunotherapy exploits the patient’s immune system to induce cellular immune responses against the tumor, for instance, by antigen-specific vaccination. Promising strategies in melanoma, renal cell carcinoma, non–small cell lung carcinoma, glioma, and other tumors include DC vaccination using autologous tumor lysate or specific proteins to load DCs ex vivo, peptide vaccination, or adoptive transfer of tumor-specific T cells (1–4). The use of tumor-associated antigens (TAAs) or tumor-specific antigens as targets is considered superior to whole tumor proteome in terms of effective induction of immune responses to overcome tolerance and to preserve integrity of healthy tissue. Vaccination against specific tumor antigens has proven to be a promising tool in therapy for various tumor entities, including gliomas. For instance, a peptide vaccine against tumor-specific neoantigens, such as the EGFR variant III, a constitutively active form that is expressed in 30% of primary gliomas and other tumor entities, is currently tested in phase III clinical trials as an adjunct to radiochemotherapy in patients with newly diagnosed glioblastoma (5–7). Classic TAAs, such as the cancer testis antigen New York esophageal 1 (NY-ESO-1), however, are intracellular proteins requiring proteasomal processing and presentation on MHC molecules to induce an antigen-specific T cell response (8).
To assess MHC binding of putative or known immunogenic epitopes, in silico binding algorithms such as NetMHC or SYFPEITHI have been established (9–12). These analyses may guide subsequent in vitro class I– and class II–binding assays and cellular binding studies, e.g., T2-binding assay. Novel epitopes presented on MHC can be identified by HLA-ligandome analyses, which elute peptides from MHC-peptide complexes from an in vitro system or tumor tissue (13). These analyses have been successfully applied to develop multipeptide vaccines, which are currently in clinical trials in patients with various cancers (e.g., IMA 950, ClinicalTrials.gov NCT01920191). All of these methods are limited in that they have been mostly developed for MHC class I–binding studies and are HLA-type restricted. Binding algorithms at present have limited reliability for MHC class II, and class II peptide complexes are rather instable and therefore difficult to analyze. Some of the tests are very expensive, time-consuming, or require freshly isolated tumor tissue. Moreover, they can rarely provide information about the processing and the cellular context in which MHC class II and peptide interaction takes place in situ.
We recently reported the immunogenicity of mutant isocitrate dehydrogenase type 1 (IDH1R132H) in MHC-humanized HLA-A*0201 HLA-DRA*0101 HLA-DRB1*0101 transgenic mice devoid of mouse MHC (A2.DR1 mice) and IDH1R132H-specific spontaneous immune responses in glioma patients with IDH1-mutated tumors in a CD4 T cell– and MHC class II–restricted manner (14). Vaccination of MHC-humanized A2.DR1 mice with a 20-mer peptide encompassing the IDH1R132H mutation resulted in control of established syngeneic IDH1R132H-positive sarcomas (14). In this context, evidence that an immunogenic IDH1R132H peptide is processed from the mutated IDH1 protein and presented on MHC class II molecules in vivo comes from whole tumor cell vaccination of A2.DR1 mice. Vaccination with irradiated tumor cells overexpressing IDH1R132H led to specific immunologic recognition of the IDH1R132H epitope (14).
Here, we approach the question of whether an IDH1R132H epitope is presented on HLA-DR in human glioma tissue. To this end, proximity ligation assay (PLA), which has until now been used as a tool to visualize endogenous protein-protein interactions or fusion proteins in situ (15, 16), was adopted to analyze binding of IDH1R132H-derived peptides to MHC class II molecules in vitro and in situ. For this purpose, an IDH1R132H-specific monoclonal antibody was used together with an HLA-DR–specific antibody. Specificity and sensitivity were validated in vitro using an HLA-DR–expressing and IDH1R132H-transfected glioma cell line as well as a primary IDH1R132H-positive glioma cell line and extended to another well-studied TAA, NY-ESO-1. Finally, we show that PLA is applicable to specifically identify colocalization of IDH1R132H peptides with HLA-DR in glioma tissue.
Results
MHC class II–restricted immunogenicity of IDH1R132H.
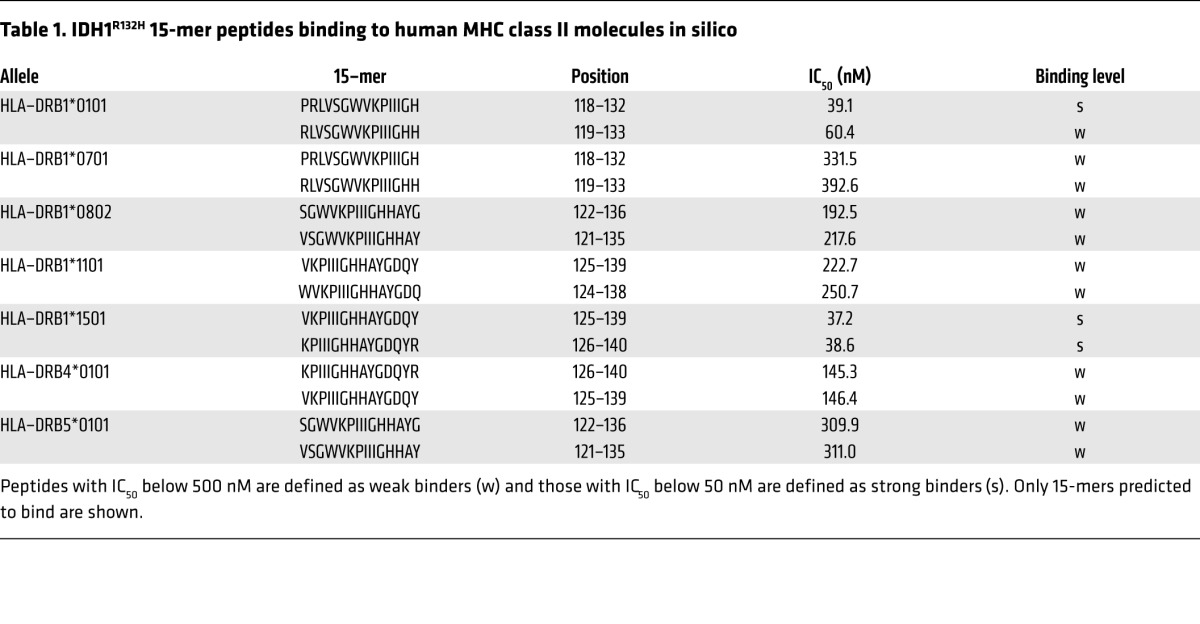
The IDH1R132H mutant protein is expressed in about 80% of diffuse grade II and III gliomas, defining a distinct glioma subtype (17–19). To assess putative immunogenicity of IDH1R132H in a human MHC class II context, epitopes were identified in silico by MHC peptide-binding predictions. Human IDH1R132H 29-mer peptide (amino acids 118–146) PRLVSGWVKPIIIGHHAYGDQYRATDFVV includes all possible processed IDH1R132H 15-mers covering the point mutation at position 132. In silico peptide-binding algorithms predicted IDH1R132H 15-mer binding to human MHC class II molecules in an HLA-DR type–dependent manner (Table 1).
Table 1.
IDH1R132H 15-mer peptides binding to human MHC class II molecules in silico
The IDH1R132H-specific antibody is suitable to detect HLA-DR epitopes.
We then sought to address IDH1R132H peptide presentation in vitro using PLA. PLA has been developed for protein-protein interaction analysis in genetically unmodified cells and tissues by applying specific primary and secondary antibodies, the latter attached to short DNA strands allowing rolling circle PCR amplification in case of close proximity, and consequently, fluorescent visualization of colocalization of native proteins in situ. Thus, in principle, this technique is applicable for the analysis of natural peptide processing, as it does not require structural modifications of the system by exogenous expression or insertion of fluorescent labels. In order to use the PLA for epitope presentation on MHC, a specific antibody detecting the epitope of interest is required. The high incidence of the IDH1R132H point mutation led to the development of a mutation-specific monoclonal antibody (H09) (20, 21), which has routine application for histological diagnostics. This mouse antibody has been generated by immunization with synthetic peptide IDH1R132H p125–137 CKPIIIGHHAYGD coupled to keyhole limpet hemocyanin and allows specific staining of diffuse infiltrating single tumor cells in gliomas (Figure 1A). For identification of putative HLA-DR epitopes that are detectable by this antibody, a peptide library was generated encompassing the IDH1R132H region (Figure 1B). Peptide-based ELISA demonstrated that the anti-IDH1R132H antibody binds to the IDH1-mutated 15-mers p122–136, p124–138, and p126–140 and the 20-mer p123–p142, whereas no binding was seen for any of the IDH1WT peptides or IDH1R132H peptides with a peripheral position of the amino acid exchange (Figure 1C). Specific binding of an IDH1R132H peptide by the used IDH1R132H-specific antibody while this peptide is bound to HLA-DR is a prerequisite for the analysis of the peptide–MHC class II interaction. We sought to address this question by an immune-competitive ELISA approach, using p123–142 IDH1R132H-loaded class II (DR1) tetramers (Figure 1D). We have used this tetramer previously to identify IDH1R132H-specific CD4+ T cells (14). Preincubation of the IDH1R132H-specific antibody (H09) with IDH1R132H-DR1 tetramer but not with control CLIP-DR1 tetramer resulted in complete inhibition of specific antibody binding to plate-coated IDH1R132H peptide p122–136 in the ELISA (Figure 1E). This result supports the hypothesis that an IDH1R132H-specific antibody recognizes an unmasked IDH1R132H epitope that is bound to MHC class II molecules.
Figure 1. Binding of soluble and MHC class II–bound IDH1R132H 15-mer and 20-mer peptides by anti-IDH1R132H antibody (H09) in peptide-based ELISA.

(A) IDH1R132H IHC using H09 on glioma tissue p001 and p018. A high magnification image of the boxed area is shown at right. Scale bar: 40 μm (left and center); 8 μm (right). (B) IDH1 15-mer and 20-mer peptide library encompassing amino acid 132. (C) IDH1 15-mer and 20-mer peptide-based H09 ELISA. MOG, negative control; DMSO, vehicle control. One-way ANOVA, Tukey corrected, n = 3. (D and E) MHC class II–bound IDH1R132H p122–136 peptide-based ELISA plates. H09 (1°) was preincubated with specific (red, IDH1R132H–HLA-DR1) and control (black, CLIP–HLA-DR1) tetramer (4-mer) and subsequently subjected to p122–136 IDH1R132H-based ELISA. 2°, secondary antibody. pIDH1R132H, p122–136 IDH1R132H. One-way ANOVA, Tukey corrected, n = 3 (E). ***P < 0.001.
Establishment of an in vitro system to detect IDH1R132H–MHC class II colocalization.
With the rationale that IDH1R132H peptides can be bound by MHC class II molecules and having verified that the IDH1R132H-specific antibody H09 can bind an MHC class II–bound epitope, we aimed to detect IDH1R132H epitope presentation on MHC class II molecules using PLA with the IDH1R132H-specific antibody and an antibody directed against the α-chain of the HLA-DR dimer (Figure 2A). Because the HLA-DR α-chain is conserved throughout HLA-DR types (22), using an anti–HLA-DR (α-chain) antibody broadens the spectrum of applicability in this system. To evaluate the applicability and specificity of the PLA for detection of IDH1R132H epitope presentation on HLA-DR, we established an in vitro system in the human glioma cell line LN229. LN229 cells endogenously express HLA-DR in a homogenous manner, as shown by single-cell analysis by flow cytometry and immunofluorescence (IF) (Figure 2B). Of note, HLA typing of these cells revealed the homozygous genotype HLA-DRB1*01 (data not shown). LN229 cells, which are IDH1WT, were stably transduced with the double mutant IDH1D252G R132H (Figure 2C). We introduced the point mutation at position 252 to abolish the neomorphic enzymatic activity of IDH1R132H. IDH1R132H produces R-2-hydroxyglutarate (R-2-HG), which can be cytotoxic when accumulating to high concentrations. Thus, IDH1D252G R132H double mutants show increased IDH1R132H expression without toxicity. Little R-2-HG was produced by LN229 transfected with IDH1D252G R132H (Figure 2D) in spite of stable IDH1D252G R132H protein expression (Figure 2, E and F; see complete unedited blots in the supplemental material; supplemental material available online with this article; doi:10.1172/JCI77780DS1). Therefore, a suitable in vitro system, together with specific tools, was established for use in IDH1R132H–HLA-DR PLA.
Figure 2. HLA-DR expression and overexpression of mutated and wild-type IDH1 in glioma cell line LN229 as analyzed by PLA, a tool for analysis of MHC class II–peptide interaction.

(A) PLA scheme using anti–HLA-DRA and H09 primary antibodies. Red represents rolling circle amplification, and α and β represent HLA-DR chains. pIDH, IDH1 epitopic peptide. (B) Flow cytometry and IF (IDH1D252G R132H) of glioma cell line LN229. Green, HLA-DR. Data are representative of 3 experiments. DG RH, IDH1D252G R132H; DG, IDH1D252G; iso, isotype control; specific, HLA-DRA–specific antibody. Scale bar: 20 μm. (C) Scheme for enzymatic activity of IDH1 mutants. α-KG, α-ketoglutarate. (D) R-2-HG measurement in IDH1D252G R132H, IDH1D252G, IDH1R132H (RH), and IDH1WT (WT) LN229 cells by enzymatic assay. Data are representative of 3 experiments. (E) Western blot and (F) and IF of LN229 cells overexpressing IDH1D252G or IDH1D252G R132H detecting IDH1. Tubulin was used as loading control. Green, IDH1R132H; blue, DAPI. Data are representative of 3 experiments. EV, empty vector. Scale bar: 20 μm.
IDH1R132H–MHC class II colocalization can be detected by PLA in vitro.
Using this in vitro system, LN229 cells transfected with IDH1D252G R132H and IDH1D252G were subjected to PLA for detection of colocalization of IDH1R132H with MHC class II. In IDH1D252G R132H-overexpressing LN229 cells, but not IDH1D252G-overexpressing cells, an epitope-specific proximity of MHC class II (HLA-DR) and IDH1R132H peptide was detected (Figure 3A). An HLA-DR–specific knockdown abolished the PLA signal in IDH1D252G R132H-overexpressing LN229 cells, confirming that PLA positivity depends on HLA-DR expression in LN229 cells (Figure 3, B and C, and Supplemental Figure 1). These results show that PLA is suitable to specifically detect colocalization of IDH1R132H epitopes and MHC class II HLA-DR and suggest that the HLA-DR–positive LN229 glioma cells are able to present these epitopes on HLA-DR. Immunofluorescent costaining with the IDH1R132H-specific antibody H09 indicated a correlation of IDH1R132H expression level and the PLA signal (Figure 3D). To further address the question of the correlation between IDH1R132H expression level and PLA intensity in detail, we made use of the LN229 glioma cell line, IVE2, which retrovirally expresses an enzymatically active IDH1R132H. IDH1R132H expression in this cell line is heterogeneous on the single-cell level (Supplemental Figure 2), thereby allowing for identification of a correlation between IDH1R132H expression level and PLA signal. Additionally, the R-2-HG–producing IDH1R132H is a less artificial system that excludes the possibility that a PLA signal is only detectable when high levels of antigen are expressed and that the introduction of the second point mutation (D252G) might influence protein processing and epitope presentation. PLA sensitivity was sufficient to visualize colocalization of IDH1R132H and HLA-DR in R-2-HG–producing LN229 IVE2 cells, which was not restricted to the cell membrane but localized within the cell, as shown by costaining of the membrane marker annexin V (annexin), and prevalently found at subcellular sites with a high density of intracellular vesicles, as shown by costaining of the endosomal-lysosomal marker Rab7 (Supplemental Figure 2). Performing PLA with IDH1R132H costaining on these cells, we measured the mean fluorescent intensity (MFI) of IDH1R132H expression and quantified PLA signals using an ImageJ algorithm (Supplemental Figure 3). Cumulative analysis of 20 images revealed a significant positive correlation between IDH1R132H expression level and PLA signal (Figure 3E).
Figure 3. Specific colocalization of IDH1R132H peptide and MHC class II in IDH1 mutant-overexpressing glioma cell line LN229 in vitro.

(A) IDH1R132H–HLA-DR PLA on IDH1D252G R132H-overexpressing LN229 cells (LN229 DG RH) and IDH1D252G-overexpressing LN229 cells (LN229 DG), respectively. Data are representative of 3 experiments. Scale bar: 20 μm. (B) IDH1R132H–HLA-DR PLA on IDH1D252G R132H-overexpressing LN229 cells after treatment with HLA-DRA–specific siRNA (siHLA-DR) or siRNA control pool (siCONTROL). Data are representative of 2 experiments. Scale bar: 15 μm. (C) Quantification of HLA-DRA knockdown in IDH1D252G R132H-overexpressing LN229 cells by flow cytometry. Numbers in the histogram indicate percentages of HLA-DR–positive cells after treatment with siCONTROL or si-HLA-DRA. Data are representative of 2 experiments. Gray, isotype control; red, siCONTROL; and blue, siHLA-DR. (D) IDH1R132H–HLA-DR PLA with IDH1R132H (H09) costaining (green) in IDH1D252G R132H-overexpressing LN229 cells. A high-magnification image of the boxed area is shown below. Data are representative of 3 experiments. Scale bar: 20 μm (top); 10 μm (bottom). (E) Correlation of IDH1R132H expression (MFI) and PLA signal in IDH1R132H-overexpressing LN229 cells; n = 152, Spearman’s rho, r = 0.6352, P < 0.0001. (A, B, and D) Red, PLA signal; blue, DAPI.
PLA demonstrates colocalization of endogenously expressed IDH1R132H in vitro.
Having demonstrated that PLA is in principle suitable to detect colocalization of endogenous MHC class II HLA-DR and the antigen IDH1R132H in a system which is genetically modified, we approached to a clinically more relevant setting in order to evaluate the opportunity to analyze IDH1R132H and HLA-DR colocalization in situ. Therefore, we utilized primary glioma cell lines, which had been established from freshly isolated patient tissue and endogenously express IDH1R132H. PLA sensitivity was sufficient to detect rare events of colocalization of IDH1R132H and HLA-DR in the IDH1R132H-positive and HLA-DR–positive cell line NCH620 (Figure 4A). No PLA signals were detected in the IDH1WT HLA-DR–negative cell line NPH001 or the IDH1R132H-positive HLA-DR–negative cell line NCH645 (Figure 4, B and C, and Supplemental Figure 4). Immunofluorescent costaining of HLA-DR with PLA revealed that only HLA-DR–expressing cells within the sphere culture were positive for PLA (Figure 4D). These results show that PLA can be used as a tool to detect colocalization of endogenous HLA-DR and IDH1R132H in primary cells that are not genetically manipulated. Moreover, PLA on these cells was performed in an immunohistochemical setting on paraffin-embedded glioma cellular spheres, closely resembling an in situ setting.
Figure 4. Specific colocalization of IDH1R132H peptide and MHC class II in primary glioma cell lines.

(A) IDH1R132H–HLA-DR PLA (scale bar: 20 μm [top]; 10 μm [bottom]), IDH1R132H IHC (scale bar: 40 μm [top]; 20 μm [bottom]), and HLA-DR IHC (scale bar: 100 μm [top]; 20 μm [bottom]) on NCH620 cells. High-magnification images of the boxed areas are shown below, with asterisks in boxed regions corresponding to those at high magnification. Data are representative of 2 or 3 experiments, respectively. (B) IDH1R132H–HLA-DR PLA on glioma cell line NPH001 and (C) on glioma cell line NCH645. Scale bar: 20 μm (left); 10 μm (right). High-magnification images of the boxed areas are shown to the right, with asterisks in boxed regions corresponding to those at high magnification. (D) IDH1R132H–HLA-DR PLA on NCH620 cells costained with anti–HLA-DR (TAL1B5). Red, PLA signal; green, HLA-DR; blue, DAPI. Scale bar: 15 μm.
PLA use can be extended to detect colocalization of MHC class II with NY-ESO-1.
To confirm the applicability of PLA as a tool to detect epitope presentation on HLA-DR, we next aimed to extend this finding to an established TAA of known functional relevance. The cancer testis antigen NY-ESO-1 (CTAG1B) represents a suitable intracellular antigen, because it has been shown to be a potent immunogenic target in various studies, inducing not only specific CD8+ T cell–mediated responses but also CD4+ T cell–mediated responses and containing several MHC class II HLA-DR–binding epitopes, e.g., p119–143, p121–138, and p123–137 for HLA-DR1 (23, 24). In order to first use the same in vitro system as for IDH1R132H, we overexpressed myc-tagged NY-ESO-1 in LN229 cells (Figure 5A and Supplemental Figures 5 and 6) and performed PLA in these cells using a specific monoclonal antibody generated against full-length human NY-ESO-1. NY-ESO-1-HLA-DR PLA showed colocalization of NY-ESO-1 and endogenous HLA-DR in NY-ESO-1–overexpressing cells but not in empty vector control cells (Figure 5, B and C). As was done for IDH1R132H, to exclude that the detected interaction between an NY-ESO-1–derived epitope and HLA-DR was a result of the forced overexpression of the antigen, we performed in vitro PLA on endogenously antigen-expressing tumor cells. The melanoma cell line SK-Mel-37 endogenously expressed NY-ESO-1 and HLA-DR (Figure 6, A and B, and Supplemental Figure 6). HLA typing revealed the heterozygous genotype HLA-DRB1*01 and HLA-DRB1*03 (data not shown). PLA demonstrated colocalization of HLA-DR and endogenous NY-ESO-1 in these cells (Figure 6C) but not in NY-ESO-1–negative SK-Mel-23 cells (Supplemental Figure 6). The signal was abolished by siRNA-mediated knockdown of either NY-ESO-1 or HLA-DR specifically in cells that were affected by the knockdown (Figure 6, D–G, and Supplemental Figure 6; see complete unedited blots in the supplemental material), confirming the capability of PLA to detect colocalization of endogenous epitopes presented on MHC class II molecules and supporting its applicability as a tool to detect antigen presentation in vitro, not only in the context of IDH1R132H but in the context of other antigens IDH1R132H is a less artificial system that excludes the possibility relevant for tumor immunotherapy.
Figure 5. Specific colocalization of NY-ESO-1 peptide and MHC class II in NY-ESO-1–overexpressing glioma cell line LN229 in vitro.

(A) IF of LN229 cells stably overexpressing myc-tagged NY-ESO-1 (LN229 NY). Green, NY-ESO-1; red, myc; blue, DAPI. Scale bar: 20 μm. (B and C) NY-ESO-1-HLA-DR PLA on (B) NY-ESO-1–overexpressing LN229 cells and (C) empty vector control cells (LN229 EV) costained with anti-myc. Red, PLA signal; green, myc; blue, DAPI. Scale bar: 20 μm. Data are representative of 3 experiments each.
Figure 6. Specific colocalization of NY-ESO-1 peptide and MHC class II in melanoma cell line SK-Mel-37 endogenously expressing NY-ESO-1 and HLA-DR in vitro.

(A) IF of SK-Mel-37 cells detecting endogenous NY-ESO-1. Green, NY-ESO-1; blue, DAPI. Scale bar: 20 μm. Data are representative of 3 experiments. (B) Flow cytometry of endogenous HLA-DR expression in SK-Mel-37 cells. Specific, HLA-DR–specific antibody. Data are representative of 3 experiments. (C) NY-ESO-1-HLA-DR PLA on SK-Mel-37 cells. A high-magnification image of the boxed area is shown to the right. Red, PLA signal, blue, DAPI. Scale bar: 15 μm (left); 5 μm (right). Data are representative of 3 experiments. (D) IF of SK-Mel-37 cells treated with siRNA control pool (siCONTROL) and NY-ESO-1 siRNA (siNY-ESO-1). Green, NY-ESO-1; blue, DAPI. Scale bar: 20 μm. Experiment was performed once. (E) Quantification of HLA-DR knockdown in SK-Mel-37 cells by flow cytometry. Gray, isotype control. Numbers in the histogram indicate percentages of HLA-DR–positive cells after treatment with siCONTROL or si-HLA-DRA. Experiment was performed once. (F) Western blot of SK-Mel-37 cells detecting HLA-DR and NY-ESO-1 after treatment with siHLA-DRA, siNY-ESO-1, and siCONTROL. Tubulin was used as loading control. Experiment was performed once. (G) NY-ESO-1-HLA-DR PLA on SK-Mel-37 cells after treatment with siCONTROL, siNY-ESO-1, and siHLA-DRA. High-magnification images of the boxed areas are shown to the right. Red, PLA signal; blue, DAPI. Scale bar: 15 μm (left); 5 μm (right). Experiment was performed once.
IDH1R132H–MHC class II PLA demonstrates colocalization in situ.
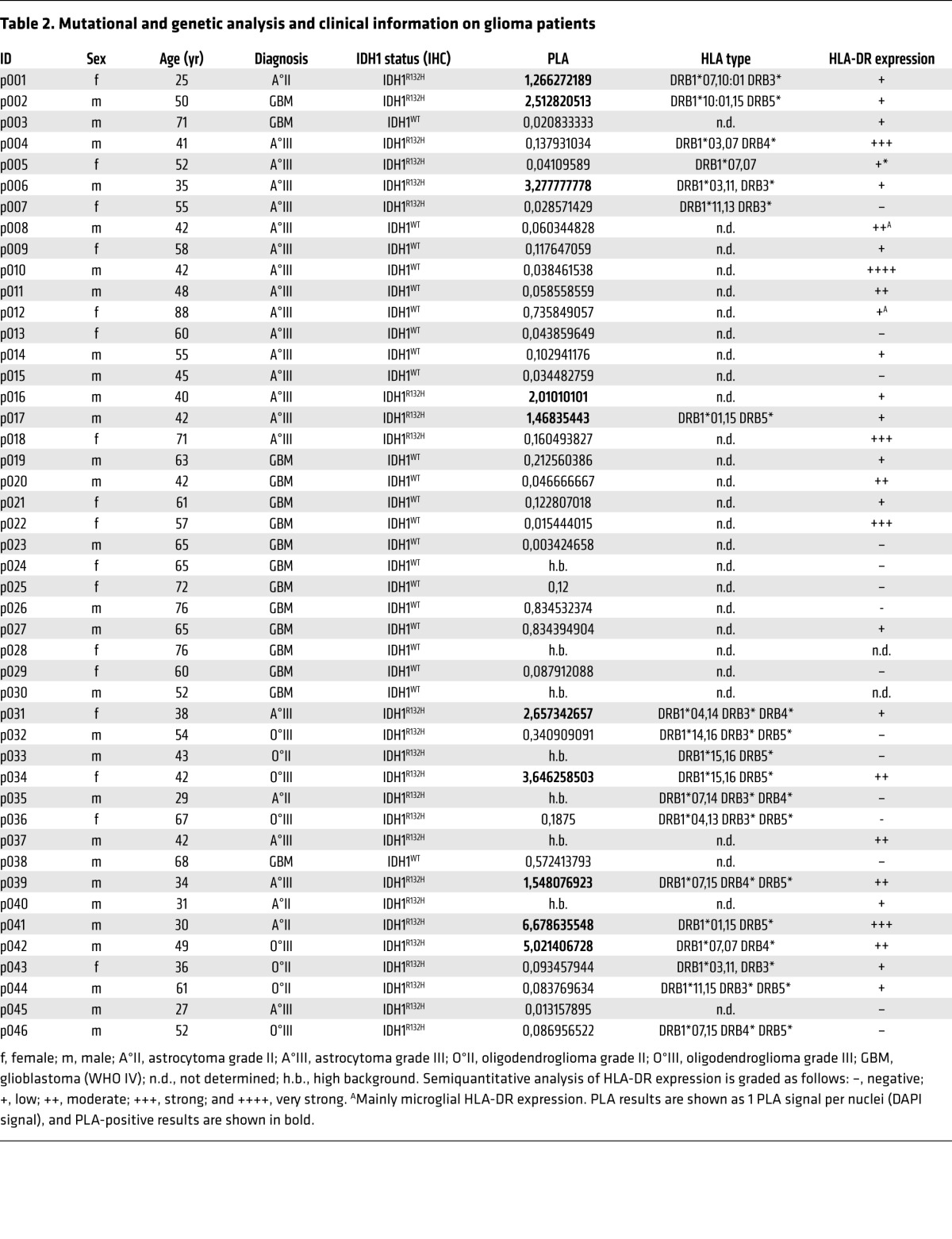
The robust suitability and sensitivity of PLA to detect colocalization of HLA-DR with endogenously expressed antigens in vitro and in paraffin-embedded cellular spheres prompted us to assess the applicability of PLA to detect IDH1R132H peptide presentation in tumor tissue in situ. To this aim, PLA was performed on paraffin-embedded glioma tissue. As demonstrated in Figure 7A, IDH1R132H–MHC class II colocalization was found specifically in IDH1R132H-positive and HLA-DR–positive (p001 and p002) glioma tissue but not in an IDH1WT HLA-DR–positive (p003) glioma tissue. We analyzed a cohort of 46 patients, including 22 patients with IDH1WT gliomas and 24 patients with IDH1R132H gliomas; the cohort included 29 patients in total with HLA-DR–positive tissue. We found that 4 of the IDH1R132H tissue samples and 3 of the IDH1WT tissue samples were not evaluable due to high background in the PLA channel. However, analysis of the remaining samples revealed that 10 of 20 IDH1R132H gliomas, but 0 of 19 IDH1WT gliomas, were positive in the IDH1R132H–MHC class II PLA system, with a threshold for positivity set to 1 PLA signal per cell (P = 0.0004 Fisher’s exact test) (Figure 7, B–D). The PLA signal was only detected in tissue that was also stained positive for HLA-DR by immunohistochemistry (IHC), but the PLA signal did not correlate with HLA-DR expression level (Figure 7E, Supplemental Figure 7, and Table 2). These results confirm the sensitivity and specificity and, hence, the applicability of PLA to detect peptide presentation on HLA-DR in situ. Subsequently, we sought to identify the IDH1R132H antigen-presenting cell (APC) types in the tumor tissue. To this end, glioma tissue was subjected to PLA and costained for tumor-specific expression of IDH1R132H. In some tissues, e.g., p039, the PLA signal was detected mainly in IDH1R132H tumor cells, suggesting that class II expressing glioma cells themselves may present IDH1R132H. However, as demonstrated in p031 tissue, PLA positivity was also detectable, albeit rarely, in IDH1R132H-negative cells (Figure 7F). We therefore sought to identify stroma cell types presenting the IDH1R132H epitope and separately costained the microglia marker Iba-1 and the endothelial marker CD34. Costaining with Iba-1 showed that PLA signals are found in but not restricted to microglia, the professional APC in the brain (Figure 7G). No PLA signal was detectable in CD34+ endothelial cells (Supplemental Figure 7). Taken together, we have shown that PLA is a suitable tool to detect antigen presentation on MHC class II molecules HLA-DR in vitro and in situ and that the IDH1R132H epitope is presented on HLA-DR in glioma tissue.
Figure 7. Specific colocalization of IDH1R132H and MHC class II in glioma tissue.

(A) IDH1R132H–HLA-DR PLA on HLA-DR+ glioma tissues p001 (IDH1R132H), p002 (IDH1R132H), and p003 (IDH1WT). Red, PLA signal; blue, DAPI. Scale bar: 20 μm. (B) Quantification of PLA signal in IDH1R132H+ (n = 20) and IDH1WT (n = 19) glioma tissues in situ. The red dashed line indicates the threshold level for PLA positivity, which was set to 1 PLA signal per nuclei (DAPI signal). ***P = 0.0004, Fisher’s exact test. (C) Quantification algorithm of exemplary glioma tissue (p041). IDH1R132H–HLA-DR PLA on exemplary glioma tissue, with stroma and tumor bulk shown as indicated. Red, PLA signal; blue, DAPI. Scale bar: 20 μm. Analysis and quantification of DAPI signals (red numbers) in stroma and tumor bulk is shown at center. Analysis and quantification of PLA signals (red numbers) in stroma and tumor bulk is shown at right. (D) Quantification of PLA signal per nuclei (DAPI signal) in exemplary glioma tissue. (E) Representative HLA-DR IHC of glioma tissues p006, p007, p011, and p012. Scale bar: 40 μm. (F) IDH1R132H–HLA-DR PLA on HLA-DR+IDH1R132H+ glioma tissues p039 and p031 costained with IDH1R132H (H09). Green, IDH1R132H; red, PLA signal, blue, DAPI. Scale bar: 15 μm. (G) PLA on HLA-DR+ glioma tissues p006 (IDH1R132H) and p012 (IDH1WT) costained with IBA-1. Green, IBA-1; red, PLA signal. Scale bar: 20 μm. Each patient was analyzed once. Insets show high-magnification images of the boxed areas.
Table 2.
Mutational and genetic analysis and clinical information on glioma patients
Discussion
With the increased interest in targeting true tumor antigens, which typically are mutated antigens, by active immunotherapy (25–27), there is an increasing requirement to evaluate whether these antigens are indeed presented in the tumor tissue. In contrast to TAAs, mutated antigens have not undergone central tolerance; however, they are often minor antigens presented on MHC class II molecules rather than MHC class I (28). While the latter has been considered a disadvantage for a long time, there is increasing evidence that an antigen-specific CD4+ T cell response is capable of executing an effective antitumor immunity and not just providing help for CD8+ T cells (29, 30). Potential mechanisms include direct cytotoxicity by antigen-specific CD4+ T cells toward tumor cells presenting the antigen on MHC class II molecules and activation of innate immune cells by antigen-specific CD4+ T cells stimulated through intratumoral professional APC-presenting tumor antigens. We have previously demonstrated that IDH1, which is frequently mutated in gliomas but also other tumor entities, such as acute myeloid leukemia (31–33), chondrosarcomas (34, 35), and intrahepatic cholangiocarcinomas (36), represents a novel tumor neoantigen (14). The IDH1R132H mutation, which represents the most common mutation in IDH1 in gliomas, is capable of inducing a mutation-specific class II–restricted CD4+ T cell response in patients with IDH1R132H-mutated gliomas and in MHC-humanized A2.DR1 mice after vaccination (14). Two lines of evidence in our previous study suggest that IDH1R132H is endogenously processed and presented on MHC class II molecules (14). (a) In patients with IDH1R132H-positive gliomas but not IDH1WT gliomas, mutation-specific CD4+ T cells can be detected after ex vivo stimulation with IDH1R132H (p123–142) in an MHC class II–restricted manner. (b) Mutation-specific CD4+ T cells can be detected after vaccination of MHC-humanized mice with irradiated syngeneic sarcomas expressing human IDH1R132H. Here, we present PLA, a combined IF and PCR approach to detect presentation of antigenic epitopes in situ.
While this approach offers the advantage of detecting presentation in situ on paraffin-embedded tissue slides, it is restricted in its use by the requirements for the antibody targeting the antigen. (a) The antibody targeting the antigen must recognize the presented epitope bound to MHC (class II) in a native state and in situ. (b) In case of mutated antigens, the antibody needs to be mutation specific. For IDH1R132H both requirements are fulfilled. The mutation specificity has been established in numerous studies (20, 21, 37). In fact, IDH1R132H IHC is now implemented in routine diagnostics. In this study, we demonstrate that the antibody recognizes the same epitope, which is presented on MHC class II molecules, which is part of IDH1R132H p123–142 and the shorter peptides p122–136, p124–138, and p126–140 (Figure 1B). Importantly, binding of the anti-IDH1R132H antibody to these epitopes can be blocked by IDH1R132H class II tetramers, further supporting the evidence that H09 recognizes the IDH1R132H epitope bound to MHC class II molecules in a native state (Figure 1E). We further extended this observation to a classic TAA, NY-ESO-1. PLA using an NY-ESO-1–specific and an HLA-DR–specific antibody detected colocalization of an NY-ESO-1 epitope with HLA-DR in SK-Mel-37 cells endogenously expressing NY-ESO-1 and HLA-DR1 and HLA-DR3 (Figure 6). While there have been several class II epitopes characterized for NY-ESO-1, the epitope recognized by the NY-ESO-1 antibody is unknown to us. The observation that this antibody recognizes an epitope, which is also presented on MHC class II molecules, is likely to be not just a coincidence but due to the fact that class II–restricted CD4 epitopes and B cell epitopes sometimes overlap (38). In fact, screening of antibodies using PLA on tumor tissue may be a useful tool to map relevant epitopes presented in the tumor tissue.
We also present evidence that IDH1R132H is presented in human IDH1R132H-mutated gliomas. While larger prospective series are required to determine the biological relevance of PLA positivity, it is conceivable that this method may find application in preselecting patients for a targeted immunotherapy. With respect to deciphering the cellular context of antigen presentation in the tumor tissue, there is room for improvement with respect to multilabeling. The attempts to do so in this study suggest that IDH1R132H is not only presented by tumor cells themselves, which can be MHC class II positive (Figure 3) (39, 40), but also presented by microglial cells (Figure 7).
In summary, we present a method to detect presentation of antigens in situ, which may find application in but is not restricted to the identification of relevant tumor (associated) antigens.
Methods
Peptides.
Human IDH1WT and IDH1R132H amino acid sequences IDH1 p118–146 PRLVSGWVKPIIIGRHAYGDQYRATDFVV and PRLVSGWVKPIIIGHHAYGDQYRATDFVV, respectively, cover the amino acid exchange from Arg to His at position 132, including all possible 15-mer IDH1 peptides containing position 132, and are identical to the mouse sequence except for position 122 (Thr in mouse). Peptide libraries for ELISA of IDH1WT and IDH1R132H 15-mers contained the following peptides: IDH1WT p118–132: PRLVSGWVKPIIIGR; IDH1WT p120–134: LVSGWVKPIIIGRHA; IDH1WT p122–136: SGWVKPIIIGRHAYG; IDH1WT p124–138: WVKPIIIGRHAYGDQ; IDH1WT p126–140: KPIIIGRHAYGDQYR; IDH1WT p128–142: IIIGRHAYGDQYRAT; IDH1WT p130–144: IGRHAYGDQYRATDF; IDH1WT p132–146: RHAYGDQYRATDFVV; IDH1R132H p118–132: PRLVSGWVKPIIIGH; IDH1R132H p120–134: LVSGWVKPIIIGHHA; IDH1R132H p122–136: SGWVKPIIIGHHAYG; IDH1R132H p124–138: WVKPIIIGHHAYGDQ; IDH1R132H p126–140: KPIIIGHHAYGDQYR; IDH1R132H p128–142: IIIGHHAYGDQYRAT; IDH1R132H p130–144: IGHHAYGDQYRATDF; and IDH1R132H p132–146: HHAYGDQYRATDFVV. 20-mers were IDH1WT p123–142 GWVKPIIIGRHAYGDQYRAT and IDH1R132H p123–142 GWVKPIIIGHHAYGDQYRAT. Negative control peptide for ELISA and in vitro stimulation was mouse myelin oligodendrocyte glycoprotein (MOG) p35–55 MEVGWYRSPFSRVVHLYRNGK. MOG peptide was synthesized by GenScript, and IDH1 (IDH1WT, IDH1R132H) 15- and 20-mers were synthesized by Bachem Distribution Services GmbH. Peptides were diluted in PBS 10% DMSO at 2.5 mg/ml and stored at –80°C.
In silico MHC class II peptide-binding prediction.
Human IDH1R132H 29-mer peptide PRLVSGWVKPIIIGHHAYGDQYRATDFVV includes all possible processed IDH1R132H 15-mers, including the point mutation at codon 132, which leads to the amino acid change from arginine (R) to histidine (H). Binding of 15-mer IDH1R132H peptides to available HLA-DR types was predicted by the NetMHCII 2.2 algorithm (10, 41).
IDH1R132H and IDH1WT peptide-based ELISA.
ELISA plates (Costar, Corning Life Sciences) were coated with IDH1R132H and IDH1WT p118–132, p120–134, p122–136, p124–138, p126–140, p128–142, p130–144, p132–146, and p123–142 (10 μg per well in PBS); washed with PBS 0.05% Tween 20; and blocked with 3% FBS (Sigma-Aldrich) in PBS 0.05% Tween 20. As negative controls, MOG p35–55 was used at equal concentrations and peptide diluent PBS 10% DMSO was used at equal volume. As primary antibody, monoclonal mouse anti-IDH1R132H (1:1,000, H09, Dianova) was used. HRP-conjugated secondary antibody was sheep anti-mouse IgG (1:5,000, Amersham, GE Healthcare). HLA-DRB1*01:01 MHC class II tetramer bound to IDH1R132H p123–142 and HLA-DRB1*01:01 MHC class II tetramer control bound to CLIP were provided by NIH tetramer core facility and used 1:200 during preincubation with H09 for competitive ELISA. The substrate was TMB (eBioscience), and the reaction was stopped with 1 M H2SO4. OD at 450 nm was measured with an ELISA reader (Thermo Fisher).
Glioma tissue.
Assays were performed with glioma tissue from patients diagnosed at the Department of Neuropathology at Heidelberg University. IDH1 mutation status was routinely diagnosed by IHC.
Cell lines and modification of gene expression.
Glioma cell line LN229 endogenously expressing HLA-DR1 was obtained from ATCC (distributed by LGC Standards); kept in DMEM with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptavidin (all Sigma-Aldrich); and was lentivirally transduced with full-length cDNA of human IDH1R132H or IDH1WT (NCBI GenBank CR641695.1) in pLenti6.2/V5-DEST for stable expression. Virus was produced by transfection of HEK293T packaging cell line, which was obtained from ATCC. Since IDH1R132H protein dimerizes and produces the oncometabolite R-2-HG, which affects proliferation and thereby impairs stable IDH1R132H expression, a second point mutation leading to the amino acid exchange D252G was introduced into IDH1R132H- and IDH1WT-coding sequences by site-directed mutagenesis. This dimerization-deficient mutation results in an inert enzyme and, therefore, increased expression stability. Transduced cells were selected with 10 μg/ml blasticidin (Sigma-Aldrich) for stable overexpression. For heterogeneous IDH1R132H expression, LN229 cells were transfected with full-length cDNA of human IDH1R132H or IDH1WT in the retroviral vector pMXs-IRES-BsdR (Cell Biolabs Inc.) using FuGene HD transfection reagent (Promega). Cells were selected with 9 μg/ml blasticidin 72 hours after transfection. LN229 oligo-clones were generated by limiting dilution, and outgrowing oligo-clones were screened for IDH1R132H expression by IF. Oligo-clone IVE2 was selected due to heterogeneous IDH1R132H expression. Stable overexpression of cancer testis antigen CTAG1B (NY-ESO-1) in LN229 cells was done by transfection with full-length NY-ESO-1 cDNA (NCBI Genbank BC160040), provided by the DKFZ Genomics Facility, in the vector MYC-N, which was generated by exchanging the 6xHis tag in pDEST26 (Invitrogen) via site-directed mutagenesis-PCR, using FuGene HD transfection reagent (Promega). Cells were selected with 1 mg/ml Geneticin (Calbiochem) 72 hours after transfection. Melanoma cell lines SK-Mel-23 and SK-Mel-37 were provided by J. Utikal (Clinical Cooperation Unit Dermato-Oncology, German Cancer Research Center, Heidelberg, Germany) and P. Krammer (Immunogenetics, German Cancer Research Center, Heidelberg, Germany), respectively, and kept in DMEM with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptavidin. SK-Mel-37 cells endogenously expressing NY-ESO-1 and HLA-DR1 and HLA-DR3 were used for analysis of endogenous presentation of NY-ESO-1–derived peptides. For knockdown of HLA-DR in LN229 cells and HLA-DR and NY-ESO-1 in SK-Mel-37 cells, ON-TARGET SMARTpool siRNA (Dharmacon RNA Technologies) was used. The siRNA target sequences for CTAG1B (NY-ESO-1) were as follows: CUGAAUGGAUGCUGCAGAU, CCGGCAACAUACUGACUAU, CGCCAUGCCUUUCGCGACA, and GCUGGAGGAGGACGGCUUA. The siRNA target sequences for HLA-DRA were as follows: UGACAAAGCGCUCCAACUA, UGACCAAUCAGGCGAGUUU, GGAAUCAUGGGCUAUCAAA, and CAACUGAGGACGUUUACGA.
As negative control, ON-TARGETplus siCONTROL Non-Targeting Pool (D-001810-10-05, Dharmacon) was used. The transfection was performed with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocol.
Primary glioma cell lines.
IDH1R132H mutant cell lines NCH620 and NCH645 were established from secondary glioblastoma at the Department of Neurosurgery, University Hospital Heidelberg, as described previously (42). They were cultivated as floating neurospheres in DMEM/F-12 medium containing 20% BIT serum-free supplement, BFGF, and EGF at 20 ng/ml each (all PELOBiotech).
IDH1WT cell line NPH001 was established from primary astrocytoma WHO grade III tissue, which was dissected using a scalpel, washed in PBS containing 4.5% glucose, and digested in HBSS without Mg2+ and Ca2+ (Gibco, Life Technologies) supplemented with 0.01% Papain (Sigma-Aldrich), 0.1% Dispase 2 (Roche), 0.01% DNase (Roche), and 12.4 mM MgSO4 for 30 minutes at room temperature. Tissue was washed 3 times with DMEM/F12 GlutaMAX (Gibco Life Technologies) and seeded in DMEM/F12 GlutaMAX with 2% B27 supplement, 100 U/ml penicillin, and 100 μg/ml streptavidin (all Gibco Life Technologies) as well as 20 ng/ml EGF and 20 ng/ml BFGF (both PromoCell) until spheres appeared. Dead cells and debris were removed regularly until culture was clean.
PLA.
Tumor cell lines were seeded on glass coverslips, grown until 70% to 90% confluent, and fixed and permeabilized with Cytofixx Pump Spray (Cell Path) for 30 minutes at –20°C and subsequent 4% PFA in PBS for 30 minutes at room temperature. Primary cell line spheres were embedded in 1.5% low-melting agarose (Sigma-Aldrich) and embedded in paraffin. Paraffin-embedded spheres and glioma tissue were deparaffinized with HistoClear II (National Diagnostics) and rehydrated. Antigen retrieval was performed using Cell Conditioning Solution CC1 (Ventana Medical Systems Inc.) for 30 minutes. For PLA with CD34 costaining, Cell Conditioning Solution CC2 (Ventana Medical Systems Inc.) was used instead of CC1. PLA was performed using Detection Reagents Red, PLA Probe anti-mouse PLUS, PLA Probe anti-rabbit MINUS, and Wash Buffers Fluorescence (all Duolink, Olink Bioscience) according to manufacturer’s instructions. Briefly, blocking was done with blocking solution for 30 minutes at 37°C, and primary antibodies monoclonal mouse anti-human IDH1R132H (1:100, H09, Dianova) or mouse anti-human monoclonal NY-ESO-1 (1:100, E978, Sigma-Aldrich) with monoclonal rabbit anti-human HLA-DR (1:100 in vitro, 1:200 in situ, EPR3692, Abcam) in antibody diluent were incubated overnight at 4°C. PLA Probe anti-mouse PLUS and PLA Probe anti-rabbit MINUS were incubated for 1 hour at 37°C. Ligation and amplification were performed using the Detection Reagents Red. IF costaining was performed as described below after amplification of PLA signal. Vectashield HardSet Mounting Medium with DAPI (Vector Laboratories) was used for mounting and nuclear staining. PLA on spheres was additionally performed using Detection Reagents Brightfield, PLA Probe anti-mouse PLUS, PLA Probe anti-rabbit MINUS, and Wash Buffers Brightfield (all Duolink, Olink Bioscience). Counterstaining was performed using hemalum (Carl Roth).
Immunofluorescent staining.
For IF staining, cells were seeded on glass coverslips, grown until 70% to 90% confluent, and fixed and permeabilized as described above. For blocking and staining, blocking solution (Duolink, Olink Bioscience) and antibody diluent (Duolink, Olink Bioscience) were used as for PLA, respectively. Anti-IDH1R132H was used as primary antibody as described above. Additional IF stainings were performed using rabbit monoclonal anti–myc-tag (1:200, 71D10, Cell Signaling), mouse monoclonal anti–HLA-DR (1:200, TAL1B5, Abcam), mouse monoclonal anti–annexin V (1:100, E-10, Santa Cruz), mouse monoclonal anti-CD34 (1:100, ICO115, Cell Signaling), rabbit monoclonal anti-Rab7 (1:100, D95F2, Cell Signaling), and rabbit polyclonal anti-human IBA-1 (1:100, Wako, 019-19741), and secondary antibodies used were donkey anti-mouse Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 546 (all 1:300, Molecular Probes, Invitrogen, A-21202 and A-11010). DAPI staining and mounting were performed as described above. For PLA costaining, secondary antibody donkey anti-mouse Alexa Fluor 488 was used for IDH1R132H, HLA-DR, CD34, and annexin V detection and donkey anti-rabbit Alexa Fluor 488 (A-21206; all 1:300, Molecular Probes, Invitrogen) was used for IBA-1, myc-tag, and Rab7 detection.
IHC.
Glioma tissue was deparaffinized with HistoClear II and rehydrated. Antigen retrieval was performed using Cell Conditioning Solution CC1 for 30 minutes. Endogenous peroxidase was blocked with 3% hydrogen peroxide in PBS. Blocking was performed with 5% FBS for 1 hour. Primary antibodies (monoclonal mouse anti-human IDH1R132H [1:100, H09, Dianova] and monoclonal rabbit anti-human HLA-DR [1:100, EPR3692, Abcam]) were incubated overnight at 4°C. Color reaction was performed using Polymer-Envision for 30 minutes at room temperature and liquid DAB+ Substrate Chromogen System (both DAKO). Counterstaining was performed using hemalum (Carl Roth).
Western blot.
Total protein was isolated by cell lysis with ice-cold Tris-HCl, 50 mM, pH 8.0 (Carl Roth), containing 150 mM NaCl (J.T. Baker, Deventer, The Netherlands), 1% Nondiet P-40 (Genaxxon Bioscience), 10 mM EDTA (GerbuBiotechnik), 200 mM dithiothreitol (Carl Roth), 100 μM PMSF, and complete EDTA-free (1:50, Roche) for 20 minutes, and centrifuged to pellet debris. Protein concentrations were measured via the Bio-Rad protein assay (Bio-Rad, Hercules) at 595 nm, and 30 μg of protein diluted in Laemmli sample buffer was denatured at 95°C for 5 minutes and electrophoretically separated on 12% acrylamide-polyacrylamide SDS-containing gels. Proteins were blotted onto nitrocellulose membranes by wet blot at 1.5 mA/cm2 for 1 hour. After blocking with 5% milk powder in 0.5 M TBS, pH 7.4, 1.5 M NaCl, 0.05% Tween 20, membranes were incubated consecutively with primary monoclonal mouse anti-IDH1R132H (1:500, H09, Dianova), monoclonal rat anti-panIDH1 (1:500, W09, Dianova) for detection of IDH1WT and IDH1R132H, monoclonal rabbit anti-myc tag (1:1,000, 71D10, Cell Signaling), mouse anti–NY-ESO-1 (1:500, E978, Sigma-Aldrich), and rabbit anti–HLA-DR (1:500, TAL1B5, Abcam) overnight at 4°C and mouse anti–α-tubulin (1:5,000, AA13, Sigma-Aldrich) as loading control for 1 hour at room temperature. Staining with secondary HRP-conjugated rabbit anti-rat (1:1,000xF, Dako, P0450), anti-mouse, or anti-rabbit (both 1:5,000, GE Healthcare, NA931 and NA934) antibodies was performed at room temperature for 1 hour and was followed by chemiluminescent development using ECL or ECL prime (both Amersham, GE Healthcare).
Flow cytometry.
Cells were harvested; washed once in PBS, 3% FBS, and 2 mM EDTA; and blocking was done with human sera obtained from peripheral blood mononuclear cell density gradient centrifugation. Surface HLA-DR was stained using eFluor-450–conjugated mouse anti–HLA-DR antibody (1:100, L243, eBioscience). Cells were acquired on a FACS Canto II (Becton Dickinson) and analyzed using FlowJo software.
HLA typing.
Genomic DNA was isolated from patient blood or tumor samples using the FFPE LEV DNA Purification Kit AS1130 (Promega) and from cell lines using the QIAamp DNA Mini Kit (Qiagen). PCR-based typing was performed using HLA-DR type–specific primer pairs lyophilized in a 96-well plate (HLA-DRB1* CTS-PCR-SSP Minitray Kit) and Mastermix 5.0% for HLA-DRB1* (all from Department of Transplantation Immunology, University Clinic Heidelberg) with Taq-polymerase (Fermentas, Thermo Scientific). PCR was performed according to the manufacturer’s instructions. PCR products were separated on a 1.5% agarose gel containing GelRed (1:10,000, Genaxxon Bioscience). Analysis was done according to the manufacturer’s instructions.
R-2-HG measurement.
R-2-HG production in cells was analyzed as described previously (43). As a control for R-2-HG production, enzymatically competent retrovirally transfected IDH1R132H-overexpressing LN229 cells were used.
Image analysis.
IF images were taken on a LEICA DM IRB microscope, using ×40 and ×63 objectives for PLA, detecting PLA signal with N2.1 filter, signal of IF costaining with GFP filter, and ×20 and ×40 objectives for IF images. IHC images were taken on a Zeiss Axioplan microscope. Contrast and brightness of images were linearly optimized with Adobe Photoshop CS3. For quantification of IDH1R132H Alexa Fluor 488 MFI and PLA signals, an algorithm was programmed using ImageJ (see Supplemental Figure 3) and a minimum number of 10 pictures per sample were analyzed.
Statistics.
Data are expressed as mean ± SEM and analysis of significance was performed using the 1-way ANOVA, Tukey corrected (GraphPad Prism 6.0) (Figure 1, C and E). PLA positivity in situ was set to 1 PLA signal per nuclei (DAPI signal), and analysis of significance was performed using Fisher’s exact test, (Table 2, R version 2.15.2.). Correlation between IDH1R132H MFI and PLA signal was analyzed by Spearman’s rho (GraphPad Prism 6.0). P values of less than 0.05 were considered significant.
Study approval.
Tissues were obtained from the archives of the Department of Neuropathology, University Hospital Heidelberg, according to the regulations of the Tissue Bank of the National Center for Tumor Diseases, University Hospital Heidelberg, and used after approval of the local regulatory authorities. All subjects provided informed consent prior to their participation in the study.
Supplementary Material
Acknowledgments
We thank D. Hartmann for technical assistance. We acknowledge support by the DKFZ Light Microscopy and Genomics and Proteomics Facilities. HLA-DRB1*01:01 MHC class II tetramer bound to IDH1R132H p123–142 was provided by NIH tetramer core facility. This work was supported by the Interdisciplinary Research Program of the National Center for Tumor Diseases Heidelberg (IFP III./2) to M. Platten and A. von Deimling, the Wilhelm Sander Foundation (2012.118.1) to M. Platten and A. von Deimling, the Helmholtz Foundation (VH-NG-306) and the Andreas Zimprich Foundation to M. Platten, and the German Research Foundation (SFB938 TPK) to M. Platten and W. Wick. T. Schumacher was supported by the Helmholtz International Graduate School. L. Bunse was supported by the Heinrich F.C. Behr Foundation and the Hartmut Hoffmann-Berling International Graduate School of Molecular and Cellular Biology MD/PhD program, University Heidelberg. F. Sahm was supported by a postdoctoral fellowship of the University Hospital Heidelberg.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(2):593–606. doi:10.1172/JCI77780.
References
- 1.Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 2.Mocellin S, Mandruzzato S, Bronte V, Lise M, Nitti D. Part I: Vaccines for solid tumours. Lancet Oncol. 2004;5(11):681–689. doi: 10.1016/S1470-2045(04)01610-9. [DOI] [PubMed] [Google Scholar]
- 3.Mocellin S, Semenzato G, Mandruzzato S, Riccardo Rossi C. Part II: Vaccines for haematological malignant disorders. Lancet Oncol. 2004;5(12):727–737. doi: 10.1016/S1470-2045(04)01649-3. [DOI] [PubMed] [Google Scholar]
- 4.Prins RM, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17(6):1603–1615. doi: 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sampson JH, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8(10):2773–2779. doi: 10.1158/1535-7163.MCT-09-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013;341(6151):1192–1198. doi: 10.1126/science.1241145. [DOI] [PubMed] [Google Scholar]
- 7.Zhao LL, Xu KL, Wang SW, Hu BL, Chen LR. Pathological significance of epidermal growth factor receptor expression and amplification in human gliomas. Histopathology. 2012;61(4):726–736. doi: 10.1111/j.1365-2559.2012.04354.x. [DOI] [PubMed] [Google Scholar]
- 8.Jager E, et al. Monitoring CD8 T cell responses to NY-ESO-1: correlation of humoral and cellular immune responses. Proc Natl Acad Sci U S A. 2000;97(9):4760–4765. doi: 10.1073/pnas.97.9.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 2008;36(Web Server issue):W509–W512. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. 2009;10:296. doi: 10.1186/1471-2105-10-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50(3–4):213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 12.Regner M, Claesson MH, Bregenholt S, Ropke M. An improved method for the detection of peptide-induced upregulation of HLA-A2 molecules on TAP-deficient T2 cells. Exp Clin Immunogenet. 1996;13(1):30–35. [PubMed] [Google Scholar]
- 13.Dutoit V, et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain. 2012;135(pt 4):1042–1054. doi: 10.1093/brain/aws042. [DOI] [PubMed] [Google Scholar]
- 14.Schumacher T, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512(7514):324–327. doi: 10.1038/nature13387. [DOI] [PubMed] [Google Scholar]
- 15.Weiler M, et al. mTOR target NDRG1 confers MGMT-dependent resistance to alkylating chemotherapy. Proc Natl Acad Sci U S A. 2014;111(1):409–414. doi: 10.1073/pnas.1314469111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schweizer L, et al. Meningeal hemangiopericytoma and solitary fibrous tumors carry the NAB2-STAT6 fusion and can be diagnosed by nuclear expression of STAT6 protein. Acta Neuropathol. 2013;125(5):651–658. doi: 10.1007/s00401-013-1117-6. [DOI] [PubMed] [Google Scholar]
- 17.Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116(6):597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- 18.Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Capper D, et al. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2010;20(1):245–254. doi: 10.1111/j.1750-3639.2009.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Capper D, Zentgraf H, Balss J, Hartmann C, von Deimling A. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009;118(5):599–601. doi: 10.1007/s00401-009-0595-z. [DOI] [PubMed] [Google Scholar]
- 22.Andersson G. Evolution of the human HLA-DR region. Front Biosci. 1998;3:d739–d745. doi: 10.2741/a317. [DOI] [PubMed] [Google Scholar]
- 23.Ayyoub M, et al. Assessment of vaccine-induced CD4 T cell responses to the 119-143 immunodominant region of the tumor-specific antigen NY-ESO-1 using DRB1*0101 tetramers. Clin Cancer Res. 2010;16(18):4607–4615. doi: 10.1158/1078-0432.CCR-10-1485. [DOI] [PubMed] [Google Scholar]
- 24.Zarour HM, et al. NY-ESO-1 119-143 is a promiscuous major histocompatibility complex class II T-helper epitope recognized by Th1- and Th2-type tumor-reactive CD4+ T cells. Cancer Res. 2002;62(1):213–218. [PubMed] [Google Scholar]
- 25.Choi BD, et al. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol. 2009;19(4):713–723. doi: 10.1111/j.1750-3639.2009.00318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sampson JH, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–4729. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sensi M, Anichini A. Unique tumor antigens: evidence for immune control of genome integrity and immunogenic targets for T cell-mediated patient-specific immunotherapy. Clin Cancer Res. 2006;12(17):5023–5032. doi: 10.1158/1078-0432.CCR-05-2682. [DOI] [PubMed] [Google Scholar]
- 28.Accolla RS, Tosi G. Optimal MHC-II-restricted tumor antigen presentation to CD4+ T helper cells: the key issue for development of anti-tumor vaccines. J Transl Med. 2012;10:154. doi: 10.1186/1479-5876-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hunder NN, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358(25):2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quezada SA, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207(3):637–650. doi: 10.1084/jem.20091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou WC, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116(20):4086–4094. doi: 10.1182/blood-2010-05-283291. [DOI] [PubMed] [Google Scholar]
- 32.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ward PS, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amary MF, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224(3):334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 35.Arai M, Nobusawa S, Ikota H, Takemura S, Nakazato Y. Frequent IDH1/2 mutations in intracranial chondrosarcoma: a possible diagnostic clue for its differentiation from chordoma. Brain Tumor Pathol. 2012;29(4):201–206. doi: 10.1007/s10014-012-0085-1. [DOI] [PubMed] [Google Scholar]
- 36.Wang P, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32(25):3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahm F, et al. Addressing diffuse glioma as a systemic brain disease with single-cell analysis. Arch Neurol. 2012;69(4):523–526. doi: 10.1001/archneurol.2011.2910. [DOI] [PubMed] [Google Scholar]
- 38.Lehtinen M, Stellato G, Hyoty H, Nieminen P, Vesterinen E, Paavonen J. A T-helper cell epitope overlaps a major B-cell epitope in human papillomavirus type 18 E2 protein. APMIS. 1992;100(11):1022–1026. doi: 10.1111/j.1699-0463.1992.tb04035.x. [DOI] [PubMed] [Google Scholar]
- 39.Rees RC, Mian S. Selective MHC expression in tumours modulates adaptive and innate antitumour responses. Cancer Immunol Immunother. 1999;48(7):374–381. doi: 10.1007/s002620050589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takamura Y, et al. Regulation of MHC class II expression in glioma cells by class II transactivator (CIITA). Glia. 2004;45(4):392–405. doi: 10.1002/glia.10343. [DOI] [PubMed] [Google Scholar]
- 41.Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinformatics. 2007;8:238. doi: 10.1186/1471-2105-8-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campos B, et al. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin Cancer Res. 2010;16(10):2715–2728. doi: 10.1158/1078-0432.CCR-09-1800. [DOI] [PubMed] [Google Scholar]
- 43.Balss J, et al. Enzymatic assay for quantitative analysis of (D)-2-hydroxyglutarate. Acta Neuropathol. 2012;124(6):883–891. doi: 10.1007/s00401-012-1060-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.