

Abstract
The current treatment for patients with hypothyroidism is levothyroxine (L-T4) along with normalization of serum thyroid-stimulating hormone (TSH). However, normalization of serum TSH with L-T4 monotherapy results in relatively low serum 3,5,3′-triiodothyronine (T3) and high serum thyroxine/T3 (T4/T3) ratio. In the hypothalamus-pituitary dyad as well as the rest of the brain, the majority of T3 present is generated locally by T4 deiodination via the type 2 deiodinase (D2); this pathway is self-limited by ubiquitination of D2 by the ubiquitin ligase WSB-1. Here, we determined that tissue-specific differences in D2 ubiquitination account for the high T4/T3 serum ratio in adult thyroidectomized (Tx) rats chronically implanted with subcutaneous L-T4 pellets. While L-T4 administration decreased whole-body D2-dependent T4 conversion to T3, D2 activity in the hypothalamus was only minimally affected by L-T4. In vivo studies in mice harboring an astrocyte-specific Wsb1 deletion as well as in vitro analysis of D2 ubiquitination driven by different tissue extracts indicated that D2 ubiquitination in the hypothalamus is relatively less. As a result, in contrast to other D2-expressing tissues, the hypothalamus is wired to have increased sensitivity to T4. These studies reveal that tissue-specific differences in D2 ubiquitination are an inherent property of the TRH/TSH feedback mechanism and indicate that only constant delivery of L-T4 and L-T3 fully normalizes T3-dependent metabolic markers and gene expression profiles in Tx rats.
Introduction
Hypothyroidism is a prevalent condition, affecting more than 10 million Americans (1, 2). Historically, the standard of care for treating hypothyroid patients was administration of thyroid extracts to resolve symptoms. The discovery that the main thyroid product thyroxine (T4) is largely activated to 3,5,3′-triiodothyronine (T3) outside of the thyroid parenchyma made treatment with levothyroxine (L-T4) and normalization of serum thyroid-stimulating hormone (TSH) levels the new, and current, standard of care (1, 2). However, patients on L-T4 monotherapy exhibit a relatively higher serum T4/T3 ratio (3), with unknown long-term consequences to thyroid hormone signaling and general health. This is particularly relevant given that clinical studies indicate that 5%–10% of L-T4–treated hypothyroid patients with normal serum TSH have persistent symptoms that can be related to the disease (4).
While the molecular basis for the residual hypothyroid symptoms is lacking, it is generally hypothesized that these patients suffer from tissue-specific states of hypothyroidism and that serum TSH might not adequately reflect thyroid status at the level of different tissues (5). This scenario is partially supported by 2 factors:
First, serum T3 is below the lower limit of the reference range in approximately 15% of the hypothyroid patients treated with L-T4 monotherapy, despite normal serum levels of TSH and a relatively higher serum free T4 (6, 7). In many cases, normalization of serum T3 with L-T4 monotherapy can only be achieved at the expense of having an elevated serum T4 and low/suppressed serum TSH (7). However, it is not clear that the relatively lower serum T3 is clinically relevant or plays a causal role in the residual hypothyroid-like symptoms, which can be nonspecific in nature, such that some investigators have implicated other prevalent conditions, such as perimenopause or depression, as possible contributing factors in these symptomatic patients (8).
Second, in many organs and tissues the type 2 deiodinase (D2, encoded by DIO2) pathway makes a substantial contribution to the local T3 content, as much as approximately 50% in some tissues (9). For instance, in both the brain and the brown adipose tissue (BAT), thyroid hormone signaling depends on both plasma T3 and T3 locally generated via the D2 pathway (10–12). In the brain, D2 is expressed in astrocytes (13, 14), while thyroid hormone receptors and type 3 deiodinase (D3), which inactivates T3, are found in neurons (15). D2-generated T3 exits the glial cells and acts in a paracrine fashion to modulate the expression of T3-responsive genes in the neighboring neurons (16). In a mouse with astrocyte-specific D2 inactivation (Dio2fl/fl crossed with GFAP-Cre) there is loss of more than 95% in brain D2 activity (17). D2 is a type I endoplasmic reticulum–resident thioredoxin fold–containing selenoprotein with a variable half-life that depends on the level of its natural substrate, T4. In the presence of T4, D2 is inactivated with an approximately 20-minute half-life, whereas in the absence of T4, its half-life is prolonged to hours (9). This provides a mechanism through which the production of T3, the biologically active thyroid hormone, can be regulated according to the availability of T4. For example, an accumulation of D2 in cells increases the fractional conversion of T4 to T3 when serum T4 levels are low, such as in the case of iodine deficiency or hypothyroidism. In contrast, because T4 inactivates D2 so efficiently, it is conceivable that thyroid hormone signaling is dampened if T4 levels are high (18). In other words, a high serum T4/T3 ratio in L-T4–treated patients could actually reduce thyroid hormone signaling in D2-expressing tissues such as brain and BAT.
D2 ubiquitination is the molecular mechanism that modifies D2 half-life where binding to ubiquitin inactivates the enzyme and targets it for degradation in the proteasomes (19, 20). Ubiquitination is thought to inactivate D2 by disrupting the conformation of the D2:D2 dimer, critical for enzyme activity. A unique 18–amino acid loop confers intrinsic metabolic instability to D2, facilitating binding to proteins involved in the ubiquitination process (21, 22). This loop is also the site where a prevalent genetic polymorphism in DIO2 causes a single amino acid substitution, Thr to Ala, in position 92 in humans (23). While the kinetic properties of the polymorphic D2 remain unaffected, the fact that it is associated with a large number of diseases and conditions, including obesity and intolerance to glucose, suggests that the Thr92AlaD2 compromises the ability of this pathway to mediate thyroid hormone signaling (24).
The ubiquitin-activating enzymes UBC6 (UBE2J) and UBC7 (UBE2G1) participate in the process of D2 ubiquitination (25, 26), as well as 2 ubiquitin ligases, the hedgehog-inducible WSB-1 (22) and TEB4, a ligase involved in the degradation of proteins in the endoplasmic reticulum (27). Ubiquitinated D2 (UbD2) is not immediately taken up by the proteasomes. Instead, UbD2 can be reactivated by deubiquitination, a process catalyzed by two USP-class D2-interacting deubiquitinases (DUBs), USP-20 and USP-33 (28). D2 ubiquitination occurs via K48-linked ubiquitin chains, and exposure to its natural substrate, T4, accelerates UbD2 formation (29). UbD2 is retrotranslocated to the cytoplasm via interaction with the p97-ATPase complex and deubiquitination by the p97-associated DUB ataxin-3. Once in the cytosol, D2 is delivered to the proteasomes and irreversibly degraded (29).
These observations that a lower serum T3 and higher serum T4 could independently dampen thyroid hormone signaling support a modified approach to treatment of hypothyroidism that includes combination therapy with L-T4 and liothyronine (L-T3), through administration of either L-T4 plus L-T3 or desiccated thyroid extracts, which inherently contain both compounds. Replacing some of the L-T4 given to patients with L-T3 would raise/normalize serum T3 while preserving serum TSH within the normal range. This rationale is supported by studies in rodents in which normalization of serum T4, T3, and TSH as well as tissue content of T4 and T3 can only be achieved if combination therapy with L-T4 and L-T3 was used (30, 31). Nevertheless, multiple randomized controlled clinical trials in hypothyroid patients have addressed objective and subjective clinical outcomes comparing L-T4 monotherapy and L-T4 plus L-T3 combination therapy. The large majority of these trials found that both forms of treatment are equivalent, despite elevation in serum T3 in the patients on combination therapy (32). A large study also considered the DIO2 polymorphism but only found a weak statistical association with preference for the combination therapy versus monotherapy among hypothyroid patients (33, 34). These trials have led major professional societies in the US and Europe to label the situation as “controversial” and to continue the recommendation that L-T4 monotherapy remain the standard of care for hypothyroid patients (1, 2, 4).
Insight into 3 basic questions is needed for a better understanding of the molecular basis for the treatment of hypothyroidism: (a) What is the mechanistic explanation for the lack of normalization of serum T3 in L-T4–treated hypothyroid individuals exhibiting a normal serum TSH? (b) Do these relatively low serum T3 levels and/or relatively elevated serum T4 levels affect thyroid hormone signaling? (c) Can thyroid hormone homeostasis and T3-dependent markers be normalized by L-T4 and L-T3 combination therapy?
To address these questions, the present studies modeled the situation in an animal system using a large number of thyroidectomized (Tx) rats that were treated with L-T4 alone or a combination of L-T4 and L-T3 for 7 weeks. Here we report that due to hypothalamus-specific differences in D2 ubiquitination there is a sensitivity gradient in the loss of D2 activity in response to T4 between the hypothalamus and the rest of the brain and body. This explains why treatment with L-T4 alone fails to normalize serum TSH and T3 simultaneously. The lack of normalization of serum T3 and the higher serum T4 levels in L-T4–treated Tx rats have clear metabolic implications, including persistent hypercholesterolemia and relatively lower mitochondria content in liver and skeletal muscle. Furthermore, the relatively high serum T4 levels reduce D2 activity in different areas of the CNS, which, combined with lower serum T3 levels, results in local hypothyroidism. Only combined therapy with constant delivery of both L-T4 and L-T3 fully normalized T3-dependent metabolic markers and gene expression in Tx rats. These findings have important implications that may support the role of combination therapy in the treatment strategy for humans with hypothyroidism and thus may drive the need for development of improved pharmacologic modes for L-T3 administration and for high-quality randomized controlled trials in humans.
Results
Serum thyroid function tests in Tx rats treated with different thyroid hormone replacement regimens
Placebo-Tx animals exhibited the expected elevation in serum TSH and decrease in serum T4, T3, and reverse T3 (rT3) levels, with a reduction in the serum T4/T3 ratio when compared with placebo-control animals (Table 1). At the same time, T4-mono animals had normal serum TSH (Table 1 and Figure 1A), but serum T4 was found to be higher, and serum T3 lower, in comparison with placebo-control animals (Table 1); thus the elevated serum T4/T3 ratio (Table 1 and Figure 1B). In contrast, T4/T3-comb animals exhibited serum levels of TSH, T4, T3, and T4/T3 ratio that were indistinguishable from those in placebo-control animals (Table 1). Notably, T4/T3-inj-comb animals exhibited normal serum T4 but serum TSH was approximately doubled, serum T3 slightly decreased, and serum T4/T3 ratio increased in comparison with placebo-control animals (Table 1). Serum rT3 levels were normalized by treatment with thyroid hormone independently of the replacement strategy used (Table 1).
Table 1.
Serum TSH, T4, and T3 in Tx rats treated with different thyroid hormone replacement regimens

Figure 1. Parameters of thyroid economy in placebo-control and T4-mono rats plotted as a function of serum T4.

(A) Serum TSH. (B) Serum T4/T3 ratio. (C) T4-to-T3 fractional conversion. (D) Absolute drop in serum T3 48 hours after daily PTU administration; the percentage drop is also indicated for each group. ***P < 0.001 vs. placebo-control rats. (E) Liver D1 activity. (F) BAT D2 activity. All entries in A–F are the mean ± SD (n = 49–94 per group).
Fractional T4-to-T3 conversion and contribution of D1 versus D2 pathways to serum T3 in T4-mono rats
The thyroid function tests of the Tx rats kept on different thyroid hormone replacement regimens suggest that impaired conversion of T4 to T3 might be a limiting factor in achieving normal serum T3. This was studied by first measuring the total-body fractional conversion of T4 to T3 in placebo-control rats and T4-mono rats and plotting the results versus serum T4 levels (Figure 1C). In placebo-control rats the fractional conversion of T4 to T3 was approximately 25% per day. In contrast, in the T4-mono rats the fractional conversion was lower at approximately 20%. Given that D2 is inhibited by T4 via ubiquitination, these findings suggest that the relative D2-mediated T3 production decreases because of the elevated serum T4 in the T4-mono rats.
To test this hypothesis, we obtained a baseline blood sample from T4-mono rats that were subsequently treated with 2 mg propylthiouracil (PTU) per 100 g body weight (BW) per day and euthanized 48 hours later. In the placebo-control animals, the drop in serum T3 — as defined by the difference between before and after treatment with PTU — was about 35%, whereas in the T4-mono rats, the drop in serum T3 was approximately 60% (Figure 1D). These figures indicate that the contribution of the D1 pathway to serum T3 is much greater and that of the D2 pathway much smaller in the T4-mono animals compared with placebo-control animals. This greater D1 versus D2 contribution to serum T3 was verified by measurement of D1 and D2 activities in the liver and BAT, respectively. Whereas liver D1 activity was decreased by approximately 10% in T4-mono rats (likely as a result of the lower serum T3 levels), BAT D2 activity was reduced by about 40% in the same group of animals (likely as a result of the elevated serum T4) (Figure 1, E and F).
Systemic impact of lower serum T3 in T4-mono rats: analysis of T3-dependent biological parameters in liver and skeletal muscle
Mitochondrial content and α-glycerolphosphate dehydrogenase activity in liver and skeletal muscle.
Tissue mitochondrial content is positively regulated by T3, and thus it was used as an index of thyroid hormone action in liver and skeletal muscle. Accordingly, there was an about 50% reduction in mitochondrial content in placebo-Tx animals in both liver and kidney (Table 2) as well as in the activity of the mitochondrial α-glycerolphosphate dehydrogenase (α-GPD), a typical T3-responsive enzyme in both of these tissues (Table 2). Notably, despite a significant improvement in serum thyroid function tests, T4-mono animals failed to normalize both mitochondrial content and α-GPD activity in liver and skeletal muscle, with values remaining significantly below those in placebo-control animals (Table 2). Only in the T4/T3-comb animals was there normalization of the mitochondrial content and α-GPD activity in liver and skeletal muscle (Table 1). Remarkably, in the T4/T3-inj-comb animals both parameters remained below the values observed in the placebo-control animals (Table 2).
Table 2.
Skeletal muscle and liver mitochondrial content, α-GPD activity, and lipid levels in the serum of Tx rats treated with different thyroid hormone replacement regimens
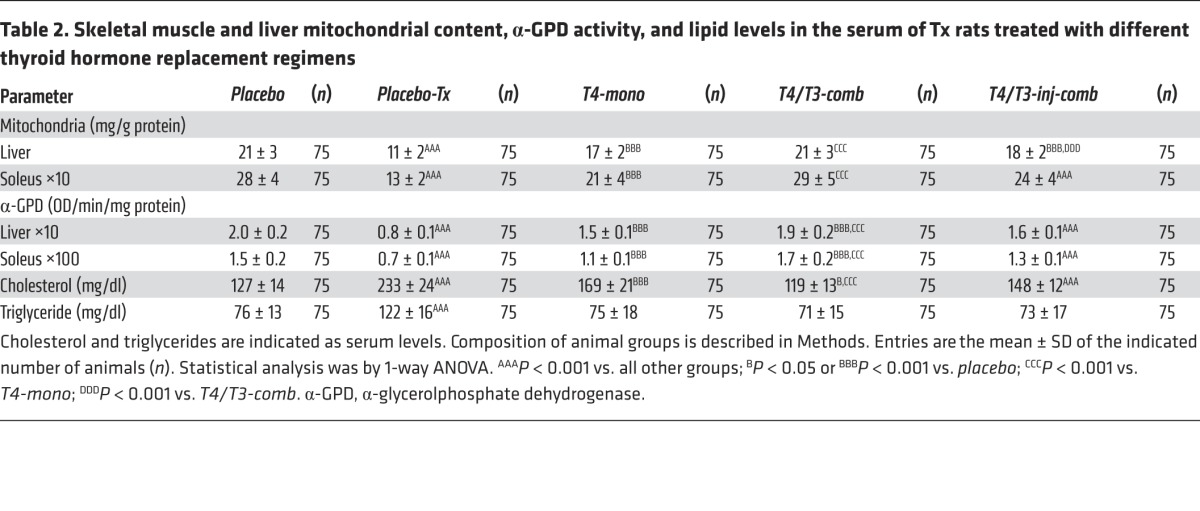
Serum cholesterol and triglyceride levels.
Serum cholesterol is negatively regulated by T3 and therefore almost doubled in the placebo-Tx rats (Table 2). At the same time, serum cholesterol was reduced in the T4-mono animals, although not to the levels observed in the placebo-control animals (Table 2). Only in the T4/T3-comb animals did serum cholesterol return to the level observed in the placebo-control animals (Table 2). In contrast, serum cholesterol remained elevated in the T4/T3-inj-comb animals (Table 2). Serum triglycerides were also elevated in placebo-Tx rats, but, in this case, all forms of replacement therapy resulted in normalization of their values, not different from in the placebo-control animals (Table 2).
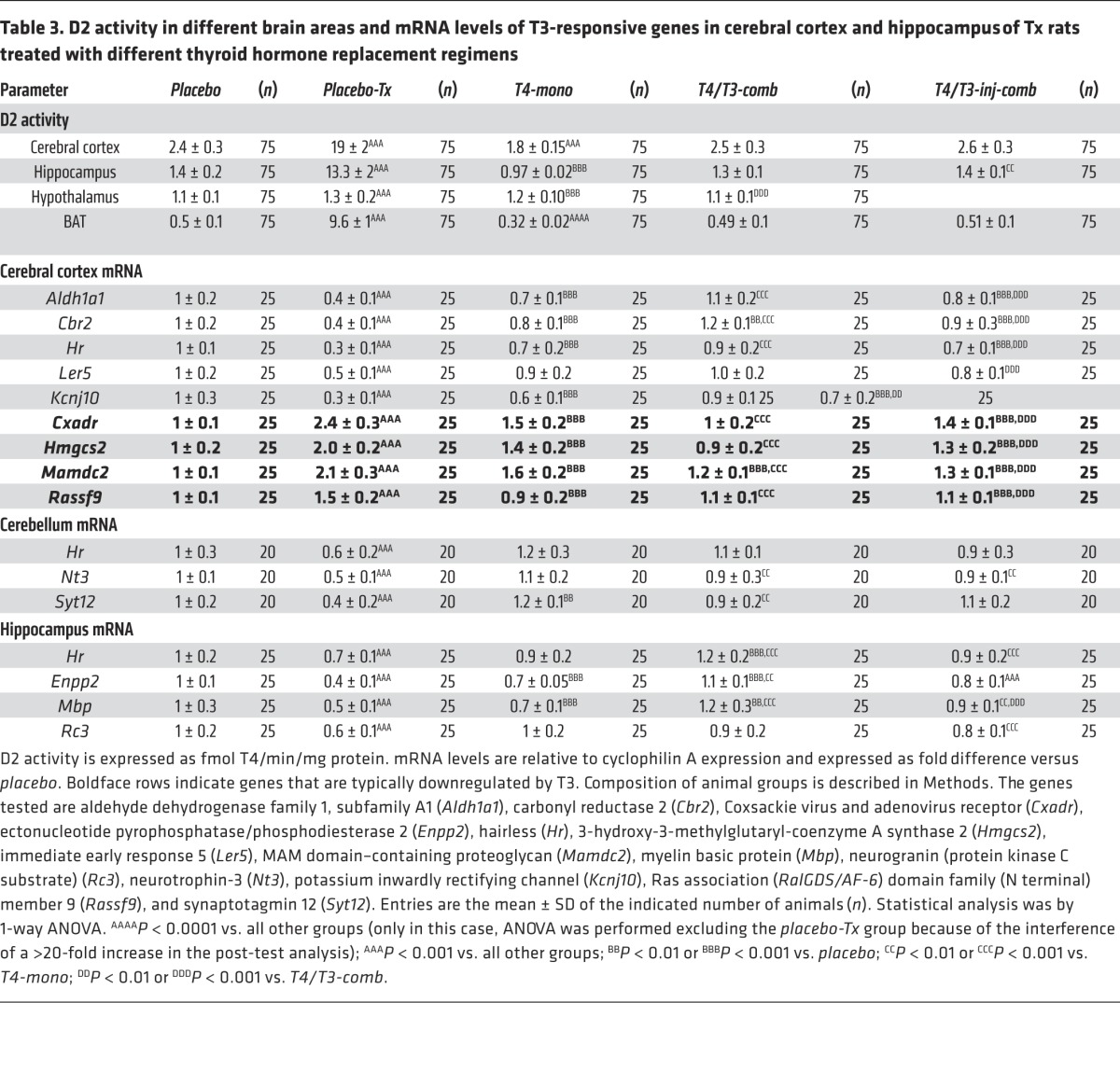
D2 activity in the rat cerebral cortex, hippocampus, and hypothalamus
It is clear that T4-mono rats exhibit lower BAT D2 activity reflecting their higher serum T4 levels. Is this true for D2 activity in the brain as well? To learn more about how D2 in different tissues responds to hypothyroidism and to the chronically elevated serum T4 levels, D2 activity was measured in the cerebral cortex, hippocampus, and hypothalamus. There was an 8- to 20-fold increase in D2 activity in the cerebral cortex, hippocampus, and BAT of the placebo-Tx rats (Table 3). D2 activity in the hypothalamus was notably less affected, increasing only by 18% in the placebo-Tx group (Table 3). Treatment with T4 (T4-mono animals) markedly reduced D2 activity in most areas of the brain and BAT, reaching levels that were significantly lower than those in placebo-control animals (Table 3). Again, hypothalamic D2 activity was less affected by T4-mono therapy, remaining 9% above levels seen in the placebo-control group (Table 3). Both T4/T3-comb and T4/T3-inj-comb animals exhibited D2 activity levels that were similar to those in placebo-control animals, while D2 activity in the hypothalamus was only minimally affected by either form of treatment (Table 3).
Table 3.
D2 activity in different brain areas and mRNA levels of T3-responsive genes in cerebral cortex and hippocampus of Tx rats treated with different thyroid hormone replacement regimens
WSB-1 modulates D2 activity through ubiquitination in discrete brain areas
As it became clear that there is tissue-specific regulation of D2 activity by T4, we hypothesized that this was due to differences in D2 ubiquitination via WSB-1, the D2-interacting ubiquitin ligase that mediates T4-induced D2 inactivation. Thus, we next studied the Astro-WSB-1KO (GFAP-Cre Wsb1fl/fl) mouse with selective disruption in WSB-1 expression in the astrocytes to study D2 regulation by T4 in the brain. As planned, these animals exhibited a reduction of about 50% in Wsb1 mRNA levels in the cerebral cortex, 80% in the hippocampus, and 65% in the cerebellum (Figure 2B), which was associated with doubling in D2 activity in the cerebral cortex and hippocampus, but not in the cerebellum (Figure 2D). Dio2 mRNA levels were not affected in any of these regions given the posttranscriptional nature of D2 regulation (Figure 2C). Unexpectedly, no changes in Wsb1 expression were observed in the hypothalamus (Figure 2B), indicating that in this region WSB-1 is predominantly expressed in GFAP-negative cells, i.e., neurons and tanycytes (35). Accordingly, D2 activity remained stable in this area (Figure 2D). To confirm that the D2 pathway is not affected in the hypothalamic hypophysiotropic area of the Astro-WSB-1KO mouse, we documented that the paraventricular nucleus Trh mRNA levels assessed by in situ hybridization, which are very sensitive to D2 activity, were indistinguishable between littermate controls and Astro-WSB-1KO animals (Supplemental Figure 1B; supplemental material available online with this article; doi:10.1172/JCI77588DS1).
Figure 2. Studies of the Astro-WSB-1KO mouse.

(A) Scheme of the mouse Wsb1 gene showing the 2 LoxP sites flanking exons 6 and 7. When the Wsb1fl/fl mouse is crossed with the GFAP-Cre mouse, exons 6 and 7 are floxed out and a frame stop codon is formed in exon 8 that prevents translation of the SOCS box located in exon 9. (B) Wsb1 mRNA levels in the hippocampus, cerebellum, cerebral cortex, and hypothalamus of Astro-WSB-1KO and littermate control mice. (C) Same as in B, except that Dio2 mRNA levels are shown. (D) Same as in C, except that D2 activity is shown. (B–D) Results are expressed as mean ± SD of 5–6 animals per group. *P < 0.01 vs. GFAP-Cre littermate control mice.
We next studied brain D2 activity in Astro-WSB-1KO animals that were made hypothyroid, which would naturally slow down D2 ubiquitination. In both littermate controls and Astro-WSB-1KO animals a mild degree of hypothyroidism was achieved with 65%–75% reduction in serum T4 concentration (control: 82 ± 18 vs. 25 ± 10 ng/ml; n = 5–6; P < 0.01; Astro-WSB-1KO: 98 ± 20 vs. 31 ± 13 ng/ml; n = 5–6; P < 0.01) and no changes in serum T3 concentration (0.45 ± 0.2 vs. 0.33 ± 0.16 ng/ml; n = 5–6; Astro-WSB-1KO: 0.42 ± 0.2 vs. 0.36 ± 0.10 ng/ml; n = 5–6). In the control animals, hypothyroidism led to a two- to fivefold elevation in D2 activity in the hippocampus, cerebral cortex, and cerebellum (Figure 3, A–C). At the same time, D2 activity increased to the same levels in Astro-WSB-1KO animals, except that in the hippocampus and cerebral cortex the baseline values were already higher in these animals (Figure 3, A–C). The elevation in D2 activity as a result of WSB-1 inactivation is indicated with a bracket (WSB-1); the residual (WSB-1–independent) elevation in D2 activity caused by hypothyroidism is likely due to the action of another ubiquitin ligase, TEB4 (Figure 3, A–C). Notably, D2 activity in the hypothalamus was not affected by hypothyroidism in either mouse strain (Figure 3D).
Figure 3. D2 activity in different areas of the brains of Astro-WSB-1KO and GFAP-Cre littermate controls.

(A) Hippocampus. (B) Cerebral cortex. (C) Cerebellum. (D) Hypothalamus. In A–D as indicated, some animals in both groups were made hypothyroid (Hypo) and subsequently treated with L-T4 (T4) for 2 weeks; other animals remained euthyroid (Eu). Results are expressed as mean ± SD of 5–6 animals per group. *P < 0.01 vs. Eu and T4 (GFAP-Cre littermate control); **P < 0.05 vs. Eu (GFAP-Cre littermate control); +P < 0.05 vs. Eu (Astro-WSB-1KO). The lines and the brackets labeled WSB-1 or TEB4 indicate the estimated role of each ubiquitin ligase in regulating D2 activity.
Next, hypothyroid Astro-WSB-1KO and littermate control animals were treated with T4 for 2 weeks in order to restore serum T4 levels (WT: 72 ± 29 ng/ml; Astro-WSB-1KO: 86 ± 8.2 ng/ml; n = 3–5). Whereas this normalized D2 activity in the hippocampus, cerebral cortex, and cerebellum of littermate controls (Figure 3, A–C), the same treatment in Astro-WSB-1KO mice only normalized D2 activity in the cerebellum (Figure 3C). In the hippocampus and cerebral cortex, treatment with T4 only brought D2 activity back to the relatively higher levels observed in intact Astro-WSB-1KO animals (Figure 3, A and B). Once again, D2 activity in the hypothalamus in all animals did not respond to treatment with T4 (Figure 3D).
An in vitro D2 ubiquitination assay to assess susceptibility to T4-induced loss of D2 activity
To define the molecular basis of such tissue specificity in the relationship between serum T4 and D2 activity and to bypass the limitation imposed by the poor coexpression of GFAP and WSB-1 in the hypothalamus, an in vitro ubiquitination assay was created in which microsomal 35S-D2 synthesized in 35S-labeled HEK-293 cells stably expressing D2 is incubated in vitro under conditions in which D2 can be ubiquitinated in the presence of a tissue-specific fraction II (29). The products of the reaction were visualized after size fractionation in SDS-PAGE followed by autoradiography (Figure 4). The 35S-D2 input and higher molecular bands corresponding to 35S-UbD2 can be seen in all lanes containing the appropriate reagents. The pattern of 35S-D2 bands was similar whether fraction II from hippocampus or cerebral cortex was used. However, the amount of 35S-UbD2 was much less when hypothalamic fraction II was used. Remarkably, the addition of recombinant human WSB-1 protein to the hypothalamic fraction II restored the pattern of 35S-UbD2 ubiquitination. No bands were observed when no tissue extract or ubiquitin was added to the reaction (Figure 4).
Figure 4. In vitro D2 ubiquitination driven by fraction II purified from the indicated tissues.

Autoradiography of an SDS-PAGE identifying 35S-ubiquitinated D2 (UbD2) produced in vitro. Addition of different components to the reaction mixture was as indicated. Fraction II from different tissues was added as indicated. This experiment was repeated twice with similar results.
CNS impact of combined lower serum T3 and D2 activity in T4-mono rats: analysis of T3-dependent biological parameters in the brain
Thyroid hormone signaling in the brain depends on serum T3 and on mechanisms that control local production and inactivation of thyroid hormone via D2 and D3, respectively. Thus, on the basis of our previous observation in cells that high T4 levels can reduce T3 production (18), we hypothesized that the brains of the T4-mono rats would be relatively hypothyroid despite normal serum TSH levels. To test this, we took advantage of the fact that the brain expresses a number of T3-responsive genes that can be used as indices of thyroid hormone activity (36). We analyzed the expression of a set of 10 positively regulated genes (cerebral cortex, hippocampus, and cerebellum) and a set of 4 negatively regulated genes (cerebral cortex) (Table 3). In the placebo-Tx animals the expression of the positively regulated genes was reduced to 30%–70% whereas the expression of the negatively regulated genes was increased 1.5- to 2.4-fold in comparison with the placebo-control animals (Table 3). Treatment with T4 (T4-mono rats) reversed this situation partially in the cerebral cortex and hippocampus and completely in the cerebellum in comparison with placebo-control animals. T4-mono treatment increased expression of the set of positively regulated genes in the cerebral cortex and hippocampus, but levels of the 5 genes remained below those in the placebo-control group (Table 3). In the cerebellum, the expression of all 3 genes was normalized by T4-mono, with 1 gene (Syt12) remaining in fact slightly higher when compared with that in the placebo-control group (Table 3). An inverse situation was seen with the genes that are negatively regulated by T3. Levels of all 4 genes were decreased by T4-mono but remained higher when compared with the levels observed in the placebo-control animals (Table 3). Remarkably, the expression of all genes, both positively and negatively regulated sets, was normalized in the T4/T3-comb therapy (Table 3). However, this was not the case when the animals receiving T4/T3-inj-com therapy were analyzed, in which case the expression of both sets of genes remained significantly different from that in placebo-control animals (Table 3).
Discussion
These studies model different thyroid hormone replacement strategies in Tx rats and provide 5 major advances toward understanding thyroid hormone homeostasis and action (Figure 5). (a) D2 susceptibility to ubiquitination differs among specific tissues as evident in different areas of the brain. These include areas where (i) WSB-1 mediates T4-induced D2 inactivation, i.e., cerebral cortex and hippocampus; (ii) WSB-1 does not mediate T4-induced D2 inactivation, i.e., cerebellum; and (iii) T4-induced D2 regulation is minimal (almost nonexistent), i.e., hypothalamus (Figures 2–4). (b) That the D2 regulation by T4 in the hypothalamus is poor was defined by the inability of hypothalamic extracts to mediate normal D2 ubiquitination. Thus, in the hypothalamus, T4 signaling is largely preserved throughout the spectrum of serum T4 levels tested here, even as serum T4 fluctuates above the levels observed in intact placebo-control animals. This resolves the apparent paradox between the homeostatic regulation of D2 and its role in mediating the critical mechanism by which T4 triggers the TSH negative feedback. These findings also explain how the higher serum T4 compensates for the lower serum T3 to normalize serum TSH in the T4-mono rats (Table 1). (c) Because D2 is generally downregulated by T4 in all tissues that were tested (Figure 1F and Figure 3, A–C) aside from the hypothalamus (Figure 3D), the fractional conversion of T4 to T3 is decreased in T4-mono rats (Figure 1C), explaining the lower serum T3 levels in these animals (Table 1). (d) The higher serum T4/T3 ratio observed in the T4-mono rats results from the combination of high serum T4 and low serum T3 levels (Table 1) and has important consequences for thyroid hormone action, as reflected in brain, liver, and skeletal muscle, all of which exhibit indications of hypothyroidism (Tables 2 And 3) despite normal serum TSH (Table 1). (e) Only the combined administration of continuous L-T4 and L-T3 via subcutaneous pellets normalized all parameters of thyroid hormone homeostasis (Table 1). While this point was reported in 2 seminal previous publications (30, 31), here we show that the same applies to T3-dependent biological effects (Tables 2 And 3), a finding that has potentially important clinical implication.
Figure 5. Illustration depicting the fundamentals of the hypothalamic-pituitary-thyroid axis, including the different sites in which T4-induced D2 ubiquitination plays a role in thyroid hormone homeostasis.

TRH-expressing neurons release TRH in the portal blood, which is transported to the anterior pituitary. There, TSH is secreted and stimulates the thyroid to produce T4 and T4. In most tissues, exposure to T4 accelerates D2 inactivation by ubiquitination and its targeting to the proteasomal system. UbD2 can also be reactivated and rescued from proteasomal destruction by DUB-mediated deubiquitination. The present findings indicate that peripheral deiodination is very sensitive to T4-induced D2 ubiquitination, and thus a mild elevation in the serum T4/T3 ratio favors D2 inactivation and decreases fractional conversion of T4 to T3 and peripheral T3 production. A similar situation is seen in different regions of the brain where the elevated serum T4/T3 ratio results in a gene expression profile typical of hypothyroidism. In contrast, hypothalamic D2 is less susceptible to T4-induced ubiquitination, and/or deubiquitination is so effective in this tissue that T4-induced D2 inactivation is insignificant. As a result, T4 signaling via D2-mediated T3 production is very effective in the hypothalamus, whereas T3 production via D2 is easily inhibited in the periphery. The situation in the pituitary thyrotrophs is probably intermediary between these two extremes on the basis of previously published data (18). This explains the discrepancy between normalization of TSH secretion and peripheral T3 production observed in L-T4–treated Tx rats.
The observation that T4-mono rats exhibit markers of reduced thyroid hormone signaling typical of hypothyroidism in certain areas of the brain, liver, and skeletal muscle was unexpected (Tables 2 And 3). In the liver it is logical to assume that this phenotype is the result of lower serum T3 levels, as local deiodination is not known to play a role in thyroid hormone signaling in the liver (9). In contrast, thyroid hormone signaling in the brain involves local D2-mediated T4-to-T3 conversion in astrocytes, with T3 exiting these cells and triggering paracrine effects in nearby neurons (16). At least 50% of the T3 in the brain is generated via this pathway (37). Thus, the facts that (a) D2 is downregulated by T4 in some brain areas and not others, and (b) the loss of D2 activity is mediated by WSB-1 in only some areas, possibly explain differences in thyroid hormone signaling within the same organ (Figures 2–4). This is likely to be aggravated by the underlying reduction in serum T3 levels (Table 1).
A similar scenario could also be present in other D2-expressing tissues such as skeleton and skeletal muscle given the bone and skeletal muscle phenotype of the Dio2 KO mouse (38, 39). However, in skeletal muscle, the D2 activity level is approximately 3 orders of magnitude lower than in BAT or brain (40). In addition, at least half of the D2 activity in this tissue is the result of the ectopic presence of BAT (41).
The present studies challenge the dogma that reduced D2 activity caused by exposure to T4 is compensated for by increased substrate availability, thus preserving, or even increasing, T3 production. While logical, this rationale requires experimental testing for each specific D2-expressing tissue, because the total-body T3 production via the D2 pathway is reduced by the higher serum T4/T3 ratio (Figure 1, B–D). In this regard, previously we observed in D2-expressing cells that T3 production trails T4 concentration in the medium only to a limited extent (18). There is an inflection point at which further elevations in T4 concentration reduce D2 activity and T3 production falls precipitously; the level of T4 at the inflection point depends on the cell type, presumably because of intrinsic differences in D2 ubiquitination (18). This phenomenon is likely to be exaggerated in the brain because of the concomitant induction of D3 expression by T3, which in turn accelerates local clearance of T3.
Inactivation of D2 by ubiquitination has been described in cell models but not in animal models (19, 20). It was, however, logical to expect that D2 ubiquitination occurs in vivo as well given that T4 is known to inactivate D2 in multiple tissues, including the brain (42). Here we used 2 approaches to prove that D2 undergoes WSB-1–mediated ubiquitination in different tissues. First, we developed the Astro-WSB-1KO mouse (Figure 2), which exhibits an elevation in brain D2 activity (without changes in Dio2 mRNA) that is not responsive to T4 administration (Figure 3). Notably, WSB-1–mediated D2 ubiquitination does not account for all of the elevation in D2 activity observed during hypothyroidism (Figure 3), the remainder being possibly explained by D2 ubiquitination via another ubiquitin ligase(s) such as TEB4 (27). Second, we used an in vitro system in which tissue-specific fraction II was prepared and used to drive D2 ubiquitination (Figure 4). This revealed that the hypothalamus is an area in the brain not so efficient at D2 ubiquitination when compared with all other areas studied. It is not clear why UbD2 does not accumulate in the hypothalamus given the local presence of Wsb1 mRNA, including in tanycytes (35). That the addition of recombinant WSB-1 drives D2 ubiquitination indistinguishable from that in the other brain areas (Figure 4) indicates perhaps that insufficient WSB-1 levels are present in these cells or that the endogenous WSB-1 molecules are complexed with other substrates, or that in the hypothalamus UbD2 deubiquitination predominates. In this respect, a major difference between tanycytes and astrocytes with respect to the posttranslational processing of D2 is that only tanycytes express the D2-interacting DUB USP-33 (35).
It is logical to assume that poor D2 ubiquitination in the hypothalamus explains why D2 in this area is only minimally sensitive to hypothyroidism and T4 administration (Table 3 and Figure3D). A similar lack of responsiveness to hypothyroidism was previously reported for hypothalamic Dio2 mRNA (14) and D2 activity (43) compared with marked elevation in the cortex and pituitary. Contrary to other regions in the brain, no increase in D2 activity in the hypothalamus has been observed in association with iodine deficiency (44).
Whatever the nature of the mechanism that preserves D2 in the hypothalamus, the end result is that the hypothalamus is exquisitely sensitive to serum T4 levels. This in fact has been documented in 2 independent studies. The first shows that systemic infusion of high doses of T4 into hypothyroid animals increases tissue and nuclear T3 in the hypothalamus rather than maintaining normal T3 levels (45). The second shows that a 40% elevation in serum T4, without changes in serum T3, is sufficient to reduce TRH expression by 50% (17). Thus, the hypothalamus stands in contrast to the other D2-expressing areas inside and outside the brain, in which the D2 pathway is downregulated by T4 (Table 3), limiting T3 production. In fact, it is difficult to conceive how the TRH/TSH feedback mechanism would function if an elevation in local D2 activity could compensate for the drop in serum T4; if this were the case, serum TSH would never respond to a drop in serum T4.
The present study explains why it is difficult to normalize serum T3 in Tx rats (and probably humans) just by increasing the replacement dose of L-T4. As the dose of L-T4 is increased and serum levels of T4 are raised, T3 is much more efficiently produced in the hypothalamus than in other tissues. As a result of the imbalance between T3 production in the hypothalamus and that in the rest of the body, TSH secretion is normalized before the L-T4 dose is sufficiently raised to normalize serum T3. This also explains why serum TSH is generally below normal in Tx patients in whom serum T3 was normalized by elevation of the dose of L-T4 (7). In addition, different patterns of response to the elevated serum T4/T3 ratio in the brain (Table 3) and possibly other tissues could explain residual “hypothyroid-like” symptoms exhibited by patients with normal serum TSH.
Previous studies in rats indicated that only the combined therapy with L-T4 and L-T3 normalizes all thyroid function parameters at the same time (30, 31). Here, we expanded these studies to demonstrate that not only thyroid function tests (Table 1) but also thyroid hormone–dependent biological parameters (Tables 2 and 3) can only be normalized by the stable administration of both hormones. It is remarkable that injections of L-T3 were not sufficient to normalize TH-dependent parameters (Tables 2 and 3); only the pellets with steady release did. This indicates that chronic fluctuations in serum T3 levels that are inherent to the subcutaneous injections (or oral administration) may not be well tolerated by T3-dependent tissues.
As the implications of these studies have significant potential impact on the treatment strategies for patients with hypothyroidism, studies should be repeated by other groups and extended to include human subjects as well. The fact that about 1,100 rats were studied for periods of up to 2 months provides unusual strength to the present observations. While the potential translational application to clinical practice is logical and much of what we know from human thyroid physiology was initially developed in rodents, these analogies should be done with utmost care. In rats the thyroid gland contributes with approximately 40% of the daily T3 production, while in humans it is approximately 20% (46); this makes rat thyroid hormone economy particularly dependent on thyroidal T3 production, relatively more than in humans. Thus, it would not be unexpected to see less dramatic effects if these studies were repeated in humans.
In conclusion, the present studies revealed that D2 ubiquitination is not universal in all tissues and that an inability of the hypothalamus to drive D2 ubiquitination is a pivotal element in the TRH/TSH negative-feedback regulation. This allows circulating thyroid hormone to function as a regulatory signal for hypophysiotropic TRH, and perhaps also for the control of appetite/satiety centers in the hypothalamus. Even in tissues where D2 is ubiquitinated and inactivated by exposure to T4, dependence on WSB-1 is not universal as well. Avoiding a higher serum T4/T3 ratio through chronic combination therapy with L-T4 and L-T3 through slow-release subcutaneous pellets was key to normalize thyroid hormone–dependent biological parameters in the brain, liver, and skeletal muscle.
Methods
Animals and treatments
Thyroidectomized rats.
All experimental protocols were performed according to official guidelines by the American Thyroid Association (47), and carried out on intact or surgically thyroidectomized (Tx) adult male Sprague Dawley rats (Taconic Farms Inc.) weighing 150–200 g.
Intact animals were housed in standard plastic cages with 4 rats per cage under standard environmental conditions (light between 0600 and 1800 hours, temperature 21°C ± 1°C, rat chow and water ad libitum). Tx rats were maintained similarly except that drinking water contained 0.05% methimazole (MMI; Sigma-Aldrich).
Some animals were implanted subcutaneously with slow-release pellets containing placebo, L-T4, or L-T3 (Innovative Research of America), designed to release the doses indicated below for 60 days. Pellets were implanted with the help of a stainless steel reusable precision trocar around the interscapular region. Other animals, as indicated, received daily subcutaneous injections of T3 dissolved in saline at the indicated dose.
Intact or Tx rats were treated for 50 ± 3 days as follows: (a) intact animals implanted with a subcutaneous placebo pellet (placebo-control group); (b) Tx animals implanted with a subcutaneous placebo pellet (placebo-Tx group); (c) Tx animals implanted with a subcutaneous L-T4 pellet to deliver T4 at the constant rate of 1 μg per 100 g BW per day (T4-mono group); (d) Tx animals implanted with a subcutaneous L-T4 plus L-T3 pellet to deliver T4 at the constant rate of 0.72 μg per 100 g BW per day and T3 at 0.12 μg per 100 g BW per day (T4/T3-comb group); or (e) Tx animals implanted with a subcutaneous L-T4 pellet to deliver T4 at the constant rate of 0.72 μg per 100 g BW per day and treated daily with a subcutaneous injection of T3 at 0.12 μg per 100 g BW per day (T4/T3-inj-comb group).
Generation of WSB-1 tissue-specific KO mice.
To establish an in vivo mouse model for cell type–specific deletion of WSB-1, first we generated floxed WSB-1 mice (Wsb1fl/fl) by inserting LoxP sites (48, 49) encompassing exons 6 and 7 of the Wsb1 gene (Figure 2A). When exons 6 and 7 are floxed out, an in-frame stop codon is formed in exon 8 that prevents translation of the more C-terminal portion, resulting in a partial disruption of the WD-40 repeat and in complete loss of the SOCS box located in exon 9. The Wsb1fl/fl mice were crossed with transgenic mice expressing Cre-recombinase under the astrocyte-specific glial fibrillary acidic protein promoter (GFAP-Cre) [FVB-Tg(GFAP-cre)25Mes/J; Jackson Laboratories] (50). This strategy was used to knock out WSB-1 in the GFAP-expressing astrocytes (Astro-WSB-1KO). In all experiments, Cre littermates were used as controls. Adult male mice were used between 9 and 12 weeks of age at the time of the studies. All mice were housed in a facility with a 12-hour light/dark cycle at 22°C and food and water ad libitum. Experiments were conducted according to protocols approved by the Animal Care and Use Committee of University of Miami Miller School of Medicine. All mice were genotyped by PCR of DNA isolated from tails. Astro-WSB-1KO animals exhibit normal serum TSH, T4, and T3 levels (Supplemental Figure 1A) and are systemically euthyroid with normal postnatal development and growth and metabolic rate — both parameters very sensitive to thyroid status — when compared with littermate controls (VO2: 264 ± 43 vs. 276 ± 68 l/kg/h). In some experiments, Astro-WSB-1KO and control mice were rendered hypothyroid by administration of 0.1% methimazole and 1% sodium perchlorate in the drinking water for 4 weeks. Control mice were kept with tap water. After 2 weeks, some mice received a daily injection of T4 (2 μg/kg BW, i.p.; Sigma Chemicals) or saline for the next 2 weeks.
In vivo fractional conversion of T4 to T3 and deiodinase-mediated T3 production in rats
In vivo fractional conversion of T4 to T3 was estimated in rats as described previously (11, 12, 51). Animals were placed under light anesthesia and the left jugular vein surgically exposed to inject [125I]T4 (PerkinElmer Life and Analytical Sciences Inc.) intravenously. Animals were sacrificed 8 hours later at the equilibrium time point (Tm), i.e., the time at which production and clearance rates of serum [125I]T3 are similar. Serum [125I]T3 was quantified after concentration by affinity chromatography with anti-T3 antiserum. At the Tm, the fractional conversion is calculated from the serum [125I]T3/[125I]T4 ratio. To estimate the contribution of the D1 and D2 pathways to serum T3 levels, blood samples were obtained by jugular puncture under light anesthesia before and after treatment with 2 mg intraperitoneal PTU per 100 g BW per day for 2 days. Liver samples were routinely obtained and processed for D1 activity, which was consistently more than 90% inhibited.
Indirect calorimetry in mice
Indirect calorimetry was performed in a comprehensive laboratory animal monitoring system (CLAMS; Columbus Instruments), a computer-controlled open-circuit calorimetry system. Animals were placed individually in metabolic cages where they were acclimatized at 22°C for 2 days with ad libitum access to water and food. Data were collected during the following 48 hours, as described previously (52). Metabolic profiles were generated based on real-time data obtained in successive 14-minute cycles. Sensors were calibrated against a standard gas mixture containing defined quantities of O2 and CO2 (Airgas). VO2 was expressed as milliliters per minute per kilogram of body weight.
Euthanasia and postmortem analyses
At the end of the experimental period, rats were euthanized by aortic exsanguination under light anesthesia. Mice were euthanized by asphyxiation in a CO2 chamber. The liver, quadriceps skeletal muscle, and BAT were harvested. The brain was also harvested and further dissected under a microscope to obtain hypothalamus, hippocampus, cerebellum, and cerebral cortex. All tissue samples were frozen and stored in the liquid nitrogen. Blood was collected and centrifuged for 10 minutes at 1,000 g within 30 minutes of blood collection. Serum was separated and stored at –80°C.
In situ hybridization and analysis
In situ hybridization histochemistry was performed on every fourth section through the paraventricular nucleus using a 741-base single-stranded [35S]UTP-labeled cRNA probe for mouse TRH (53). In vitro transcription was performed using SP6 (Promega Corp.) and [35S]UTP (1,250 Ci/mmol; PerkinElmer). The hybridization was performed under plastic coverslips in a buffer containing 50% formamide, a twofold concentration of standard sodium citrate (2XSSC), 10% dextran sulfate, 0.5% SDS, 250 μg/ml denatured salmon sperm DNA, and 5 × 105 cpm of radiolabeled probe for 16 hours at 56°C. Slides were dipped into Kodak NTB autoradiography emulsion (Eastman Kodak) diluted 1:1 in distilled water, and the autoradiograms developed after 3–7 days of exposure at 4°C. Autoradiograms were visualized under dark field illumination using a COHU 4910 video camera (COHU Inc.). The images were captured with a color PCI frame grabber board (Scion Corp.) and assembled in Photoshop (53).
Quantitative mRNA analysis
Total RNA was extracted from adipose tissue samples using the RNeasy kit (Qiagen). The extracted RNA was analyzed by a NanoDrop spectrophotometer, and 1.0 μg of total RNA was reverse transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Genes of interest as described in the legend to Table 3 were measured by quantitative RT-PCR (BioRad iCycler iQ Real-Time PCR Detection System) using the iQ SYBR Green Supermix (BioRad) with the following conditions: 15 minutes at 94°C (Hot Start), 30–50 seconds at 94°C, 30–50 seconds at 55°C–60°C, and 45–60 seconds at 72°C for 40 cycles. A final extension at 72°C for 5 minutes was performed as well as the melting curve protocol to verify the specificity of the amplicon generation. Standard curves consisting of 4–5 points of serial dilution of mixed experimental and control group cDNA were prepared for each assay. Cyclophilin A was used as a housekeeping internal control gene. The coefficient of correlation (r2) was greater than 0.98 for all standard curves, and the amplification efficiency varied between 80% and 110%. Results are expressed as ratios of test mRNA/cyclophilin A mRNA.
Analytical procedures
Serum was processed for measurement of TSH, T4, T3, and rT3 levels using a MILLIPLEX rat thyroid hormone panel kit following the instructions of the manufacturer (Millipore Corp.) and read on a BioPlex (BioRad). Serum cholesterol and triglyceride levels were measured using commercially available kits (Sigma-Aldrich). Liver and quadriceps mitochondrial fraction was isolated as previously described (54) and processed for determination of activity of α-GPD, a T3-sensitive nonlipogenic enzyme, as described previously (55). Mitochondrial α-GPD activity was expressed as OD units change per minute per milligram protein. Protein concentration was measured by the method of Bradford (56).
Deiodinase assays
D1 and D2 assays were performed in tissue sonicates in the presence of 10 mM DTT and 0.25 M sucrose as described previously (57). D1 and D2 assays were performed using 500 nM 125I-(5′)rT3 or 0.1 nM 125I-(5′)T4 (PerkinElmer Life and Analytical Sciences Inc.) as substrate, respectively.
Microsomal isolation and in vitro ubiquitination
The in vitro ubiquitination reaction was performed using a ubiquitin conjugation kit (K-960; Boston Biochem). The substrate was microsomal fraction of HEK-293 cells stably expressing YFP-D2 containing approximately 1,000,000 cpm of 35S-labeled proteins (22, 29). Conjugation Fraction A provided with the kit contains predominantly E1 and E2 enzymes. Instead, this was replaced with 300 μg of microsomal fraction II prepared from cerebral cortex, hippocampus, or hypothalamus as described and performed according to the manufacturer’s instructions in the presence of purified ubiquitin molecules (58). Shortly, the indicated area of the brains of 20 rats was dissected, cleaned, and kept in ice-cold PBS. Pools from each specific brain area were then lysed and separated on DEAE-cellulose into fraction I (unadsorbed material) and fraction II (0.5 M KCl eluate). Bacterially expressed GST or GST-WSB-1 was added to the reaction mixture as indicated (22), which also contained 1 μM MG132. Incubation lasted for 4 hours and was followed by immunoprecipitation (IP) with α-YFP and size-fractionation in SDS-PAGE followed by autoradiography. α-YFP IP was performed using the MACS GFP-tagged protein isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions (29).
Statistics
All data were analyzed using PRISM software (GraphPad Software) and are expressed as means ± SD. One-way ANOVA was used to compare more than 2 groups, followed by the Tukey’s test to detect differences between 2 groups. The Student’s t test was used to compare the differences between 2 groups. P < 0.05 was used to reject the null hypothesis.
Study approval
All studies were reviewed and approved by the local Institutional Animal Care and Use Committees at University of Miami Miller School of Medicine and at Rush University Medical Center.
Supplementary Material
Acknowledgments
The authors thank James Fagin for continued motivation and insightful suggestions during the course of these studies. Studies partially supported by the NIDDK and the National Brain Research Program (to B. Gereben).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(2):769–781. doi:10.1172/JCI77588.
References
- 1.Garber JR, et al. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Thyroid. 2012;22(12):1200–1235. doi: 10.1089/thy.2012.0205. [DOI] [PubMed] [Google Scholar]
- 2.Singer PA, et al. Treatment guidelines for patients with hyperthyroidism and hypothyroidism. Standards of Care Committee, American Thyroid Association. JAMA. 1995;273(10):808–812. [PubMed] [Google Scholar]
- 3.Stock JM, Surks MI, Oppenheimer JH. Replacement dosage of L-thyroxine in hypothyroidism. A re-evaluation. N Engl J Med. 1974;290(10):529–533. doi: 10.1056/NEJM197403072901001. [DOI] [PubMed] [Google Scholar]
- 4.Wiersinga WM, Duntas L, Fadeyev V, Nygaard B, Vanderpump MP. 2012 ETA Guidelines: The Use of L-T4 + L-T3 in the Treatment of Hypothyroidism. Eur Thyroid J. 2012;1(2):55–71. doi: 10.1159/000339444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alevizaki M, Mantzou E, Cimponeriu AT, Alevizaki CC, Koutras DA. TSH may not be a good marker for adequate thyroid hormone replacement therapy. Wien Klin Wochenschr. 2005;117(18):636–640. doi: 10.1007/s00508-005-0421-0. [DOI] [PubMed] [Google Scholar]
- 6.Gullo D, Latina A, Frasca F, Le Moli R, Pellegriti G, Vigneri R. Levothyroxine monotherapy cannot guarantee euthyroidism in all athyreotic patients. PLoS One. 2011;6(8):e22552. doi: 10.1371/journal.pone.0022552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito M, et al. TSH-suppressive doses of levothyroxine are required to achieve preoperative native serum triiodothyronine levels in patients who have undergone total thyroidectomy. Eur J Endocrinol. 2012;167(3):373–378. doi: 10.1530/EJE-11-1029. [DOI] [PubMed] [Google Scholar]
- 8.Panicker V, Evans J, Bjoro T, Asvold BO, Dayan CM, Bjerkeset O. A paradoxical difference in relationship between anxiety, depression and thyroid function in subjects on and not on T4: findings from the HUNT study. Clin Endocrinol (Oxf). 2009;71(4):574–580. doi: 10.1111/j.1365-2265.2008.03521.x. [DOI] [PubMed] [Google Scholar]
- 9.Gereben B, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev. 2008;29(7):898–938. doi: 10.1210/er.2008-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crantz FR, Silva JE, Larsen PR. Analysis of the sources and quantity of 3,5,3′-triiodothyronine specifically bound to nuclear receptors in rat cerebral cortex and cerebellum. Endocrinology. 1982;110(2):367–375. doi: 10.1210/endo-110-2-367. [DOI] [PubMed] [Google Scholar]
- 11.Bianco AC, Silva JE. Nuclear 3,5,3′-triiiodothyronine (T3) in brown adipose tissue: receptor occupancy and sources of T3 as determined by in vivo techniques. Endocrinology. 1987;120(1):55–62. doi: 10.1210/endo-120-1-55. [DOI] [PubMed] [Google Scholar]
- 12.Bianco AC, Silva JE. Cold exposure rapidly induces virtual saturation of brown adipose tissue nuclear T3 receptors. Am J Physiol. 1988;255(4):E496–E503. doi: 10.1152/ajpendo.1988.255.4.E496. [DOI] [PubMed] [Google Scholar]
- 13.Guadano-Ferraz A, Obregon MJ, St Germain DL, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci U S A. 1997;94(19):10391–10396. doi: 10.1073/pnas.94.19.10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tu HM, et al. Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology. 1997;138(8):3359–3368. doi: 10.1210/endo.138.8.5318. [DOI] [PubMed] [Google Scholar]
- 15.Tu HM, Legradi G, Bartha T, Salvatore D, Lechan RM, Larsen PR. Regional expression of the type 3 iodothyronine deiodinase messenger ribonucleic acid in the rat central nervous system and its regulation by thyroid hormone. Endocrinology. 1999;140(2):784–790. doi: 10.1210/endo.140.2.6486. [DOI] [PubMed] [Google Scholar]
- 16.Freitas BC, et al. Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J Clin Invest. 2010;120(6):2206–2217. doi: 10.1172/JCI41977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fonseca TL, et al. Coordination of hypothalamic and pituitary T3 production regulates TSH expression. J Clin Invest. 2013;123(4):1492–1500. doi: 10.1172/JCI61231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christoffolete MA, et al. Atypical expression of type 2 iodothyronine deiodinase in thyrotrophs explains the thyroxine-mediated pituitary thyrotropin feedback mechanism. Endocrinology. 2006;147(4):1735–1743. doi: 10.1210/en.2005-1300. [DOI] [PubMed] [Google Scholar]
- 19.Sagar GD, et al. Ubiquitination-induced conformational change within the deiodinase dimer is a switch regulating enzyme activity. Mol Cell Biol. 2007;27(13):4774–4783. doi: 10.1128/MCB.00283-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gereben B, Goncalves C, Harney JW, Larsen PR, Bianco AC. Selective proteolysis of human type 2 deiodinase: a novel ubiquitin-proteasomal mediated mechanism for regulation of hormone activation. Mol Endocrinol. 2000;14(11):1697–1708. doi: 10.1210/mend.14.11.0558. [DOI] [PubMed] [Google Scholar]
- 21.Zeold A, et al. Metabolic instability of type 2 deiodinase is transferable to stable proteins independently of subcellular localization. J Biol Chem. 2006;281(42):31538–31543. doi: 10.1074/jbc.M604728200. [DOI] [PubMed] [Google Scholar]
- 22.Dentice M, et al. The Hedgehog-inducible ubiquitin ligase subunit WSB-1 modulates thyroid hormone activation and PTHrP secretion in the developing growth plate. Nat Cell Biol. 2005;7(7):698–705. doi: 10.1038/ncb1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mentuccia D, et al. Association between a novel variant of the human type 2 deiodinase gene Thr92Ala and insulin resistance: evidence of interaction with the Trp64Arg variant of the β-3-adrenergic receptor. Diabetes. 2002;51(3):880–883. doi: 10.2337/diabetes.51.3.880. [DOI] [PubMed] [Google Scholar]
- 24.Bianco AC, Casula S. Thyroid hormone replacement therapy: three ‘simple’ questions, complex answers. Eur Thyroid J. 2012;1(2):88–98. doi: 10.1159/000339447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Botero D, Gereben B, Goncalves C, De Jesus LA, Harney JW, Bianco AC. Ubc6p and ubc7p are required for normal and substrate-induced endoplasmic reticulum-associated degradation of the human selenoprotein type 2 iodothyronine monodeiodinase. Mol Endocrinol. 2002;16(9):1999–2007. doi: 10.1210/me.2002-0135. [DOI] [PubMed] [Google Scholar]
- 26.Kim BW, et al. Endoplasmic reticulum-associated degradation of the human type 2 iodothyronine deiodinase (D2) is mediated via an association between mammalian UBC7 and the carboxyl region of D2. Mol Endocrinol. 2003;17(12):2603–2612. doi: 10.1210/me.2003-0082. [DOI] [PubMed] [Google Scholar]
- 27.Zavacki AM, et al. The E3 ubiquitin ligase TEB4 mediates degradation of type 2 iodothyronine deiodinase. Mol Cell Biol. 2009;29(19):5339–5347. doi: 10.1128/MCB.01498-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curcio-Morelli C, et al. Deubiquitination of type 2 iodothyronine deiodinase by von Hippel-Lindau protein-interacting deubiquitinating enzymes regulates thyroid hormone activation. J Clin Invest. 2003;112(2):189–196. doi: 10.1172/JCI18348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arrojo EDR, Egri P, Jo S, Gereben B, Bianco AC. The type II deiodinase is retrotranslocated to the cytoplasm and proteasomes via p97/Atx3 complex. Mol Endocrinol. 2013;27(12):2105–2115. doi: 10.1210/me.2013-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Escobar-Morreale HF, Rey F, Obregon MJ, Escobar GM. Only the combined treatment with thyroxine and triiodothyronine ensures euthyroidism in all tissues of the thyroidectomized rat. Endocrinology. 1996;137(6):2490–2502. doi: 10.1210/endo.137.6.8641203. [DOI] [PubMed] [Google Scholar]
- 31.Escobar-Morreale HF, Obregon MJ, Escobar del Rey F, Morreale de Escobar G. Replacement therapy for hypothyroidism with thyroxine alone does not ensure euthyroidism in all tissues, as studied in thyroidectomized rats. J Clin Invest. 1995;96(6):2828–2838. doi: 10.1172/JCI118353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grozinsky-Glasberg S, Fraser A, Nahshoni E, Weizman A, Leibovici L. Thyroxine-triiodothyronine combination therapy versus thyroxine monotherapy for clinical hypothyroidism: meta-analysis of randomized controlled trials. J Clin Endocrinol Metab. 2006;91(7):2592–2599. doi: 10.1210/jc.2006-0448. [DOI] [PubMed] [Google Scholar]
- 33.Panicker V, et al. Common variation in the DIO2 gene predicts baseline psychological well-being and response to combination thyroxine plus triiodothyronine therapy in hypothyroid patients. J Clin Endocrinol Metab. 2009;94(5):1623–1629. doi: 10.1210/jc.2008-1301. [DOI] [PubMed] [Google Scholar]
- 34.Dayan CM, Panicker V. Novel insights into thyroid hormones from the study of common genetic variation. Nat Rev Endocrinol. 2009;5(4):211–218. doi: 10.1038/nrendo.2009.19. [DOI] [PubMed] [Google Scholar]
- 35.Fekete C, et al. Expression patterns of WSB-1 and USP-33 underlie cell-specific posttranslational control of type 2 deiodinase in the rat brain. Endocrinology. 2007;148(10):4865–4874. doi: 10.1210/en.2007-0448. [DOI] [PubMed] [Google Scholar]
- 36.Hernandez A, Morte B, Belinchon MM, Ceballos A, Bernal J. Critical role of types 2 and 3 deiodinases in the negative regulation of gene expression by t3 in the mouse cerebral cortex. Endocrinology. 2012;153(6):2919–2928. doi: 10.1210/en.2011-1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider M, Clark AS, Germain DL. Life without T4 to T3 conversion: studies in mice devoid of the 5′-deiodinases. Endocrinology. 2009;150(6):2957–2963. doi: 10.1210/en.2008-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dentice M, et al. The FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration. J Clin Invest. 2011;120(11):4021–4030. doi: 10.1172/JCI43670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bassett JH, et al. Optimal bone strength and mineralization requires the type 2 iodothyronine deiodinase in osteoblasts. Proc Natl Acad Sci U S A. 2010;107(16):7604–7609. doi: 10.1073/pnas.0911346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramadan W, Marsili A, Huang S, Larsen PR, Silva JE. Type-2 iodothyronine 5’deiodinase in skeletal muscle of C57BL/6 mice. I. Identity, subcellular localization, and characterization. Endocrinology. 2011;152(8):3082–3092. doi: 10.1210/en.2011-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Werneck de Castro JP, et al. Both skeletal myocytes and adipocytes express the type 2 deiodinase within skeletal muscle. Presented at: 83rd Annual Meeting of the Americam Thyroid Association; October 17, 2013; San Juan, Porto Rico. [Google Scholar]
- 42.Peeters R, et al. Regional physiological adaptation of the central nervous system deiodinases to iodine deficiency. Am J Physiol Endocrinol Metab. 2001;281(1):E54–E61. doi: 10.1152/ajpendo.2001.281.1.E54. [DOI] [PubMed] [Google Scholar]
- 43.Diano S, Naftolin F, Goglia F, Horvath TL. Fasting-induced increase in type II iodothyronine deiodinase activity and messenger ribonucleic acid levels is not reversed by thyroxine in the rat hypothalamus. Endocrinology. 1998;139(6):2879–2884. doi: 10.1210/endo.139.6.6062. [DOI] [PubMed] [Google Scholar]
- 44.Serrano-Lozano A, Montiel M, Morell M, Morata P. 5′ Deiodinase activity in brain regions of adult rats: modifications in different situations of experimental hypothyroidism. Brain Res Bull. 1993;30(5):611–616. doi: 10.1016/0361-9230(93)90090-x. [DOI] [PubMed] [Google Scholar]
- 45.Broedel O, et al. Effects of hyper- and hypothyroidism on thyroid hormone concentrations in regions of the rat brain. Am J Physiol Endocrinol Metab. 2003;285(3):E470–E480. doi: 10.1152/ajpendo.00043.2003. [DOI] [PubMed] [Google Scholar]
- 46.Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. 2002;23(1):38–89. doi: 10.1210/edrv.23.1.0455. [DOI] [PubMed] [Google Scholar]
- 47.Bianco AC, et al. American thyroid association guide to investigating thyroid hormone economy and action in rodent and cell models. Thyroid. 2014;24(1):88–168. doi: 10.1089/thy.2013.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abremski K, Hoess R. Bacteriophage P1 site-specific recombination. Purification and properties of the Cre recombinase protein. J Biol Chem. 1984;259(3):1509–1514. [PubMed] [Google Scholar]
- 49.Hoess RH, Abremski K. Mechanism of strand cleavage and exchange in the Cre-lox site-specific recombination system. J Mol Biol. 1985;181(3):351–362. doi: 10.1016/0022-2836(85)90224-4. [DOI] [PubMed] [Google Scholar]
- 50.Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31(2):85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 51.Carvalho SD, Kimura ET, Bianco AC, Silva JE. Central role of brown adipose tissue thyroxine 5′-deiodinase on thyroid hormone-dependent thermogenic response to cold. Endocrinology. 1991;128(4):2149–2159. doi: 10.1210/endo-128-4-2149. [DOI] [PubMed] [Google Scholar]
- 52.Castillo M, et al. Disruption of thyroid hormone activation in type 2 deiodinase knockout mice causes obesity with glucose intolerance and liver steatosis only at thermoneutrality. Diabetes. 2011;60(4):1082–1089. doi: 10.2337/db10-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kadar A, et al. Distribution of hypophysiotropic thyrotropin-releasing hormone (TRH)-synthesizing neurons in the hypothalamic paraventricular nucleus of the mouse. J Comp Neurol. 2010;518(19):3948–3961. doi: 10.1002/cne.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carvalho SD, Negrao N, Bianco AC. Hormonal regulation of malic enzyme and glucose-6-phosphate dehydrogenase in brown adipose tissue. Am J Physiol. 1993;264(6):E874–E881. doi: 10.1152/ajpendo.1993.264.6.E874. [DOI] [PubMed] [Google Scholar]
- 55.Gardner RS. A sensitive colorimetric assay for mitochondrial alpha-glycerophosphate dehydrogenase. Anal Biochem. 1974;59(1):272–276. doi: 10.1016/0003-2697(74)90033-5. [DOI] [PubMed] [Google Scholar]
- 56.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 57.Curcio-Morelli C, et al. In vivo dimerization of types 1, 2, and 3 iodothyronine selenodeiodinases. Endocrinology. 2003;144(3):937–946. doi: 10.1210/en.2002-220960. [DOI] [PubMed] [Google Scholar]
- 58.Ciechanover A, Heller H, Elias S, Haas AL, Hershko A. ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc Natl Acad Sci U S A. 1980;77(3):1365–1368. doi: 10.1073/pnas.77.3.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.