

Abstract
Anabolic biosynthesis requires precursors supplied by the Krebs cycle, which in turn requires anaplerosis to replenish precursor intermediates. The major anaplerotic sources are pyruvate and glutamine, which require the activity of pyruvate carboxylase (PC) and glutaminase 1 (GLS1), respectively. Due to their rapid proliferation, cancer cells have increased anabolic and energy demands; however, different cancer cell types exhibit differential requirements for PC- and GLS-mediated pathways for anaplerosis and cell proliferation. Here, we infused patients with early-stage non–small-cell lung cancer (NSCLC) with uniformly 13C-labeled glucose before tissue resection and determined that the cancerous tissues in these patients had enhanced PC activity. Freshly resected paired lung tissue slices cultured in 13C6-glucose or 13C5,15N2-glutamine tracers confirmed selective activation of PC over GLS in NSCLC. Compared with noncancerous tissues, PC expression was greatly enhanced in cancerous tissues, whereas GLS1 expression showed no trend. Moreover, immunohistochemical analysis of paired lung tissues showed PC overexpression in cancer cells rather than in stromal cells of tumor tissues. PC knockdown induced multinucleation, decreased cell proliferation and colony formation in human NSCLC cells, and reduced tumor growth in a mouse xenograft model. Growth inhibition was accompanied by perturbed Krebs cycle activity, inhibition of lipid and nucleotide biosynthesis, and altered glutathione homeostasis. These findings indicate that PC-mediated anaplerosis in early-stage NSCLC is required for tumor survival and proliferation.
Introduction
Accelerated glycolysis under aerobic conditions (the “Warburg effect”) has been a hallmark of cancer for many decades (1). It is now recognized that cancer cells must undergo many other metabolic reprogrammings (2) to meet the increased anabolic and energetic demands of proliferation (3, 4). It is also becoming clear that different cancer types may utilize a variety of metabolic adaptations that are context dependent (5–11), commensurate with the notion that altered metabolism is a hallmark of cancer (12). Enhanced glucose uptake and aerobic glycolysis generates both energy (i.e., ATP) and molecular precursors for the biosynthesis of complex carbohydrates, sugar nucleotides, lipids, proteins, and nucleic acids (3, 13, 14). However, increased glycolysis alone is insufficient to meet the total metabolic demands of proliferating cancer cells. The Krebs cycle is also a source of energy via the oxidation of pyruvate, fatty acids (15), and amino acids such as glutamine (10, 16, 17). Moreover, several Krebs cycle intermediates are essential for anabolic and glutathione metabolism, including citrate, oxaloacetate, and α-ketoglutarate (Figure 1A).
Figure 1. PC is activated in human NSCLC tumors.

(A) PC and GLS1 catalyze the major anaplerotic inputs (blue) into the Krebs cycle to support the anabolic demand for biosynthesis (green). Also shown is the fate of 13C from 13C6-glucose through glycolysis and into the Krebs cycle via PC (red). (B) Representative Western blots of PC and GLS1 protein expression levels in human NC lung (N) and NSCLC (C) tissues. (C) Pairwise PC and GLS1 expression (n = 86) was normalized to α-tubulin and plotted as the log10 ratio of CA/NC tissues. For PC, nearly all log ratios were positive (82 of 86), with a clustering in the 0.5–1 range (i.e., typically 3- to 10-fold higher expression in the tumor tissue; Wilcoxon test, P < 0.0001). In contrast, GLS1 expression was nearly evenly distributed between positive and negative log10 ratios and showed no statistically significant difference between the CA and NC tissues (Wilcoxon test, P = 0.213). Horizontal bar represents the median. (D) In vivo PC activity was enhanced in CA tissue compared with that in paired NC lung tissues (n = 34) resected from the same human patients given 13C6-glucose 2.5–3 hours before tumor resection. PC activity was inferred from the enrichment of 13C3-citrate (Cit+3), 13C5-Cit (Cit+5), 13C3-malate (Mal+3), and 13C3-aspartate (Asp+3) as determined by GC-MS. *P < 0.05 and **P < 0.01 by paired Student t test. Error bars represent the SEM.
Continued functioning of the Krebs cycle requires the replenishment of intermediates that are diverted for anabolic uses or glutathione synthesis. This replenishment process, or anaplerosis, is accomplished via 2 major pathways: glutaminolysis (deamidation of glutamine via glutaminase [GLS] plus transamination of glutamate to α-ketoglutarate) (18, 19) and carboxylation of pyruvate to oxaloacetate via ATP-dependent pyruvate carboxylase (PC) (EC 6.4.1.1) (refs. 3, 20, 21, and Figure 1A). The relative importance of these pathways is likely to depend on the nature of the cancer and its specific metabolic adaptations, including those to the microenvironment (20, 22).
For example, glutaminolysis was shown to be activated in the glioma cell line SF188, while PC activity was absent (17), despite the high PC activity present in normal astrocytes (23, 24). However, SF188 cells use PC to compensate for GLS1 suppression or glutamine restriction (20), and PC, rather than GLS1, was shown to be the major anaplerotic input to the Krebs cycle in primary glioma xenografts in mice (22). It is also unclear as to the relative importance of PC and GLS1 in other cancer cell types or, most relevantly, in human tumor tissues in situ. Our preliminary evidence from 5 non–small-cell lung cancer (NSCLC) patients indicated that PC expression and activity are upregulated in cancerous (CA) compared with paired noncancerous (NC) lung tissues (21), although it was unclear whether PC activation applies to a larger NSCLC cohort or whether PC expression was associated with the cancer and/or stromal cells. Nor has the relationship between PC activation and glutaminase status been explored in human patient studies. Moreover, the role of PC in cell survival and proliferation and whether glutaminolysis can compensate for this role under PC suppression in lung cancer cells is unknown.
Here, we have greatly extended our previous findings (21) in a larger cohort (n = 86) by assessing glutaminase 1 (GLS1) status and analyzing in detail the biochemical and phenotypic consequences of PC suppression in NSCLC. We found PC activity and protein expression levels to be, on average, respectively, 100% and 5- to 10-fold higher in cancerous (CA) lung tissues than in paired NC lung tissues resected from NSCLC patients, whereas GLS1 expression showed no significant trend. We have also applied stable isotope–resolved metabolomic (SIRM) analysis to paired freshly resected CA and NC lung tissue slices in culture (analogous to the Warburg slices; ref. 25) using either [U-13C] glucose or [U-13C,15N] glutamine as tracers. This novel method provided information about tumor metabolic pathways and dynamics without the complication of whole-body metabolism in vivo. We used immunohistochemical analysis to verify the specific localization of PC in cancer cells within the tumor tissue. We further determined the functional role of PC in NSCLC cell lines using shRNA, which showed that attenuation of PC activity inhibited cell proliferation, colony formation, and tumor growth in mouse xenografts, while compromising the cells’ ability to quench oxygen radicals. These phenotypic effects can be linked to several important metabolic outcomes of PC suppression in the lung adenocarcinoma A549 cell line cultured in vitro and in vivo, including reduced lipid and nucleotide biosynthesis and altered glutathione homeostasis.
Results
PC expression and activity, but not glutaminase expression, are significantly enhanced in early stages of malignant NSCLC tumors.
PC protein expression was significantly higher in primary NSCLC tumors than in paired adjacent NC lung tissues (n = 86, P < 0.0001, Wilcoxon test) (Figure 1, B and C). The median PC expression was 7-fold higher in the tumor, and the most probable (modal) overexpression in the tumor was approximately 3-fold higher (see Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI72873DS1). We found that PC expression was also higher in the tumor tissue compared with that detected in the NC tissue in 82 of 86 patients. In contrast, GLS1 expression was not significantly different between the tumor and NC tissues (P = 0.213, Wilcoxon test) (Figure 1C and Supplemental Table 1).
To estimate in vivo PC activity, the production of 13C3-Asp from 13C6-glucose (Figure 1A) infused into NSCLC patients was determined by gas chromatography–mass spectrometry (GC-MS). A bolus injection of 10 g 13C6-glucose in 50 ml saline led to an average of 44% 13C enrichment in the plasma glucose immediately after infusion (Supplemental Table 2). Because the labeled glucose was absorbed by various tissues over the approximately 2.5 hours between infusion and tumor resection, plasma glucose enrichment dropped to 17% (Supplemental Table 2). The labeled glucose in both CA and NC lung tissues was metabolized to labeled lactate, but this occurred to a much greater extent in the CA tissues (Supplemental Figure 1A), which indicates accelerated glycolysis in these tissues.
The enrichment of 13C3-Asp (a PC marker, Figure 1A) was on average 117% higher (n = 34, P < 0.005) in NSCLC tumors than in NC lung tissue (Figure 1D). 13C3-Asp can also be produced after the second turn of the Krebs cycle via 13C4-citrate, which is the condensation product of 13C2-acetyl CoA and 13C2-oxaloacetate (OAA). Since the 13C enrichment of the 13C4-citrate precursor was lower than that of the 13C3-Asp product (Supplemental Figure 1, B and C, and Figure 1D), it argues against a significant contribution of this route to 13C3-Asp enrichment. We also measured PC activity by the fractional 13C enrichment of other markers, 13C3-citrate, 13C5-citrate, and 13C3-malate, which were, respectively, 27%, 88%, and 113% higher in CA lung tissues than in NC lung tissues (Figure 1D). We also observed that 13C3-citrate was 5- to 6-fold higher in fractional enrichment than was 13C5-citrate for both CA and NC lungs. This could be attributed to the expectedly low 13C enrichment in pyruvate and thus acetyl CoA due to a large dilution of the 13C-glucose tracer in the human body, leading to a lower probability of condensation of 13C3-OAA with 13C2-acetyl CoA than with unlabeled acetyl CoA to produce less 13C5-citrate than 13C3-citrate. Moreover, the fractional enhancement of 13C3-citrate in CA over NC lungs was less than that of 13C5-citrate (Figure 1D). This could be a result of increased flux through the pyruvate dehydrogenase–initiated (PDH-initiated) Krebs cycle reactions in CA lung compared with that in NC lung, as evidenced by the elevated fraction of 13C2 isotopologs of Krebs cycle metabolites in CA versus NC lung (Supplemental Figure 1B). This could in turn lead to a relatively higher enrichment of 13C2-acetyl CoA and thus its product (13C5-citrate) of condensation with 13C3-OAA in CA lung compared with that in NC lung. Altogether, the significant increase in the fractional enrichment of these PC markers reflects increased PC activity (which we refer to herein) in the cancer tissue.
In addition to enhanced production of labeled PC markers, we observed a higher percentage of enrichment in the 13C2-isotopologs of Krebs cycle metabolites (citrate+2, glutamate+2, succinate+2, fumarate+2, malate+2, and aspartate+2 in Supplemental Figure 1B) in NSCLC tumors than in NC lung tissue, which is indicative of enhanced glucose oxidation in the Krebs cycle via PDH.
Fresh tissue (Warburg) slices confirm enhanced PC and Krebs cycle activity in NSCLC.
To further assess PC activity relative to GLS1 activity in human lung tissues, thin (<1 mm thick) slices of paired CA and NC lung tissues freshly resected from 13 human NSCLC patients were cultured in 13C6-glucose or 13C5,15N2-glutamine for 24 hours. These tissues maintain biochemical activity and histological integrity for at least 24 hours under culture conditions (Figure 2A, Supplemental Figure 2, A and B, and ref. 26). When the tissues were incubated with 13C6-glucose, CA slices showed a significantly greater percentage of enrichment in glycolytic 13C3-lactate (3 in Figure 2B) than did the NC slices, indicative of the Warburg effect. In addition, the CA tissues had significantly higher fractions of 13C4-, 13C5-, and 13C6-citrate (4, 5, and 6 of citrate, respectively, in Figure 2B) than did the NC tissues. These isotopologs require the combined action of PDH, PC, and multiple turns of the Krebs cycle (Figure 2C). Consistent with the labeled citrate data, the increase in the percentage of enrichment of 13C3-, 13C4-, and 13C5-glutamate (3, 4, and 5 of glutamate, respectively, in Figure 2B) in the CA tissues indicates enhanced Krebs cycle and PC activity.
Figure 2. Ex vivo CA lung tissue slices have enhanced oxidation of glucose through glycolysis and the Krebs cycle with and without PC input compared with that of paired NC lung slices.

Thin slices of CA and NC lung tissues freshly resected from 13 human NSCLC patients were incubated with 13C6-glucose for 24 hours as described in the Methods. The percentage of enrichment of lactate, citrate, glutamate, and aspartate was determined by GC-MS. (A) 1H{13C} HSQC NMR showed an increase in labeled lactate, glutamate, and aspartate. In addition, CA tissues had elevated 13C abundance in the ribose moiety of the adenine-containing nucleotides (1′-AXP), indicating that the tissues were viable and had enhanced capacity for nucleotide synthesis. (B) CA tissue slices (n = 13) showed increased glucose metabolism through glycolysis based on the increased percentage of enrichment of 13C3-lactate (“3”), and through the Krebs cycle based on the increased percentage of enrichment of 13C4–6-citrate (“4–6”) and 13C3–5-glutamate (“3–5”) (see 13C fate tracing in C). *P < 0.05 and **P < 0.01 by paired Student’s t test. Error bars represent the SEM. (C) An atom-resolved map illustrates how PC, PDH, and 2 turns of the Krebs cycle activity produced the 13C isotopologs of citrate and glutamate in B, whose enrichment were significantly enhanced in CA tissue slices.
When tissue slices were incubated with 13C5,15N2-glutamine, 13C5,15N1-glutamate (6 of glutamate in Supplemental Figure 2C) was produced in both CA and NC tissues, indicating active glutaminase (see below). In addition, the 13C4-isotopologs of succinate, fumarate, malate (data not shown), and citrate (Supplemental Figure 2C) were detected, which confirms glutaminase action. However, the enrichment patterns of all but 1 isotopolog of the Krebs cycle intermediates derived from 13C5,15N2-glutamine were comparable between CA and NC tissues (Supplemental Figure 2C). These data, along with the GLS1 expression data (Figure 1C), indicate that GLS1 is active and likely a significant player in the anaplerosis of the Krebs cycle in both types of tissues. But unlike PC, GLS1 was not significantly activated in NSCLC tumors relative to that seen in NC lung tissues.
PC is localized in the cancer cells within NSCLC tumors.
NSCLC tumors are very heterogeneous and comprise various cell types including cancerous epithelial cells, pneumocytes, fibroblasts, endothelial cells, and immune cells such as macrophages. To determine whether PC is expressed specifically in cancer cells, we performed immunohistochemical analysis of paired CA and NC tissues from 9 patient samples. Figure 3 and Supplemental Figure 3 illustrate elevated PC levels in 4 NSCLC tumors compared with levels in the paired NC lung tissue, which was also evident in 5 other pairs of samples (data not shown) and is consistent with the Western blot data (Figure 1C). Moreover, PC was localized in the perinuclear region of the cancerous epithelial cells but not in stromal cells in the tumor tissue (Figure 3, bottom left panel, and Supplemental Figure 3D). Such a perinuclear staining pattern is consistent with the localization of PC in the mitochondria, which is supported by the perinuclear costaining of PC and the mitochondrial marker in NSCLC A549 cells (Supplemental Figure 3, C and D).
Figure 3. Immunohistochemical localization of PC in lung tissues.
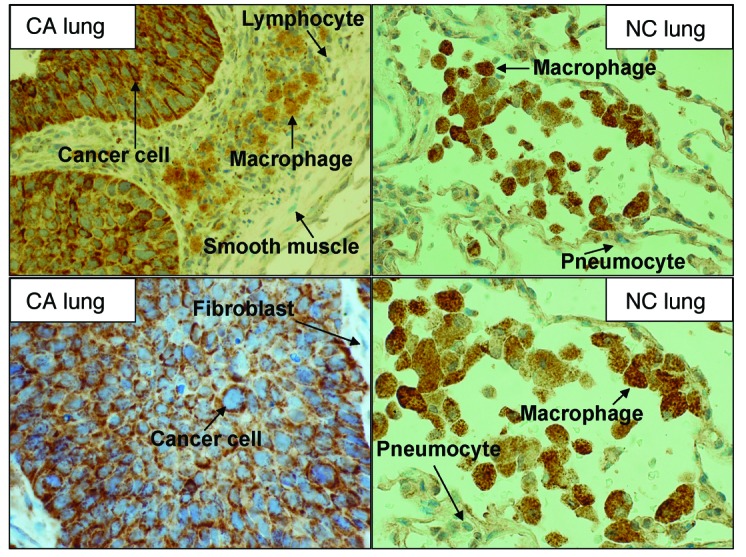
Pairs of CA and NC tissues obtained from NSCLC patients were stained with anti-PC antibody. Shown are representative pairs of micrographs from 1 patient. Original magnification, ×400 (top panels) and ×600 (bottom panels). Additional patient data are provided in Supplemental Figure 3. In CA tissues, viable cancer cells stained intensely for PC, while macrophages stained weakly, and other stromal cells showed no staining for PC. In adjacent NC tissues, macrophages stained strongly for PC, while pneumocytes stained very weakly for the protein.
In contrast, in the NC lung tissue, only macrophages were enriched in PC, while PC in other cell types stained weakly or was absent (Figure 3 and Supplemental Figure 3). Thus, PC is preferentially localized in the cancer cells of heterogeneous lung tumor tissues.
PC suppression by shRNA in lung cancer cells leads to growth arrest and decreased tumor burden in vivo.
In order to assess the role of PC in the survival and growth of lung cancer cells, we used PC-specific shRNA to suppress PC mRNA expression in human A549 adenocarcinoma cells. As shown in Supplemental Figure 4A, vector shPC54 reduced PC protein levels to 20% of the control levels, whereas PC expression was barely detected in cells transduced with shPC55. To verify the inhibition of PC activity in cells, we incubated empty vector– (shEV), shPC54-, and shPC55-transduced A549 cells in 13C6-glucose for 24 hours and measured the markers for PC activity by GC-MS (Figure 1A). The levels of 13C3-citrate, 13C3-aspartate, and 13C3-malate were reduced by both constructs; however, shPC55 was more effective than shPC54 (Supplemental Figure 4B). It is interesting to note that increased PC activity may occur in other unrelated transformed cell lines (3, 27). PC knockdown slowed cell growth, again with shPC55 being more effective than shPC54 (Supplemental Figure 4C). This corroborates the greater extent of PC knockdown by shPC55 (Supplemental Figure 4A). Furthermore, PC knockdown greatly decreased the number of colonies formed in soft agar (Supplemental Figure 4D), reduced cell viability (Supplemental Figure 4E), and altered morphology (Supplemental Figure 4F).
Since shPC55 was the more effective construct in suppressing both PC expression and activity, we used it to probe the effects of PC suppression on the additional NSCLC adenocarcinoma cell lines H1299, H2030, HCC827, and PC9. PC knockdown inhibited the net growth of all cell lines tested (Figure 4A). HCC827 and PC9 cells were more sensitive to PC knockdown than were A549, H2030, or H1299 cells. HCC827 and PC9 cells also expressed more PC than did A549 and H1299 (Supplemental Figure 5A). The colony-formation assay revealed that PC knockdown significantly inhibited the ability of A549 and PC9 cells to form colonies in soft agar (Figure 4B). Although PC suppression did not reduce the total number of colonies produced by H1299 cells (Figure 4B), it dramatically affected the size of each colony (Supplemental Figure 5B), which is consistent with slower cell growth (Figure 4A). Moreover, PC-suppressed cells became large and multinucleated, and the degree of multinucleation increased over time (Supplemental Figure 6, A–C).
Figure 4. PC suppression via shRNA inhibits proliferation and tumorigenicity of human NSCLC cell lines in vitro and in vivo.

Proliferation and colony-formation assays were initiated 1 week after transduction and selection with puromycin. A549 xenograft in NSG mice was performed 8 days after transduction. *P < 0.01, **P < 0.001, ***P < 0.0001, and ****P < 0.00001 by Student t test, assuming unequal variances. Error bars represent the SEM. (A) NSCLC cells lines were transduced with shPC55 or shEV. Proliferation assays (n = 6) revealed substantial growth inhibition induced by PC knockdown in all 5 cell lines after a relatively long latency period. (B) Colony-formation assays indicated that PC knockdown reduced the capacity of A549 and PC9 cells to form colonies in soft agar (n = 3). (C) Tumor xenografts from shPC55-transduced A549 cells showed a 2-fold slower growth rate than did control shEV tumors (P < 0.001 by the unpaired Welch version of the t test). Tumor size was calculated as πab/4, where a and b are the x,y diameters. Each point represents an average of 6 mice. The solid lines are the nonlinear regression fits to the equation: size = a + bt2, as described in the Methods. (D) The extent of PC knockdown in the mouse xenografts (n = 6) was lesser than that in cell cultures, leading to less attenuation of PC expression (30%–60% of control) and growth inhibition. In addition, PC expression in the excised tumors correlated with the individual growth rates, as determined by Pearson’s correlation coefficient.
We then determined whether PC-suppressed A549 cells are compromised in tumor growth in immune-deficient mice. We followed the rate of tumor growth after injecting NSG mice with A549 cells transduced with an empty vector (shEV) or shPC55 (shPC). PC knockdown resulted in a 2-fold slower rate of tumor growth (P = 0.001), as assessed by quadratic curve fitting of the tumor growth kinetics (Figure 4C and Supplemental Table 3). We found that decreased tumor growth was clear by day 14 (P = 0.009). In addition, endpoint analysis of tumor weights showed a 30% smaller tumor in the shPC55 xenografts compared with that observed in the shEV xenografts (P = 0.0073). However, the knockdown did not remain as effective in vivo as in cell culture. We found that PC expression in the knockdown tumors after 36 days had increased to 30% to 60% of that in tumors with control empty vector (Supplemental Figure 7A). Interestingly, the growth rate of individual tumors correlated significantly (P = 0.006) with their level of PC expression (Figure 4D). Therefore, residual PC expression is likely to account for the limited ability of the shPC55 tumors to grow. Altogether, these data indicate that PC expression is important for in vivo tumor growth.
PC knockdown perturbs Krebs cycle activity, the anabolic capacity of the cell, and glutathione homeostasis.
To investigate the metabolic consequences of PC knockdown, A549 cells were transduced with shPC55, grown in 13C6-glucose or 13C5,15N2-glutamine, and incorporation of the label was analyzed by NMR and MS. In addition, the mice engrafted with shPC55- or shEV-transduced A549 cells were injected with 13C6-glucose. With the glucose tracer, we found no evidence that PC affects aerobic glycolysis, as PC-knockdown cells had similar glucose consumption and production of total and labeled lactate in the media (Supplemental Figure 7B). In addition, the lactate released into the media had the same percentage of enrichment pattern in PC-knockdown and control cells (Supplemental Figure 7C). Likewise, mouse tumors bearing shEV or shPC55 had comparable concentrations of total lactate, labeled lactate derived from 13C6-glucose (Supplemental Figure 7D), and percentage of lactate enrichment patterns (Supplemental Figure 7E).
In contrast, PC knockdown reduced the oxidation of 13C6-glucose through the Krebs cycle. As expected, entry of glycolytic 13C3-pyruvate into the Krebs cycle via PC was inhibited, as indicated by the reduced production of various 13C3 isotopologs and 13C5-citrate (highlighted by blue circles in Figure 5A). Interestingly, pyruvate entry via PDH was also hindered by PC knockdown (highlighted by red circles in Figure 5A). We observed the same trend in mouse tumors initiated by PC-knockdown cells (Supplemental Figure 8), although the effects were less severe than those seen in cell cultures. This is again consistent with the finding that A549 cells with PC knockdown were able to recover PC expression to some extent in vivo (Supplemental Figure 7A). Reduced glucose oxidation through the Krebs cycle in cells and tumors was also observed by 1H-{13C} heteronuclear single-quantum coherence (HSQC) NMR (Supplemental Figure 9, A and C), which showed reduced incorporation of 13C from the glucose tracer into citrate, aspartate, glutamate, and the glutamate residue of glutathione.
Figure 5. PC knockdown perturbs glucose and glutamine flux through the Krebs cycle.

13C Isotopolog concentrations were determined by GC-MS (n = 3). Values represent the averages of triplicates, with standard errors. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by Student’s t test, assuming unequal variances. The experiments were repeated 3 times. (A) A549 cells were transduced with shPC55 for 10 days before incubation with 13C6-glucose for 24 hours. As expected, the 13C isotopologs of Krebs cycle metabolites produced via PC and Krebs cycle activity were depleted in PC-deficient cells (tracked by blue dots in the atom-resolved map and blue circles in the bar graphs; see also Figure 2C). In addition, 13C6-glucose metabolism via PDH was also perturbed (indicated by red dots and circles). (B) Treatment of PC-knockdown cells with 13C5,15N2-glutamine revealed that anaplerotic input via GLS did not compensate for the loss of PC activity, since GLS activity was attenuated, as inferred from the activity markers (indicated by red dots and circles). Decarboxylation of glutamine-derived malate by malic enzyme (ME) and reentry of glutamine-derived pyruvate into the Krebs cycle via PC or PDH (shown in blue and green, respectively) were also attenuated. Purple diamonds denote 15N; black diamonds denote 14N.
Since glutaminolysis is another major anaplerotic pathway, we asked whether this pathway could compensate for PC deficiency in NSCLC cells. shPC55- or shEV-transduced A549 cells were incubated with 13C5,15N2-glutamine to track glutaminolysis. As shown in Figure 5B, GLS1 activity can produce 13C5,15N1-glutamate (6) from 13C5,15N2-glutamine (7), which subsequently enters the Krebs cycle to generate 13C4-succinate (4), 13C4- (4) or 13C4,15N1-aspartate (5), and 13C4-citrate (4). A549 cells readily took up and metabolized 13C5,15N2-glutamine into these labeled metabolites. However, the levels of all these labeled isotopologs were significantly reduced in the PC-knockdown cells (highlighted by red circles in Figure 5B), which is consistent with reduced GLS activity. Decreased GLS activity was also evident in the 1H-{13C} HSQC spectra, which showed that PC-suppressed cells incorporated less glutamine carbon into glutamate, aspartate, and citrate (Supplemental Figure 9B). It should be noted that amidotransferase activity can also contribute to the production of the labeled products shown in Figure 5B and Supplemental Figure 9B. However, changes in amidotransferase activity are unlikely to account for the observed blockage of Gln metabolism induced by PC knockdown. This is because PC knockdown in A549 cells or in mouse xenografts led to suppressed GLS1 expression (Supplemental Figure 10A). In addition, A549 cells exhibited high GLS1 expression and GLS activity in cell lysates, suggesting that GLS1 expression is important for A549 cell growth (see below).
To further probe the importance of glutamine utilization in PC-suppressed cells, A549 cells were incubated with either 2 or 0.2 mM glutamine. We found that glutamine restriction significantly reduced the growth rate of control cells by approximately 30%, but had no influence on PC-knockdown cells (Supplemental Figure 10, B and C). Thus, glutaminolysis does not compensate for the loss of PC as an anaplerotic supplier of precursors to the Krebs cycle intermediates in NSCLC cells.
Beyond supplying reducing equivalents and energy (ATP and GTP), the Krebs cycle also supplies various precursors for anabolism. For instance, citrate is exported from the mitochondria to supply acetyl CoA for fatty acid synthesis; OAA is transaminated to aspartate for pyrimidine biosynthesis; α-ketoglutarate is aminated to glutamate for de novo glutathione synthesis; and malate is decarboxylated to pyruvate, providing NADPH for lipid biosynthesis and glutathione reduction (Figure 1A). To determine how perturbation of the Krebs cycle by PC suppression affects these anabolic pathways, we used SIRM to follow the biosynthesis of nucleotides, lipids, and glutathione in A549 cells grown in 13C6-glucose or 13C5-glutamine tracers.
PC knockdown reduced the levels of uracil and adenine nucleotides both in cell culture and in vivo (Supplemental Figure 11, A and B). These changes were at least in part attributed to attenuated biosynthesis of these nucleotides by PC knockdown, as evidenced by the reduced incorporation of labeled glucose into the C1′ ribose carbon observed in HSQC NMR analysis of 1H{13C} (Supplemental Figure 9A and Supplemental Figure 11A). This can be accounted for by the reduced production of the uracil and cytosine ring via decreased synthesis of their precursor aspartate from glucose and glutamine both in cells (Figure 5) and in vivo (Supplemental Figure 8). Consistently, we observed less incorporation of the glucose and glutamine carbon into the ring of UTP and CTP by Fourier transform ion cyclotron resonance (FT-ICR-MS) analysis (Figure 6A).
Figure 6. PC suppression hinders Krebs cycle–fueled biosynthesis.

(A) 13C atom–resolved pyrimidine biosynthesis from 13C6-glucose and 13C5-glutamine is depicted with a 13C5-ribose moiety (red dots) produced via the pentose phosphate pathway (PPP) and 13C1-3 uracil ring (blue dots) derived from 13C2-4-aspartate produced via the Krebs cycle or the combined action of ME and PC (blue dots). A549 cells transduced with shPC55 or shEV were incubated with 13C6-glucose or 13C5-glutamine for 24 hours. Fractional enrichment of UTP and CTP isotopologs from FT-ICR-MS analysis of polar cell extracts showed reduced enrichment of 13C6-glucose–derived 13C5-ribose (the “5” isotopolog) and 13C6-glucose– or 13C5-glutamine–derived 13C1-3-pyrimidine rings (the “6–8” or “1–3” isotopologs, highlighted by dashed green rectangles; for the “6–8” isotopologs, 5 13Cs arose from ribose and 1–3 13Cs from the ring) (10, 45). These data suggest that PC knockdown inhibits de novo pyrimidine biosynthesis from both glucose and glutamine. (B) Glucose and glutamine carbons enter fatty acids via citrate. FT-ICR-MS analysis of labeled lipids from the nonpolar cell extracts showed that PC knockdown severely inhibited the incorporation of glucose and glutamine carbons into the fatty acyl chains (even) and fatty acyl chains plus glycerol backbone (odd >3) of phosphatidylcholine lipids. However, synthesis of the 13C3-glycerol backbone (the “3” isotopolog) or its precursor 13C3-α-glycerol-3-phosphate (αG3P, m+3) from 13C6-glucose was enhanced rather than inhibited by PC knockdown. These data suggest that PC suppression specifically hinders fatty acid synthesis in A549 cells. Values represent the averages of triplicates (n = 3), with standard errors. *P < 0.05, **P < 0.01, and ***P < 0.001 by Student’s t test, assuming unequal variances.
Lipid biosynthesis was assessed in the nonpolar fractions of the same cell extracts by FT-ICR-MS, which can resolve a large number of intact lipids and determine their 13C enrichments in the glycerol and fatty acyl components (28). PC knockdown greatly inhibited synthesis of the major cell membrane components phosphatidylcholines from both glucose and glutamine tracers (Figure 6B). Using 13C6-glucose as a tracer, 13C was incorporated extensively into the glycerol backbone (3 13C atoms), fatty acyl chains (even 13C), and glycerol plus fatty acyl residues (odd >3 13C) in shEV control cells (28). With PC suppression, almost all phosphatidylcholine species were either unlabeled (as 0) or labeled at the glycerol backbone only (as 3), while the 13C isotopologs reflecting fatty acyl synthesis from glucose (even and odd >3) were greatly diminished in A549 cells. In addition, the glycerol backbone precursor α-glycerol-3-phosphate (αG3P) (such as 13C3-αG3P derived from 13C6-glucose) accumulated in the PC-knockdown cells (Figure 6B). Likewise, fatty acyl synthesis from 13C5-glutamine (“Even” in Figure 6B) via glutaminolysis and the Krebs cycle was greatly attenuated in PC-suppressed cells. Taken together, these results suggest that PC knockdown severely inhibits lipid production by blocking the biosynthesis of fatty acyl components but not the glucose-derived glycerol backbone. This is consistent with decreased Krebs cycle activity (Figure 5), which in turn curtails citrate export from the mitochondria to supply the fatty acid precursor acetyl CoA in the cytoplasm.
Finally, de novo glutathione synthesis was analyzed by 1H{13C} HSQC NMR. Glutathione synthesis from both glucose and glutamine was suppressed by PC knockdown (Supplemental Figure 9, A and B). Reduced de novo synthesis led to a large decrease in the total level of reduced glutathione (GSH; Supplemental Figure 12, A and B). At the same time, PC-knockdown cells accumulated slightly more oxidized GSH (GSSG; Supplemental Figure 12, A and B), leading to a significantly reduced GSH/GSSG ratio both in cell culture and in vivo (Supplemental Figure 12C). To determine whether this perturbation of glutathione homeostasis compromises the ability of PC-suppressed cells to handle oxidative stress, we measured ROS production by DCFDA fluorescence. PC-knockdown cells had over 70% more basal ROS than did control cells (0 mM H2O2; Supplemental Figure 12D). When cells were exposed to increasing concentrations of H2O2, the knockdown cells were less able to quench ROS, as they produced up to 300% more ROS than did control cells (Supplemental Figure 12D). However, N-acetylcysteine (NAC) at 10 mM did not rescue the growth of PC-knockdown cells, suggesting that such a growth effect is not simply related to an inability to regenerate GSH from GSSG. Altogether, these results show that PC suppression compromises anaplerotic input into the Krebs cycle, which in turn reduces the activity of the Krebs cycle, while limiting the ability of A549 cells to synthesize nucleotides, lipids, and glutathione. These downstream effects of PC knockdown were also evident when comparing the metabolism of shPC55-transduced A549 cells against that of A549 cells transduced with a scrambled vector (shScr) (Supplemental Figure 13), which suggests that they are on-target effects of PC knockdown.
Glutaminolysis is also a functionally significant source of anaplerosis in NSCLC.
To determine whether GLS1 is functionally important for NSCLC cells, we suppressed GLS1 expression by shRNA in both A549 and H1299, which resulted in reduced growth rates in both cells (Supplemental Figure 14, A and B). The growth of A549, H1299, and PC9 cells also decreased in response to BPTES (a known GLS inhibitor) treatment (Supplemental Figure 14C). Further, 13C isotopolog analysis of these 3 cell lines after incubation with 13C5,15N2-Gln showed that they used glutamine as a significant source of carbon for the Krebs cycle (Supplemental Figure 14D). Therefore, it appears that both GLS1 and PC are important for supporting proliferation and Krebs cycle activity in NSCLC cells. 13C5,15N2-Gln as a substantial carbon source for the Krebs cycle was also recapitulated in the human tissue slice data (Supplemental Figure 2C). However, unlike PC-based anaplerosis (Figure 2B), GLS-fueled anaplerotic activity was comparable between CA and NC tissue slices (Supplemental Figure 2C), which is consistent with the lack of discernible distinction in GLS1 protein expression between bulk CA and NC tissues (Figure 1C).
Discussion
Prior to our earlier report (21), the upregulation of PC in NSCLC was an undocumented phenomenon. Most research on anaplerosis in cancer cells has focused on the upregulation of glutamine metabolism via GLS1 in established cell lines, which cannot recapitulate the influence of the tumor microenvironment (17, 29–32). To gain insights into metabolic reprogramming in human cancer, it is imperative to study human tumors in situ and to compare paired CA and surrounding NC tissues from the same patient to circumvent cell culture artifacts, while eliminating confounding intrinsic genetic and physiological variables. It is also important to implement a fast and consistent liquid N2–quenching protocol to minimize metabolic artifacts during tissue sampling after surgical resection.
By addressing the two aspects of our study, we found that although PC expression and activity in lung tissues varied substantially from patient to patient, 94% of the NSCLC tumors (82 of 86) overexpressed PC, and 73% (25 of 34) showed elevated PC activity in vivo relative to that in their paired NC lung tissues. This contrasts with the insignificant difference observed in the expression of GLS1 between CA and NC tissues (Figure 1C). PC was highly expressed exclusively in cancer cells of early-stage NSCLC tumor tissues. In contrast, PC was not expressed in the pneumocytes, but its expression was evident in macrophages of paired NC tissue (Figure 3 and Supplemental Figure 3). We surmise that the latter may be related to inflammation, which is common in the lungs of NSCLC patients.
The requirement of PC for NSCLC proliferation and tumorigenesis was demonstrated by PC suppression in several lung cancer cell lines (Figure 4, A and B) and in vivo for A549 cells (Figure 4, C and D) using specific shRNAs. The long latency period between mRNA knockdown and phenotypic effects (Figure 4A) may be related to the long half-life of PC (1–2 d, ref. 33). The intracellular level of PC may need several cell divisions to be attenuated sufficiently to compromise anaplerosis, leading to reduced cell proliferation. We have verified that inhibition of cell proliferation by PC suppression is accompanied by a decrease in anaplerotic input into the Krebs cycle (Figure 5) and the consequent reduction of nucleotide (Figure 6A, Supplemental Figure 11A, and Supplemental Figure 13C) and lipid biosynthesis (Figure 6B and Supplemental Figure 13D). The correlation of the severity of these metabolic effects with the efficiency of the knockdown and the inhibition of cell proliferation both in vitro and in vivo (Figure 4 and Supplemental Figure 4) is consistent with PC knockdown being a significant contributor to the reduced proliferation. Although we have clearly demonstrated the importance of PC for cancer cell proliferation and that enzymic activity is an important aspect of the role of PC, we cannot rule out the possibility that the protein has alternative nonenzymic functions in the cells, as has been observed for several other metabolic enzymes (34).
In addition, PC knockdown in A549 cells greatly depletes the GSH pool and perturbs glutathione homeostasis (Supplemental Figure 12C) by blocking de novo glutathione synthesis (Supplemental Figure 12B and Supplemental Figure 13B), which presumably leads to greater ROS production (Supplemental Figure 12D) and a compromised ability to survive oxidative stress. Exogenous reductants such as NAC in some circumstances can reduce cellular GSSG to GSH, but NAC cannot overcome the depleted GSH pool in PC-suppressed cells, thus accounting for the failure of NAC to rescue PC-suppressed cells from growth inhibition (Supplemental Figure 12E).
Although active, anaplerotic glutaminolysis could not compensate for lost PC-based anaplerosis in A549 cells (Figure 5B and Supplemental Figure 10), despite the abundant supply of glutamine. This is presumably due to the reduced expression of GLS1 in PC-knockdown cells or in xenograft tumors (Supplemental Figure 10A). Moreover, we noted that in the human NSCLC tumors, in which PC was underexpressed, GLS1 was also underexpressed. These results suggest that PC and GLS1 are not compensatory in their functions in human NSCLC, in contrast to some glioma cells (20).
Finally, the improved variant of Warburg’s tissue slices (25) that we have introduced enables detailed ex vivo analysis of patient-specific tumor metabolism (Figure 2 and Supplemental Figure 2), while maintaining most of the tissue microenvironment and cellular heterogeneity without systemic complications from the blood supply. This ex vivo system also affords versatility of experimental manipulations (e.g., tracer choices or drug treatments) similar to that of the pure cell culture system. In our case, enhanced in vivo PC activity (Figure 1D) in human NSCLC tumor tissues was confirmed by the corresponding ex vivo tracer studies (Figure 2B). Equally important is that the ex vivo system enables unequivocal determination of anaplerotic glutaminolysis using the labeled glutamine tracer (Supplemental Figure 2C), thereby corroborating the lack of in vivo upregulation of GLS1 in human tumor tissues (Figure 1C). This would have been difficult to demonstrate in vivo in human patients due to cost considerations. Thus, the ex vivo tissue culture system can complement existing model systems for translating bench-to-bedside understanding, particularly in an individualized manner (26).
In conclusion, our results in primary human lung tumors and ex vivo tissue slices show that PC protein expression and activity are enhanced in early stages of human NSCLC tumor tissues compared with that seen in NC tissues from the same patient and that the PC protein is localized in the cancer cells within the tumor tissue. Silencing PC expression and activity compromises NSCLC cell growth, tumorigenicity, and antioxidation capacity. These phenotypic effects of PC suppression were related to the vital metabolic functions of PC in NSCLC, including sustaining mitochondrial Krebs cycle activity, nucleotide and lipid biosynthesis, and redox homeostasis.
Methods
Patient accrual.
Eighty-six patients with suspected primary lung cancer but without diagnosed diabetes were recruited on the basis of their surgical eligibility. The patients were then randomly grouped into 2 arms. In the first arm, the patients were overnight fasted (>8 hours) before 10 grams of uniformly labeled 13C glucose was administered i.v. and again 2.8 ± 0.5 hours prior to video-assisted thorascopic surgical (VATS) wedge resection, as previously described in detail (35), to enable tracking of metabolic pathways (21, 36–38). Patients in the second arm did not receive a glucose injection. We and others have previously discussed the relative advantages of bolus injections versus slow-infusion protocols (21, 36–41). The bolus injection leads to an initially high blood glucose concentration, which then decays to the baseline level, thus limiting the length of time that effective labeling can be achieved but more closely approximating an initial rate condition. In comparison, a prolonged slow infusion can lead to a higher degree of isotopic labeling and approach isotopic steady-state conditions, but is a much more involved process, and a true isotopic steady state may be difficult and cost prohibitive to achieve.
The extent of resection was determined by the surgeon according to clinical criteria. The majority of the specimens were obtained from wedge resections to minimize surgical times, while the remainder were acquired in less than 5 minutes after the pulmonary vein was clamped; both practices helped avoid development of significant ischemia in resected tissues. Immediately after resection, the tumor was transected, and sections of CA tissue and surrounding NC lung tissue at least 2 cm away from the tumor were biochemically quenched by flash-freezing in liquid N2 (36, 38). The margins of the tumor were initially assessed by the surgeon by visual inspection. Parallel tissue samples were sent to on-site pathologists for confirmation of the diagnosis and the cancer-free margins. The remaining specimen was preserved in buffered formalin for detailed pathological examination.
Sample preparation.
The frozen tissue samples were pulverized to a particle size of less than 10 μm in liquid N2 using a Spex freezer mill (Spex). Polar and nonpolar metabolites were extracted from the ground tissue, which also yielded insoluble protein residues (21, 42). The protein residues were extracted by homogenization in 2% SDS, 62.5 mM Tris, and 1 mM DTT buffer for protein determination using the Pierce BCA method (Thermo Fisher Scientific).
Electrophoresis, Western blotting, immunohistochemistry, cell proliferation, and colony-formation assays.
Detailed protocols are described in the Supplemental Methods.
shRNA knockdown of PC in lung cancer cells.
NSCLC cells lines and HEK 293T cells obtained from ATCC were grown in DMEM media containing 0.45% glucose and 4 mM glutamine at 37°C, 95% relative humidity, and 5% CO2. HEK 293T cells were transfected with pCMV and pMD2.G vectors carrying viral packaging (gag, pol, rev) and envelope (VSV-G) genes, along with a plasmid containing shRNA against PC (shPC54 or shPC55), shEV, or shScr (Mission shRNA; Sigma-Aldrich). The anti-PC sequences used were: shPC54: CCGGGCCCAGTTTATGGTGCAGAATCTCGAGATTCTGCACCATAAACTGGGCTTTTTG; shPC55: CCGGGCCAAGGAGAACAACGTAGATCTCGAGATCTACGTTGTTCTCCTTGGCTTTTTG.
After 48 hours, medium from the transfected cells was collected, filtered, supplemented with 16 μg/ml polybrene, and added to NSCLC cell lines in an equal volume of DMEM media. Fresh media were added to the 293T cells, and after 24 hours, transduction of the NSCLC cells was repeated. The resulting NSCLC cells bearing the plasmid with the puromycin resistance gene were selected using 1 μg/ml puromycin.
Tracer studies in cell cultures.
Cells transduced with shEV, shScr, shPC54, or shPC55 were incubated in DMEM in the presence of 10 mM 13C6-glucose or 2 mM 13C5,15N2-glutamine for 24 hours, quenched in cold acetonitrile, and extracted in acetonitrile/water/chloroform (v/v 2:1.5:1), as described previously (10).
Tracer studies in mouse xenografts of PC-knockdown cells.
To assess tumor growth of PC-knockdown A549 cells, 6-week-old female NOD/SCID gamma (NSG) mice (n = 6 mice/group) (The Jackson Laboratory) were injected s.c. with 8 × 106 cells containing either shEV or shPC55 mixed with Matrigel (BD Biosciences). The tumor diameters were determined 3 times per week by caliper. Tumor sizes were calculated as πab/4, where a and b are the lengths of the maximum and minimum dimensions. After 36 days, the mice were injected via the tail vein with 100 μl 1.075 M [U-13C]-glucose 3 times at 15-minute intervals. Blood samples were taken intraorbitally immediately after the first injection and at sacrifice 60 minutes later. The tumors were immediately excised and weighed prior to flash-freezing in liquid N2. Metabolites were extracted as described above (43).
Tracer studies in ex vivo tissue slices.
These experiments were performed on fresh slices of paired tumor and nontumorous lung tissues resected from individual patients (26). Upon lung resection, thin slices (0.5–1 mm thick) of tissue were excised using a Weck microtome in the operating room. These slices were immediately placed in T-flasks containing DMEM with the appropriate tracer (either 10 mM [U-13C]-glucose or 2 mM [U-13C,15N]-glutamine), and then transferred to a CO2 incubator set at 37°C and 5% CO2. The flasks were continuously rocked for 24 hours for aeration and to maintain constant nutrient supplies at the tissue surface, while avoiding local buildup of waste products such as acids. The slices were then washed in cold PBS, frozen in liquid N2, pulverized as described above, and extracted for polar and nonpolar metabolites and proteins as described above for cell cultures.
SIRM analysis.
Polar metabolite fractions from cells, patient tissues, and mouse tumors were analyzed by GC-MS, NMR, and FT-ICR-MS as previously described (10, 42). Nonpolar fractions were analyzed for lipids by FT-ICR-MS as previously described (10, 28). 13C fractional enrichments were calculated for metabolite isotopomers (13C at different atomic positions) and isotopologs (different numbers of 13C atoms) as the level of a given isotopomer or isotopolog divided by the summed level of all isotopomers or isotopologs. The absolute levels of labeled metabolites were obtained by normalizing the moles of metabolites to the extractable protein content.
Statistics.
Statistical analyses were conducted using SPSS 19.0 (IBM- Somers) and Kaleidagraph (Synergy Software). Differences between patient cohorts were assessed using the paired Wilcoxon test, the 2-tailed Student t test on the logarithms of the values (i.e., for fractional differences), and by examining the distribution terms of the mean, median, and modal values. The expression ratios (CA/NC) for PC or GLS were highly non-normally distributed, and the mean, median, and modal values were calculated accordingly. The modal values were estimated from the maximum in the frequency-versus-ratio histograms and were thus slightly affected by the bin size.
Mouse tumor sizes were tested using an unpaired Welch’s version of the t test (44). First, the significance was evaluated at each time point after injection. Second, all growth curves were fitted to different models, including exponential, power, and quadratic growth, and best-fit parameters with the associated standard deviations were subjected to Welch’s t test. The simplest form that gave good fitting statistics was a simple quadratic equation: size = a + bt2. In this form, a is the initial tumor size at t = 0, and b is the specific growth rate. Third, the tumor weights at necropsy were compared as 2 groups. The significance of differences between shEV and shPC groups in terms of proliferation and metabolites was determined using Welch’s t test. In all analyses, a P value of less than 0.05 was considered significant.
Study approval.
This study was approved by the IRB of the University of Louisville. Written informed consent was obtained from all subjects prior to inclusion in this study. The murine tumor protocol was performed in compliance with all relevant laws and institutional guidelines and was approved by the IACUC of the University of Louisville.
Supplementary Material
Acknowledgments
This work was supported in part by National Science Foundation EPSCoR infrastructure grants EPS-0447479 (to T.W.-M. Fan) and EPS-0132295 (to R.J. Wittebort, for the 18.8 T NMR spectrometer); NIH National Center for Research Resources (NCRR) grants 5P20RR018733 (to D.M. Miller and A.N. Lane), 1R01CA118434-01A2, 1RO1CA101199-01, 3R01CA118434-02S1, and 1R01ES022191-01 (to T.W.-M. Fan), P01CA163223-01A1 (to A.N. Lane), and 1U24DK097215-01A1 (to R.M. Higashi); the University of Louisville CTSPGP/ARRA grant 20044; the Kentucky Lung Cancer Research Program grants OGMB090354B1 and OGMB101380 (to T.W.-M. Fan and A.N. Lane); the Robert W. Rounsavall Jr. Family Foundation; and the Kentucky Challenge for Excellence and Drive Cancer Out Campaign. We thank Jin Lian Tan, Alex Belshoff, Radhika Burra, and Tao Xu for technical assistance; Pawel Lorkiewicz for help with FT-ICR-MS analysis; Ronald Bruntz for performing the scrambled vector experiment; and Melissa Hall and Bridgett Curry for clinical support.
Footnotes
Conflict of interest: Teresa W.-M. Fan, Andrew N. Lane, Richard M. Higashi, and Michael Bousamra II declare that they have filed a US patent application entitled “Methods for Detecting Cancer” (US20130109592).
Reference information:J Clin Invest. 2015;125(2):687–698. doi:10.1172/JCI72873.
See the related Commentary beginning on page 495.
References
- 1.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24(2):68–72. doi: 10.1016/S0968-0004(98)01344-9. [DOI] [PubMed] [Google Scholar]
- 3.Fan TW, et al. Rhabdomyosarcoma cells show an energy producing anabolic metabolic phenotype compared with primary myocytes. Mol Cancer. 2008;7:79. doi: 10.1186/1476-4598-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metallo CM, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481(7381):380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wise DR, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108(49):19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frezza C, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477(7363):225–228. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- 8.Mullen AR, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481(7381):385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Possemato R, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476(7360):346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le A, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuneva MO, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15(2):157–170. doi: 10.1016/j.cmet.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Serkova NJ, Spratlin JL, Eckhardt SG. NMR-based metabolomics: Translational application and treatment of cancer. Curr Opin Mol Ther. 2007;9(6):572–585. [PubMed] [Google Scholar]
- 14.Lane AN, Fan TWM, Higashi RM. Stable isotope assisted metabolomics in cancer research. IUBMB Life. 2008;60(2):124–129. doi: 10.1002/iub.17. [DOI] [PubMed] [Google Scholar]
- 15.Guppy M, Leedman P, Zu X, Russell V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J. 2002;364(1):309–315. doi: 10.1042/bj3640309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Portais JC, Voisin P, Merle M, Canioni P. Glucose and glutamine metabolism in C6 glioma cells studied by carbon 13 NMR. Biochimie. 1996;78(3):155–164. doi: 10.1016/0300-9084(96)89500-9. [DOI] [PubMed] [Google Scholar]
- 17.DeBerardinis RJ, et al. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104(49):19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazurek S, Grimm H, Oehmke M, Weisse G, Teigelkamp S, Eigenbrodt E. Tumor M2-PK and glutaminolytic enzymes in the metabolic shift of tumor cells. Anticancer Res. 2000;20(6D):5151–5154. [PubMed] [Google Scholar]
- 19.Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007;178(1):93–105. doi: 10.1083/jcb.200703099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng T, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108(21):8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan TW, et al. Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM). Mol Cancer. 2009;8:41. doi: 10.1186/1476-4598-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marin-Valencia I, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15(6):827–837. doi: 10.1016/j.cmet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan TWM, et al. Stable isotope-resolved metabolomic analysis of lithium effects on glial-neuronal metabolism and interactions. Metabolomics. 2010;6(2):165–179. doi: 10.1007/s11306-010-0208-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 2007;27(2):219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- 25.Warburg O. Versuche an überlebendem Carcinomgewebe (Methoden). Biochem Zeitschr. 1923;142:317–333. [Google Scholar]
- 26.Xie H, et al. Targeting lactate dehydrogenase-A (LDH-A) inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor initiating cells. Cell Metab. 2014195795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ochoa-Ruiz E, Diaz-Ruiz R. Anaplerosis in cancer: another step beyond the warburg effect. Am J Mol Biol. 2012;2:291–303. doi: 10.4236/ajmb.2012.24031. [DOI] [Google Scholar]
- 28.Lane AN, Fan TW, Xie Z, Moseley HN, Higashi RM. Isotopomer analysis of lipid biosynthesis by high resolution mass spectrometry and NMR. Anal Chim Acta. 2009;651(2):201–208. doi: 10.1016/j.aca.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mazurek S, Eigenbrodt E. The tumor metabolome. Anticancer Res. 2003;23(2A):1149–1154. [PubMed] [Google Scholar]
- 30.Lobo C, Ruiz-Bellido MA, Aledo JC, Marquez J, Nunez De Castro I. Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem J. 2000;348(pt 2):257–261. [PMC free article] [PubMed] [Google Scholar]
- 31.Seltzer MJ, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70(22):8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marin-Valencia I, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15(6):827–837. doi: 10.1016/j.cmet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandler CS, Ballard FJ. Regulation of the breakdown rates. of biotin-containing proteins in Swiss 3T3-L1 cells. Biochem J. 1988;251(3):749–755. doi: 10.1042/bj2510749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim JW, Dang CV. Multifaceted roles of glycolytic enzymes. Trends Biochem Sci. 2005;30(3):142–150. doi: 10.1016/j.tibs.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 35. Fan TWM, Lane AN, Higashi RM, eds. The Handbook of Metabolomics. New York, New York, USA: Humana Press; 2012. [Google Scholar]
- 36.Lane AN, Fan TW, Bousamra M, 2nd, Higashi RM, Yan J, Miller DM. Stable isotope-resolved metabolomics (SIRM) in cancer research with clinical application to nonsmall cell lung cancer. OMICS. 2011;15(3):173–182. doi: 10.1089/omi.2010.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan TW, Lane AN, Higashi M, Bousamra M, 2nd, Kloecker G, Miller DM. Erlotinib-sensitive and resistant lung tumors show radically different metabolic profiles. Exp Molec Pathol. 2009;87:83–86. doi: 10.1016/j.yexmp.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bousamra M, Day J, Fan TWM, Kloecker G, Lane AN, Miller DM. Clinical aspects of metabolomics. In: Fan TWM, Higashi RM, Lane AN, eds. The Handbook of Metabolomics. New York, New York, USA: Humana Press; 2012:29–60. [Google Scholar]
- 39.Fan TW, Lane AN, Higashi RM, Yan J. Stable isotope resolved metabolomics of lung cancer in a SCID mouse model. Metabolomics. 2011;7(2):257–269. doi: 10.1007/s11306-010-0249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Graaf RA, Rothman DL, Behar KL. State of the art direct 13C and indirect 1H-[13C] NMR spectroscopy in vivo. A practical guide. NMR Biomed. 2011;24(8):958–972. doi: 10.1002/nbm.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maher EA, et al. Metabolism of U-13C glucose in human brain tumors in vivo. NMR Biomed. 2012;25(11):1234–1244. doi: 10.1002/nbm.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fan TWM. Metabolomics-edited transcriptomics analysis (Meta). In: McQueen CA ed. Comprehensive Toxicology. 2nd ed. Oxford, United Kingdom: Academic Press; 2010:685–706. [Google Scholar]
- 43. Fan TW-M. Sample preparation for metabolomics investigation. In: Fan TW-M, Lane AN, Higashi RM, eds. The Handbook of Metabolomics: Pathway and Flux Analysis, Methods in Pharmacology and Toxicology. New York, New York, USA: Springer Science; 2012:7–27. [Google Scholar]
- 44. Rosner B. Fundamentals of Biostatistics. Belmont, California, USA: Thomson; 2006. [Google Scholar]
- 45.Lorkiewicz P, Higashi RM, Lane AN, Fan TW. High information throughput analysis of nucleotides and their isotopically enriched isotopologues by direct-infusion FTICR-MS. Metabolomics. 2012;8(5):930–939. doi: 10.1007/s11306-011-0388-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.