

Abstract
A powerful new strategy for the fabrication of high-density RNA arrays is described. A high-density DNA array is fabricated by standard photolithographic methods, the surface-bound DNA molecules are enzymatically copied into their RNA complements from a surface-bound RNA primer, and the DNA templates are enzymatically destroyed, leaving behind the desired RNA array. The strategy is compatible with 2’-fluoro-modified (2’F) rNTPs, which may be included in the polymerase extension reaction to impart nuclease resistance and other desirable characteristics to the synthesized RNAs. The use and fidelity of the arrays are explored with DNA hybridization, DNAzyme cleavage, and nuclease digestion experiments.
Keywords: Microarray, DNAzyme, Photolithography, Nucleic Acids, Maskless array synthesis
In 1991 Fodor et al. ushered in the new field of biomolecular array analysis, showing how photolithographic methods developed for integrated circuit fabrication could be adapted for the light-directed synthesis of addressed arrays of biomolecules on planar substrates. Array technologies evolved over the ensuing years into the powerful analytical platform they represent today, with DNA arrays containing millions of oligonucleotide features widely available and employed for myriad applications such as SBH (Sequencing by Hybridization),[1] Genome-wide Gene Expression Analysis,[2] ChIP-chip (Chromatin ImmunoPrecipitation and analysis by DNA ‘chip’),[3] and CSI (cognate site identification).[4]
Remarkably, while DNA arrays became mainstream and ubiquitous tools of the modern molecular biologist, the seemingly straightforward extension of the concept to RNA arrays never became practical. The fabrication of DNA arrays relies upon the phosphoramidite chemistry developed by Caruthers et al.[5a,b] This chemistry is remarkable because of its extraordinary efficiency, which manifests itself in stepwise yields for monomer addition over 99%.[6] This high efficiency is what allows DNA molecules as long as 150 nt in length[7] to be synthesized, and is the fundamental reason for the widespread availability of both individual high quality oligonucleotides and high-density DNA arrays. Although the same chemistry can be used for RNA synthesis, it does not give results of comparable quality. For RNA synthesis it is necessary to protect the 2’ hydroxyl to keep it from coupling during synthesis; in spite of much effort by many groups, it has not been possible to find a protecting group for this position that does not interfere with coupling at the adjacent 3’ hydroxyl, and/or give rise to undesired side reactions during deprotection.[8] Because of these issues, there has not existed to date any viable technology for the fabrication of high density RNA arrays, although two recent reports describe the synthesis of RNAs attached to DNA sequencing templates in a next-gen sequencing flow cell.[9a,b]
We describe here a simple yet powerful new strategy for the enzymatic synthesis of high-density RNA arrays with a maskless array synthesizer.[10] The key idea is to use RNA polymerase to copy surface-attached DNA molecules on a high-density DNA array into their RNA complements (Figure 1). The surface is first partially deprotected (e.g. light is used to effect removal of 50% of the NPPOC photolabile protecting groups covering the surface),[11] an array of the DNA complements to the eventual desired RNA sequences is synthesized by standard light-directed synthesis on the exposed sites, and the remaining surface sites are then deprotected, followed by synthesis of an RNA primer sequence. These primer sequences on the second group of sites are hybridized to their complements on the first group, whereupon they may be extended with T7 RNA polymerase to yield RNA:DNA duplexes. The DNAs are removed with DNase I, leaving behind the desired single stranded RNAs. The strategy is compatible with either unmodified ribonucleoside triphosphates (rNTPs), or alternatively, 2’-fluoro-modified (2’F) rNTPs may be included in the polymerase extension reaction to impart nuclease resistance and other desirable characteristics to the synthesized RNAs.[12] We note that the use of a very long, flexible, hydrophilic spacer (we employed a PEG 2000 moiety) between the substrate and the oligonucleotides is critical – this is not surprising, as it is necessary for the DNA complement and RNA primer sequences to anneal while both are still attached to the surface. A second key to this strategy is the ability to fabricate two different nucleic acid sequences within individual DNA features – in this case, both a primer sequence, and a template sequence.
Figure 1.

Enzymatic fabrication of high density RNA arrays.
Several approaches were employed to evaluate the fidelity and utility of the arrays: these include nuclease sensitivity, DNA hybridization, and sequence-specific DNAzyme cleavage. Figure 2 shows the results of nuclease digestion experiments on DNA, RNA, and 2’F RNA arrays. Each array contains three 31–33mer sequences arranged in a grid pattern (Supplementary Table 2). The arrays were visualized after nuclease treatment by hybridizing them with a mixture of the three corresponding oligodeoxynucleotide complements, tagged respectively with the fluorophores FAM (red), Texas Red (gray), and Cy3 (blue), followed by washing and fluorescence imaging. It is evident from the figure that while the DNA arrays are completely destroyed by DNase treatment but impervious to RNase treatment, the RNA arrays show the opposite behaviour, in that they are completely destroyed by RNase treatment but impervious to DNase treatment. As expected, the 2’F RNA arrays are resistant to both DNase and RNase treatment. These results confirm in each case the nature of the nucleic acid comprising the array elements. In addition, the experiments also show that for each array, the nucleic acid molecules on the surface hybridize specifically to fluorescently tagged solution complements, as illustrated by the correct localization of the different features in the image.
Figure 2.

Array susceptibility tests to DNase I and RNase A. The unmodified RNA and 2’-fluoro RNA array were treated with DNase I and RNase A, sequentially, while the DNA array was treated with RNase A and then DNase I. The arrays were visualized by hybridization with their DNA complements labeled with FAM (red), Texas Red (gray), and Cy3 (blue). Differences in signal intensity reflect variability associated with the array synthesis platform. A threshold was used to enhance feature clarity (settings listed in SI).
Figure 3 shows additional hybridization results further demonstrating the fidelity of the RNA arrays. In this experiment, a tiling array was synthesized containing the 161 20 mer RNA complements to a 180 bp DNA PCR amplicon from the murine IGFBP1 promoter[13] (stepping through the entire sequence in single base increments; sequence information is provided in SI). The fluorescein-labeled target amplicon was digested with T7 exonuclease and incubated with the array, following by washing and imaging. The pattern of hybridization illustrates the distribution of the exonuclease digestion products, and is consistent with results obtained previously using DNA arrays.[13]
Figure 3.
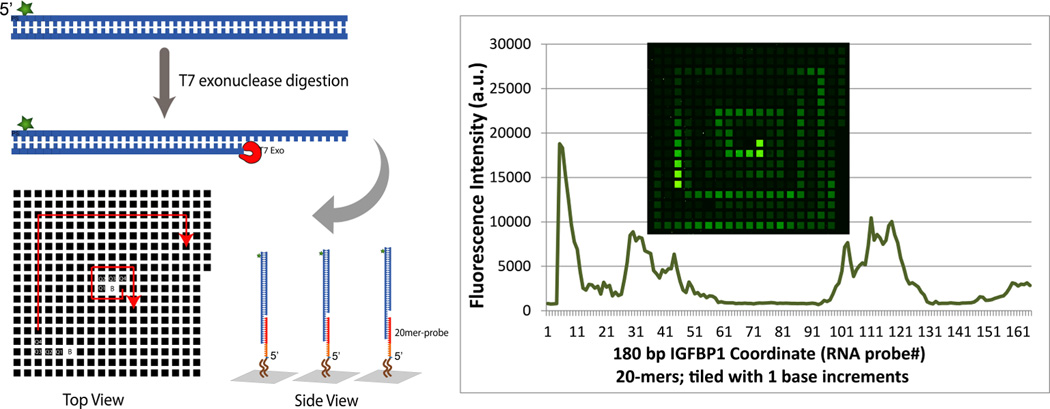
Capture of FAM-labeled 180bp IGFBP1 promoter DNA on a tiling RNA array.
The hybridization and exonuclease sensitivity results presented above provide strong evidence that the normal and modified RNA arrays have the correct nucleic acid compositions, and exhibit normal base-pairing functionality. We sought to further confirm the functionality of the sequences with an additional experiment to test the ability of the RNA sequences to serve as substrates for a RNA-specific DNAzyme.
The 10–23 DNAzyme, first described by Joyce and colleagues in 1997, consists of a catalytic core of 15 deoxynucleotides flanked by substrate-binding domains.[14] Any RNA substrate that is accessible to Watson-Crick pairing with the 10–23 DNAzyme substrate-binding domains can be cleaved at the phosphodiester linkage between purine and pyrimidine nucleobases that separate the complementary regions on the substrate (Supplementary Fig. 1A). We designed and fabricated DNA and RNA arrays encoding a “Badger Chemist” image. Each array contains three 31–33 mer sequences corresponding to the body, sweater/flask, or lab coat of a ‘Badger Chemist’ (Supplementary Table 3). The arrays were visualized by hybridizing them with a mixture of the three corresponding oligodeoxynucleotide complements, tagged respectively with the fluorophores FAM (green), Texas Red (yellow), and Cy5 (red), followed by washing and fluorescence imaging. The arrays were incubated in a Mn+2 containing buffer for 5 hr at 37°C with a 10–23 DNAzyme specific for the RNA sequences on the ‘Badger Chemist’ array that correspond to the red lab coat. As shown in Supplementary Figure 1B, the ‘lab coat’ sequences remained intact on the DNA array, whereas 70% were cleaved on the RNA array. These results show that the surface-bound RNA molecules are recognized as RNA by the DNAzyme.
In summary, we have described a novel strategy for the fabrication of high-density RNA arrays. The fidelity and functionality of the RNA elements are demonstrated in hybridization, nuclease digestion, and DNAzyme cleavage experiments. Many possible applications for RNA arrays can be envisioned, including: deciphering the binding specificities of RNA-binding proteins, as a tool to aid in the engineering of sequence-specific RNA-binding proteins,[15] for screening and characterizing RNA-based therapeutics,[16] for fabricating tiling arrays of RNA viral genomes, for fabricating miRNA arrays, engineering ribozyme arrays, discovering new ribozymes, studying ribozyme function, engineering artificial siRNAs and miRNAs, fabricating mRNA tiling arrays, and searching for miRNA ‘sponges’ (molecules that bind to and inactivate miRNAs).[17]
Supplementary Material
Acknowledgements
This publication was supported by the National Institutes of Health grants 1U54DK093467-01, 5T32GM08349, and 1R01GM108727-01A1.
References
- 1.Drmanac R, Drmanac S, Chui G, Diaz R, Hou A, Jin H, Jin P, Kwon S, Lacy S, Moeur B, Shafto J, Swanson D, Ukrainczyk T, Xu C, Little D. Advances in Biochem. Eng./Biotech. 2002;77:75–101. doi: 10.1007/3-540-45713-5_5. [DOI] [PubMed] [Google Scholar]
- 2.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Proc. Natl. Acad. Sci. U.S.A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA. Science. 2000;290(5500):2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 4.Warren CL, Kratochvil NCS, Hauschild KE, Foister S, Brezinski ML, Dervan PB, Phillips GN, Ansari AZ. Proc. Natl. Acad. Sci. U.S.A. 2006;103(4):867–872. doi: 10.1073/pnas.0509843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Beaucage SL, Caruthers MH. Tetrahedron Letters. 1981;22(20):1859–1862. [Google Scholar]; (b) Caruthers MH. Biochem. Soc. Trans. 2011;39:575–580. doi: 10.1042/BST0390575. [DOI] [PubMed] [Google Scholar]
- 6.Cheng JY, Chen HH, Kao YS, Kao WC, Peck K. Nucleic Acids Res. 2002;30(18):e93. doi: 10.1093/nar/gnf092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LeProust EM, Peck BJ, Spirin K, McCuen HB, Moore B, Namsaraev E, Caruthers MH. Nucleic Acids Research. 2010;38(8):2522–2540. doi: 10.1093/nar/gkq163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Somoza A. Chem. Soc. Rev. 2008;37(12):2668–2675. doi: 10.1039/b809851d. [DOI] [PubMed] [Google Scholar]
- 9.a) Buenrostro JD, Araya CL, Chircus LM, Layton CJ, Chang HY, Snyder MP, Greenleaf WJ. Nat Biotech. 2014;32(6):562–568. doi: 10.1038/nbt.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tome JM, Ozer A, Pagano JM, Gheba D, Schroth GP, Lis JT. Nat. Meth. 2014;11(6):683–688. doi: 10.1038/nmeth.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh-Gasson S, Green RD, Yue Y, Nelson C, Blattner F, Sussman MR, Cerrina F. Nat. Biotech. 1999;17(10):974–978. doi: 10.1038/13664. [DOI] [PubMed] [Google Scholar]
- 11.Chen S, Phillips MF, Cerrina F, Smith LM. Langmuir. 2009;25(11):6570–6575. doi: 10.1021/la9000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aurup H, Williams DM, Eckstein F. Biochemistry. 1992;31(40):9636–9641. doi: 10.1021/bi00155a016. [DOI] [PubMed] [Google Scholar]
- 13.Wu CH, Chen S, Shortreed MR, Kreitinger GM, Yuan Y, Frey BL, Zhang Y, Mirza S, Cirillo LA, Olivier M, Smith LM. PLOS One. 2011;6(10):e26217. doi: 10.1371/journal.pone.0026217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santoro SW, Joyce GF. Proc. Natl. Acad. Sci. U.S.A. 1997;94(9):4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Filipovska A, Razif MF, Nygard KK, Rackham O. Nat. Chem. Biol. 2011;7(7):425–427. doi: 10.1038/nchembio.577. [DOI] [PubMed] [Google Scholar]
- 16.Keefe AD, Pai S, Ellington A. Nat. Rev. Drug. Discov. 2010;9(7):537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebert MS, Neilson JR, Sharp PA. Nat. Meth. 2007;4(9):721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.