

Summary
Intestinal FABP (IFABP) and liver FABP (LFABP), homologous proteins expressed at high levels in intestinal absorptive cells, employ markedly different mechanisms of fatty acid transfer to acceptor model membranes. Transfer from IFABP occurs during protein-membrane-collisional interactions, while for LFABP transfer occurs by diffusion through the aqueous phase. In addition, transfer from IFABP is markedly faster than from LFABP. The overall goal of this study was to further explore the structural differences between IFABP and LFABP which underlie their large functional differences in ligand transport. In particular, we addressed the role of the αI-helix domain in the unique transport properties of intestinal FABP. A chimeric protein was engineered with the ‘body’ (ligand binding domain) of IFABP and the αI-helix of LFABP (α(I)LβIFABP), and the fatty acid transfer properties of the chimeric FABP were examined using a fluorescence resonance energy transfer assay. The results showed a significant decrease in the absolute rate of FA transfer from α(I)LβIFABP compared to IFABP. The results indicate that the αI-helix is crucial for IFABP collisional FA transfer, and further indicate the participation of the αII-helix in the formation of a protein-membrane “collisional complex”. Photo-crosslinking experiments with a photoactivable reagent demonstrated the direct interaction of IFABP with membranes and further supports the importance of the αI helix of IFABP in its physical interaction with membranes.
Keywords: fatty acid binding protein, fatty acid, chimeric proteins, lipid metabolism, small intestine, lipid transport
Introduction
In vertebrates, tissues like intestine, liver, adipose, and cardiac and skeletal muscles have metabolic pathways that demand a substantial transport of lipids, especially fatty acids (FA), both from other tissues via the blood, or from other organelles inside the cell. One of the physiological processes known to involve transport of high amounts of FA is the intestinal absorption of lipids after a meal. Small intestinal enterocytes express high levels of two homologous fatty acid-binding proteins (FABPs), liver FABP (LFABP), also named FABP1, and intestinal FABP (IFABP), also named FABP2. It is hypothesized that these FABPs are important in intracellular transport of FA, however their precise functions as well as the reason why a single cell type contains more than one distinct FABP, are only beginning to be understood. Both I- and LFABP bind long chain fatty acids with high affinity, nevertheless, it has been suggested that they are functionally distinct. LFABP is expressed in both small intestine and liver, whereas IFABP is found exclusively in the small intestine [1]. IFABP has a single binding site for long chain FA [2], while LFABP contains at least two FA binding sites [3]. LFABP binds a number of other endogenous hydrophobic ligands [4-7], whereas IFABP appears to bind exclusively long chain FA [8]. A functional comparison of these FABPs was made using an in vitro fluorescence energy transfer assay in order to examine the rate and mechanism of transfer of fluorescently tagged fatty acids from proteins to phospholipid membranes. These studies have demonstrated that transfer of fatty acids from IFABP to membranes occurs by a collisional mechanism involving a physical contact with membranes, whereas LFABP seems to employ an aqueous diffusion mechanism for ligand transfer [9].
Both I- and LFABP, as well as the other members of the family, share a common tertiary structure consisting of ten antiparallel β-strands that form a β-barrel, which is capped by two short α-helixes arranged as a helix-turn-helix segment. It is believed that this helical region is part of a “dynamic portal” that regulates fatty acid entry and exit from the internal cavity [10, 11]. The β-barrel domain contains the ligand binding site. The structural elements underlying collisional transfer of a fatty acid from IFABP to membranes could have important physiological consequences as they may dictate the fatty acid trafficking patterns within the cell. Using a helix-less variant of IFABP [12], we previously demonstrated that the α-helical region of IFABP plays a primary role in the collisional transfer of fatty acid from IFABP to membranes [11, 13]. Moreover, analysis of a pair of chimeric proteins generated by exchanging the helix-turn-helix domains between I- and LFABP, showed a significant modification of the absolute rates of fatty acid transfer of the chimeric proteins when compared to the wild types. These results further indicated that the α-helical region of LFABP is responsible for its diffusional mechanism of fatty acid transfer to membranes, as well as the importance of the α-helical region of IFABP in the determination of the collisional fatty acid transfer mechanism [14].
Despite its relatively short length, the 9 residue αI-helix of IFABP would be expected to be membrane interactive, due to its amphipathic character [15]. Amphipathic helices are well known to be important in the targeting of proteins to membranes, and the charge characteristics of the helix appear to modulate interactions with membranes [16]. Thus, we hypothesized that the charged face of the α-I helix is critical for membrane interactions which lead to the dramatic increase in FA transfer rate to anionic membranes in IFABP but not LFABP [9]. In order to test this hypothesis, in the present studies we engineered a pair of chimeric proteins by exchanging only the α-I helixes from I- and LFABP, thus generating α(I)LβIFABP and α(I)IβLFABP chimeric proteins. Analysis of the structural integrity of the chimeric proteins showed that α(I)LβIFABP folded properly and was able to bind fatty acids. α(I)IβLFABP displayed structural problems that precluded further analysis. Based on this assessment, further functional studies were conducted only with α(I)LβIFABP.
Employing a fluorescence resonance energy transfer assay, the transfer of anthroyloxy-labeled fatty acids (AOFA) from the chimeric protein to model membranes was analyzed and compared to the wild type- I- and LFABP. Direct protein-membrane interaction was assessed for the α(I)LβIFABP chimera using a photo-crosslinking assay.
The results indicate that the αI-helix of the FABPs plays an important role in determining the rate and, importantly, the mechanism of fatty acid transfer to membranes. For IFABP, the amphipathic character of the αI-helix is critical for collision-mediated FA transfer. Moreover the αI-helix seems to be important in the physical interaction of IFABP with membranes and as a sensor of membrane charge.
Materials and Methods
Materials
The mutagenic primers and Pfx polymerase were obtained from Invitrogen (Carlsbad, CA). Restriction enzymes XbaI, BamHI and AgeI, pGEM-T vector and T4 DNA ligase were from Promega (Madison, WI). Fluorescently labeled AOFA, 12-(9-anthroyloxy) oleic acid (12AO) was purchased from Molecular Probes, Inc. (Eugene, OR). Egg phosphatidylcholine (EPC), egg N-(7-nitro-2,1,3-benzoxadiaxol-4-yl)-phosphatidylcholine (NBD-PC), brain phosphatidylserine (PS) and bovine heart cardiolipin (CL) were obtained from Avanti Polar Lipids (Alabaster, AL). Lipidex-1000 was purchased from Sigma (Saint Louis, MI). [125I] NaI was from Dupont NEN Products (Boston, MA). All other chemicals were reagent grade or better.
Construction of chimeric FABPs
Recombinant rat pET11d-IFABP and pET11a-LFABP plasmids were generously provided by Drs. Alan Kleinfeld and Ron Ogata (Medical Biology Institute, La Jolla CA). A unique restriction site (AgeI) was generated in the region between αI and αII in both of the plasmids to allow subsequent separation and exchange of segments. Employing overlaping PCR methodology [17], two point mutations were introduced in position 104 and 105 of the LFABP cDNA sequence resulting in a substitution of Met for Thr 22; in the IFABP sequence three point mutations were introduced, two of them resulting in a substitution of Met for Thr 19 (positions 77 and 78 of the cDNA sequence) and the third one was a silent mutation in position 81. The sequence of the primers used to construct the restriction site mutations are the following (point mutations underlined): 5′CGGATAACAATTCCCCTCTA3′ and 5′TTCCTTTCGGGCTTTGTTAG3′ as external primers (the same external primers were used for both constructs), 5′CACGTTAATACCGGTTTTCTCCAT3′and 5′ATGGAGAAAACCGGTATTAACGTG3′ as internal primers for IFABP cDNA, and 5′CTCAGGCAGACCGGTCGCCTTCAT3′ and 5′ATGAAGGCGACCGGTCTGCCTGAG3′ as internal primers for LFABP cDNA. The mutated FABP constructs were verified by sequence analysis. Prior to the treatment with AgeI, the mutant cDNAs were subcloned into pGEM-T vectors by direct ligation of the PCR product. The mutant cDNAs were treated with restriction enzymes AgeI and BamHI in order to separate the αII and β-barrel region from the rest of the construct. The αII and β-barrel of IFABP was ligated to the rest of the construct belonging to LFABP using T4 DNA ligase, generating in this way a chimeric cDNA with βA, αI from LFABP and αII and the remaining β-barrel from IFABP. Similarly, ligation of the αII and β-barrel of LFABP to the rest of the construct belonging to IFABP generated a chimeric cDNA with βA and αI from IFABP, and αII and the remaining β-barrel from LFABP. The chimeric cDNAs were subcloned into pET11d vector by using the XbaI and BamHI restriction sites to construct the expression vectors. The chimeric FABP constructs were verified by sequence analysis.
Protein expression and purification
The wild type and chimeric proteins were overexpressed in Escherichia coli harboring pET11d-IFABP, pET11a-LFABP, pet11d-α(I)LβIFABP and pet11d-α(I)IβLFABP respectively, as detailed elsewhere [9, 11]. The wild type proteins were purified from E. coli as described previously [9]. Neither of the chimeric proteins was expressed as a soluble protein, so it was necessary to purify them from inclusion bodies. The bacterial pellet was therefore dissolved in lysis buffer (Tris.HCl 50 mM, NaCl 100mM, EDTA 1 mM; pH= 8.0), sonicated on ice for 30 sec four times and centrifuged at 10000 rpm for 10 minutes. These steps were repeated for, at least, three more times, with the addition of 0,01 mg/ml DNAseI, 1 mg/ml sodium deoxycholate and 0,5% Triton X-100 in the lysis buffer. The protein pellet was then diluted in denaturating buffer (6.5 M Urea, 5 mM Glycine, 25 mM H3PO4, pH=3.5) and loaded onto a Fast Flow Sp-Sepharose column (Pharmacia) equilibrated with the same buffer. Elution was performed with a linear gradient between the equilibrating buffer without NaCl and a 1M NaCl solution. Fractions containing the chimeric proteins were pooled and store at -70°C. Before each experiment the proteins were extensively dialyzed against K2HPO4 5 mM, KH2PO4 5 mM, KCl 150 mM, pH=7.4 buffer (phosphate buffer) pH 7.4. Both chimeras were > 95 % pure as judged by SDS-PAGE analysis.
Analysis of wild-type and chimeric FABPs
The conformation and ligand binding site integrity of the chimeric constructions were examined by several methods.
Circular Dichroism (CD). Spectra in the near and far UV were measured at 25 °C on a JASCO 810 spectropolarimeter using a 1 cm and 0.1 cm path length quartz cuvette respectively [18]. Each spectrum was obtained from five scans between 240 and 340 nm for near UV data and between 180 and 240 for far UV data. A 15 μM protein concentration solution was used. Secondary structure calculations were performed with the suite of programs provided by DICHROWEB available at the online server of the Department of Crystallography, Birkbeck College, University of London (http://www.cryst.bbk.ac.uk/cdweb).
The Stokes’ radii of the wild-type and chimeric proteins was determined by size-exclusion chromatography (SEC) as described elsewhere [19]. The following proteins were used for column calibration: bovine serum albumin (65 kDa), carbonic anhydrase (30 kDa) and cytochrome c (12 kDa). We used the Superdex 75 column (Pharmacia, Uppsala, Sweden) equilibrated with phosphate buffer and connected to a Merck-Hitachi HPLC apparatus with UV detection at 280 nm.
Fluorescent quantum yields (Qf) of 12-(9-anthroyloxy)oleic acid (12AO) bound to wild-type FABPs and the chimeras were determined using quinine sulfate in 0.1 N H2SO4 as the reference fluorophore, with Qref = 0.7 (Storch et al., 1989). Excitation was at 352 nm for quinine sulfate and 383 nm for 12AO.
- The relative partition coefficient (Kp) for AOFA partitioning between wild type and chimeric proteins and SUVs was determined by measuring AOFA fluorescence at a given molar ratio of Protein:SUV after titration of SUV into a solution containing 5 μM Protein and 0.5 μM 12AO in 40 mM Tris, 100 mM NaCl, pH 7.4 (TBS) [20, 21]
(1)
The decrease in AOFA fluorescence upon titration of AOFA-containing FABP with SUVs was related to Kp by the following equation:
| (2) |
where ΔF is the difference between the initial fluorescence of AOFA in the FABP and the AOFA fluorescence at a given Protein:SUV ratio, and ΔFmax is the maximum difference in AOFA fluorescence. A plot of 1/ΔF versus (1/ΔFmax)([SUV]/[FABP]) gives a slope of 1/Kp. The partition coefficient was used to establish AOFA transfer assay conditions so as to ensure essentially unidirectional transfer [22].
Vesicle Preparation for AOFA Transfer Experiments
Small unilamellar vesicles (SUV) were prepared by sonication and ultracentrifugation as described previously [23, 24]. The standard vesicles were prepared to contain 90 mol % of EPC and 10 mol % of NBD-PC, which served as the fluorescent quencher. To increase the negative charge density of the acceptor vesicles, either 25 mol % of PS or CL were incorporated into the SUVs in place of an equimolar amount of EPC. Vesicles were prepared in 40 mM Tris, 150 mM NaCl, pH=7,4 buffer (TBS) except for SUV containing cardiolipin which were prepared in TBS with 1 mM EDTA.
Transfer of AOFA from FABP to SUV
A fluorescence resonance energy transfer (FRET) assay was used to monitor the transfer of AOFA from the wild type and the chimeric proteins to acceptor model membranes as described in detail elsewhere [9, 11, 25]. Briefly, FABP with bound AOFA was mixed with SUV using a Stopped-Flow Spectrofluorometer DX-17MV (Applied Photophysics Limited, UK). The NBD moiety is an energy transfer acceptor of the anthroyloxy group (AO) donor, therefore the fluorescence of the AOFA is quenched when the ligand is bound to SUV which contain NBD-PC. Upon mixing, transfer of AOFA from protein to membrane is directly monitored by the time-dependent decrease in AO fluorescence. Final transfer assay conditions were 15 μM wild-type IFABP or α(I)LβIFABP with 1.5 μM 12AO and 150-600 μM SUV and 5 μM wild-type LFABP and 150 -1200 μM SUV. Transfer was monitored at 25°C. Controls to ensure that photobleaching was eliminated were performed prior to each experiment, as previously described [11]. Data were analyzed using software provided with the instrument, and all curves were well described by a single exponential function. For each experimental condition, at least five replicates were done. Average values ± S.D. for three or more separate experiments are reported, unless otherwise indicated.
Preparation of photoactivable reagents
125I-TID-PC was prepared by radioiodination of its non radioactive tin-containing precursor 1-O-hexadecanoyl-2-O-[9-[[[2-(tributylstannyl)-4-(trifluoromethyl-3H-diazirin-3-yl)benzyl]oxy]carbonyl]nonanoyl] -sn-glycero-3-phospho-choline according to Weber and Brunner [26] and our previous work [27]. The precursor was generously donated by Prof. J. Brunner from the Swiss Federal Institute of Technology, Zurich, Switzerland. The dried tin-containing precursor (~20 nmol) was dissolved in 10 μL of acetic acid in a 1 mL Reacti-Vial (Pierce, Rockford, IL). [125I]NaI (1 mCi) was added and the iodination started by the addition of peracetic acid (2 μL of a 32% solution in acetic acid). After 2 min at room temperature, the reaction was quenched with 50 μL of 10% Na2S2O5. Then, 40 μL of chloroform/methanol (2:1) were added and vortexed. The organic phase was collected and concentrated using a charcoal filter to adsorb volatile radioactivity. The residue was dissolved in 20 μL of methanol/chloroform/water (9:1:1) and subjected to reverse-phase HPLC using the same solvent and a 208HS54 C8 column (Vydac) in a Merck-Hitachi apparatus with UV detection at 254 nm. The flow rate was 1 mL/min and fractions of 0.5 mL were collected. 125I-TID-PC eluted at approximately 20 min, while the excess of tin-containing precursor eluted at approximately 40 min. An aliquot (5 μL) of each fraction in the elution region of 125I-TID-PC was analyzed by TLC on silica gel plates (LK6D, 60 Å, Whatman, Clifton, NJ) and subjected to autoradiography. Fractions containing radioactivity were pooled and concentrated by co-evaporation with toluene/ethanol (1:1). 125I-TID-PC was dissolved in ethanol/toluene (1:1) at approximately 1 mCi/mL, and stored at -20 °C.
Preparation of lipid vesicles containing I-TID-PC
Large unilamellar vesicles (LUV) of EPC, EPC/PS (3:1 mol:mol) or EPC/CL (3:1 mol:mol) were prepared (0.5 mM total PL) by extrusion through polycarbonate membranes of 100 nm pore diameter (Avestin Inc., Ottawa, Canada). To prepare the LUV containing 125I-TID-PC (200 μCi/μmol of PL), the photo reagent was mixed with the lipids in chloroform. The mixed lipids were dried under a stream of N2 and resuspended in 40 mM Tris, 100 mM NaCl, 50 mM glutathione (buffer A) by vortexing. Cardiolipin-containing vesicles also had 1 mM EDTA included in the buffer A. Then, lipid suspensions were incubated at 37°C for 30 minutes and passed 11 times through the polycarbonate filters using a Liposofast-extruder system (Avestin).
Photolabeling analysis of FABP-membrane interactions
Experiments were conducted as previously described [27]. Briefly, 120 μl of 0.5 mM photoreagent-containing LUVs (30 μCi/ml) were incubated with the 60 μg FABP in 200 μl buffer A, at room temperature for 30 sec. After the indicated incubation time, mixtures (0.32 mL) in glass cuvettes were irradiated for 30 sec with a Xenon lamp (450 Watts) at a distance of 25 cm. As a control, the photoreagent-containing LUV were irradiated prior to their mixture with IFABP. After irradiation, 3 volumes of chloroform/methanol (2:1) were added, vortexed, and the organic phase discarded. FABPs were precipitated with 5 volumes of acetone, and dissolved in 25 μl gel loading sample buffer (126 mM Tris, 20% glycerol, 4% SDS, 10% 2-β mercaptoethanol, 0.004% BrPhenolBlue) for direct analysis by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Following Coomassie blue staining, gels were dried and exposed to X-Omat film (Kodak) at -80°C for different times depending on the amount of radioactivity. Images were quantified using the program Image J (National Institutes of Health).
Results
Construction of chimeric proteins and comparison of their physical properties with those of native FABPs
To examine whether the primary determinant of the FABP fatty acid transfer mechanism resides in the α-I helix domain, we undertook the exchange of the α-I regions between I- and LFABP. To control for structural integrity of the chimeric proteins, the following methods were employed: (a) circular dichroism spectroscopy, (b) determination of the Stokes’ radii, (c) fluorescence quantum yield measurements of bound anthroyloxy fatty acid, and (d) determination of relative partition coefficient of 12AO between the FABPs and SUVs. A summary of the results for these experiments is presented in Table 1.
Table 1.
Physical parameters of Wild-Types and chimeric FABPsa
| Kp (suv:prot)b | Qf c | θ215 (grad M-1cm-1) | Stokes’ radii (Ǻ) | |
|---|---|---|---|---|
| IFABP | 6.74 ± 2.25 | 0.08 ± 0.04 | -7212.33 | 19.08 ± 0.08 |
| LFABP | 0.09 ± 0.06 | 0.31 ± 0.09 | -8528.85 | 19.28 ± 0.05 |
| α(I)LβIFABP | 2.08 ± 0.14 | 0.12 ± 0.05 | -4776.02 | 19.42 ± 0.08 |
Data are the mean ± standard deviation of values obtained in three separate experiments.
Apparent partition constant.
Quantum yield.
The CD spectra of wild-type IFABP showed the maximum at 198-200 nm and the minimum near 215 nm typical of β-proteins, and its shape and intensity agreed with previously published data [14, 18]. The α(I)LβIFABP chimeric protein exhibited the same peaks as wild-type IFABP but with an approximately 30% decreased intensity. This decrease could be explained taking into account the lower Trp content of α(I)LβIFABP and the strong influence that these aromatic residues can have on intensity in the far-UV CD signal [28]. The CD spectra of wild-type LFABP showed the maximum at 198-200 nm and the minimum near 218 nm, and its shape and intensity agreed with previously published data [14, 29]
In order to explore the tertiary structure of α(I)LβIFABP, spectra in the near-UV were taken. A direct comparison of α(I)LβIFABP spectra with the wild type proteins is not possible because of the difference in the Trp content. Nevertheless, α(I)LβIFABP showed two well defined bands with opposite signs centered at 260 and 280 nm, suggesting a well defined tertiary structure.
The hydrodynamic properties of α(I)LβIFABP were studied by size-exclusion chromatography and compared with those of I- and LFABP. The Stokes’ radius of the chimeric protein, 19.4 ± 0.1 Å, was almost identical to those of IFABP (19.1 ± 0.1 Å) and LFABP (19.3 ± 0.1 Å). The elution profile is consistent with α(I)LβIFABP adopting a monomeric conformational state.
Fluorescence quantum yields (Qf) for the AOFA are used to assess the relative hydrophobicity of the environment surrounding the fluorophore [30]. The comparison of 12AO Qf values for the chimeric and native proteins could, therefore, indicate whether the modifications introduced into the native protein may have altered the dielectric environment of its binding pocket. We found that the Qf value for α(I)LβIFABP was close to the Qf for binding to wild-type IFABP (Table 1), suggesting that the introduction of the α-I segment of LFABP does not modifiy the hydrophobicity of the ligand binding site.
An apparent partition coefficient value was also obtained for each protein, describing the relative distribution of 12AO between FABP and EPC-SUVs. Analysis of these data showed a preferential partitioning of 12AO to SUVs relative to IFABP in agreement with previous results [25] (Table 1). LFABP showed a higher relative affinity for the 12AO when compared to IFABP, in agreement with previous results [31]. On the other hand the chimeric proteins showed Kp values that were in between the ones obtained for the wild types.
Taken together, the controls suggested no major alterations in the conformation and binding site properties of the chimeric protein α(I)LβIFABP relative to its parent wild-type proteins. Therefore, the same assay conditions were employed for monitoring AOFA transfer from wild-type IFABP and the chimeric.
Effect of vesicle concentration on the transfer of AOFA from FABPs to membranes
The proportional increase in transfer rate as a function of SUV concentration is referred to as a “collisional transfer mechanism” and is different from an “aqueous diffusion mechanism”, where no effect is observed. To distinguish between these transfer mechanisms, transfer of AOFA from the chimeric proteins to model membranes was examined as a function of increasing membrane concentration, and results were compared to those for wild-type LFABP and IFABP. Figure 1 shows the results obtained when constant concentrations of these FABP-AOFA donor complexes were mixed with increasing concentrations of EPC SUVs. The typical “collisional transfer mechanism” was observed for IFABP, in agreement with previous results [9, 14, 32]. For LFABP, no effect of SUV concentration on 12AO transfer rate was found, as expected [9, 14, 33]. On the other hand a very slight but statistically significant increase (p<0.01, ANOVA) was observed for α(I)LβIFABP as a function of SUV concentration, suggesting that the collisional transfer mechanism, while markedly blunted, was not completely abolished (Fig. 1A). In addition, the absolute transfer rates of 12AO from the chimeric protein α(I)LβIFABP were slower than the obtained for IFABP.
Fig 1.
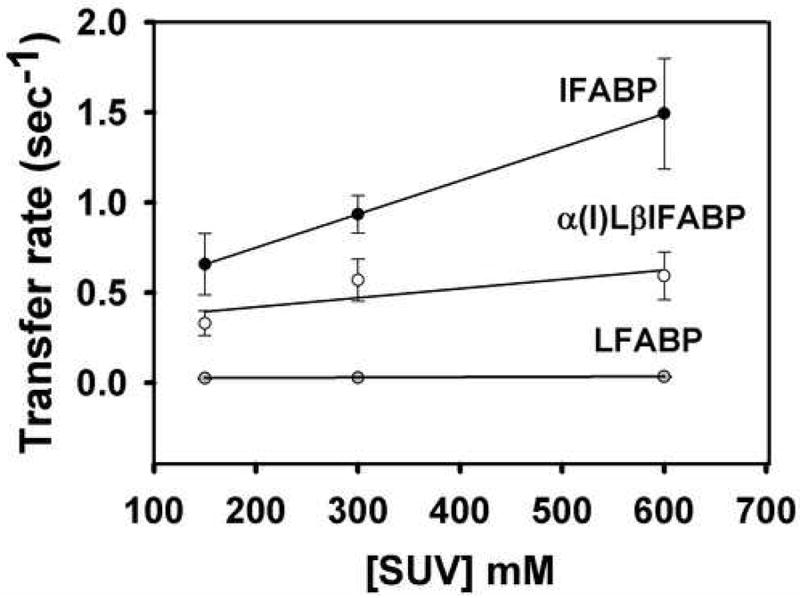
Effect of acceptor membrane concentration on AOFA transfer from IFABP (black circles), α(I)LβIFABP (white circles) and LFABP (grey circles). Transfer of 1.5 μM 12AO from 15 μM IFABP/ α(I)LβIFABP or 0.5 μM 12AO from 5 μM LAFBP to EPC/NBD-PC SUV. Averages from three different experiments ± SD are shown. Transfer rates for LFABP are 0.0249 ± 0.005 per sec, 0.0282 ± 0.003, 0.0340 ± 0.0035 for 150, 300 and 600 μM SUV, respectively.
Effect of vesicle charge on the transfer of AOFA from FABPs to membranes
Changes in the surface charge density of the acceptor vesicles can also influence ligand transfer rates if electrostatic interactions between the donor protein and acceptor membranes are involved. By contrast, in the case of aqueous diffusion, characteristics of the acceptor membrane would not be expected to modulate the transfer rate under the donor:acceptor ratios used in these experiments. Figure 2 shows that, as expected from previous studies, the rate of transfer of 12AO from IFABP is substantially increased by incorporation of 25 mol % PS or CL into EPC/NBD-PC acceptor membranes (Fig.2A) [9, 14, 32]. Transfer of 12AO from the chimeric protein α(I)LβIFABP did not show any increase when 25 % of PS was added to EPC-SUVs, however a small but significant increase was observed when 25 % of CL was incorporated in the acceptor vesicles (p<0,05, t-test) (Fig. 2). These results suggest that the chimeric protein is still somewhat charge sensitive and, hence, capable of making effective collisions with charged vesicles to certain point. LFABP was essentially unaffected by the presence of negatively charged phospholipids as expected for an aqueous transfer mechanism [9, 33].
Fig. 2.
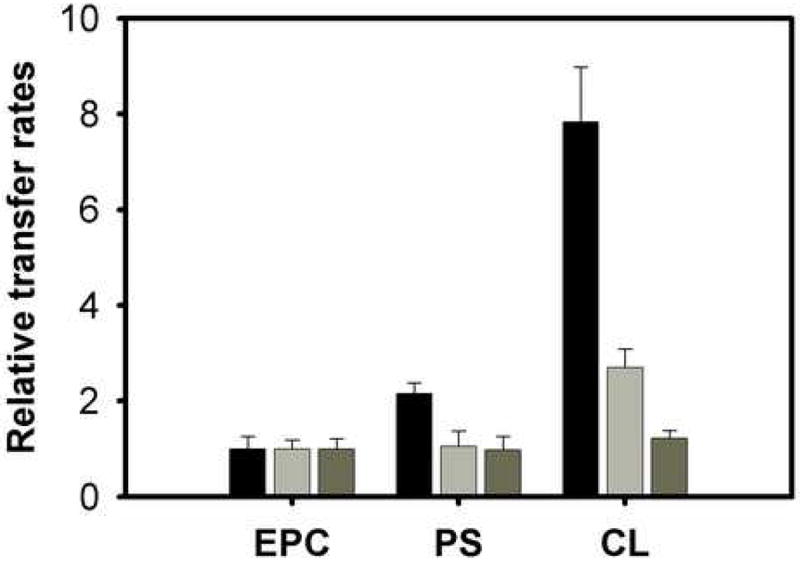
Effect of charge on AOFA transfer from IFABP (black bars), α(I)LβIFABP (grey bars) and LFABP (dark grey bars). Transfer of 1.5 μM 12AO from 15 μM IFABP/ α(I)LβIFABP to 150 μM EPC/NBD-PC SUV conatining 25 mol% brain phosphatidilserine (PS) or CL; or 0.5 μM 12AO from 5 μM LFABP protein to 150 μM EPC/NBD-PC SUV conatining 25 mol% PS or CL. Results are expressed relative to the rate of transfer of 12AO to EPC/NBD-PC membranes. Averages from three different experiments ± SD are shown.
Interaction of chimeric proteins with PL membranes
To analyze directly the interaction of FABPs with membranes, we conducted a series of experiments with vesicles containing the photoactivable reagent 125I-TID-PC. Upon photolysis, the trifluoromethyl diazirine group of this reagent is capable of reacting with protein segments inserted into or in contact with the hydrophobic region of the phospholipid membrane. These experiments were conducted to determine whether the α-I region is involved in the interaction of intestinal FABP with membranes. We examined wild-type IFABP as well as the chimeric protein α(I)LβIFABP for their interactions with zwitterionic and acidic phospholipid vesicles. As can be seen in Figure 3, α(I)LβIFABP showed a lower level of interaction when compared with IFABP. A helix-less variant of IFABP was also assayed as a negative control of interaction; as expected from our previous results [25], the helix-less IFABP did not show interaction with either the zwitterionic or anionic vesicles (results not shown). These results indicate that even in the presence of the α-I region of LFABP, the chimeric IFABP protein is still capable of interacting with LUVs. Overall, these results suggest that, despite the importance of the α-I region, the α-II or other parts of the protein also participate in the physical interaction with membranes during FA transfer from IFABP. On the other hand, while IFABP showed a higher level of interaction with the negatively charged acceptor vesicles, α(I)LβIFABP did not show any increase. This loss of sensitivity of the chimeric protein to membrane charge density is in agreement with results of the 12AO transfer assays.
Fig. 3.
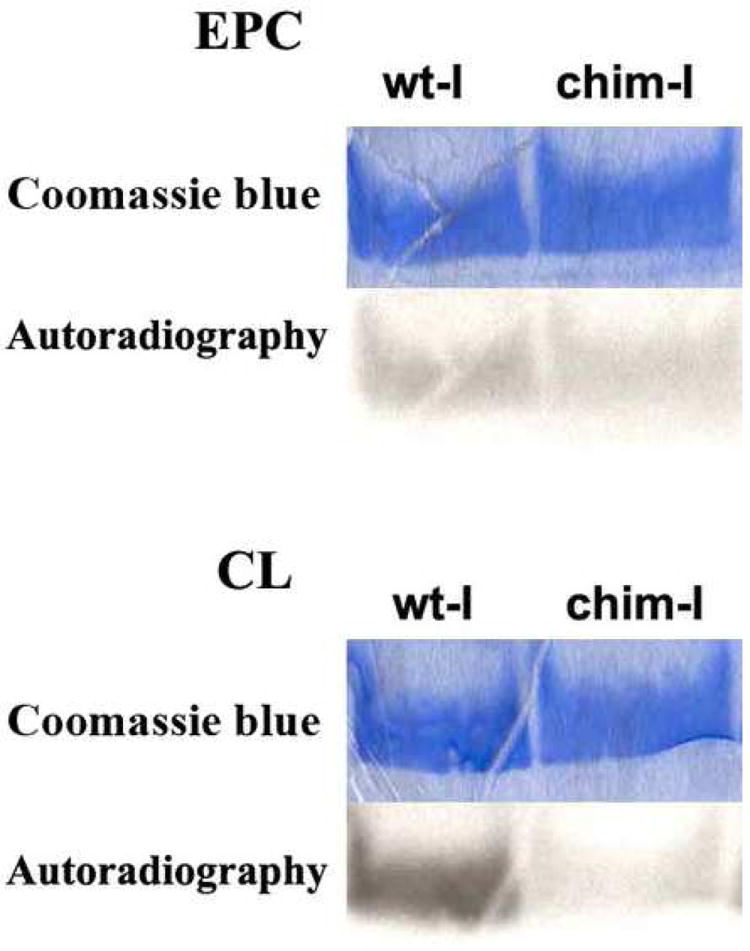
Photolabeling of the apo forms of IFABP and α(I)LβIFABP by incubation with zwitterionic (EPC) and acidic (25% CL) membranes containing 125I-TID-PC, as described in Methods. Results are representative of 2 sets of experiments. IFABP shows a 1.3 fold greater interaction than α(I)LβIFABP when assayed with EPC vesicles. This difference increased to 3 fold when CL vesicles were used.
Discussion
Employing a resonance energy transfer assay we have demonstrated that movement of AOFA from LFABP to acceptor vesicles is regulated, mostly, by FA solubility [9, 33], according to a diffusional FA transfer mechanism. On the other hand it has also been shown that IFABP transfers the ligand via a collisional FA transfer mechanism, where a protein-membrane complex is formed [9]. This collisional FA transfer mechanism has also been observed with other members of the FABP family, including H-, A, and KFABP [34-36].
We have also demonstrated that cationic residues on IFABP’s surface play a primary role in establishing electrostatic interactions with phospholipid membranes [32], and the same has been observed for other members of the family that present a collisinal mechanism [36]. “Collisional FABPs” show a very similar surface electrostatic potential topology, they have a net positive potential in the α-helical region supporting the importance of this region in the interaction with membranes [37]. On the other hand LFABP and toad liver basic fatty acid binding protein (Lb-FABP), “diffusional FABPs”, show both positive and negative electrostatic potential in the α-helical region [37, 38]. When compared to collisional FABPs, this region could be seen as a net neutral one and hence have a diminished capability of forming strong electrostatic interactions with charged phospholipid membranes. Moreover, site directed mutagenesis studies demonstrated that all the Lys residues of the alpha-helical domain of IFABP (but not the β-barrel) contribute, to different degrees, to the collisional mechanism of FA transfer from IFABP to model membranes [25]. These studies also suggested that the alpha-II segment is of particular importance in the establishment of charge-charge interactions between IFABP and membranes.
The construction of chimeric proteins has been used to reveal the importance and function of isolated domains in several proteins [39, 40]. The highly similar tertiary structures of I- and LFABP, despite their low primary sequence homology, indicated that this pair of proteins were very good candidates for the construction of distinct chimeric proteins that would reveal the specific functions of different domains in the transfer of AOFA to membranes.
As we have demonstrated, the entire α-helical domain plays an important role in the interaction of IFABP with membranes and in the mechanism of FA transfer to acceptor membranes [11, 13]. Intestinal and liver FABP have very similar overall conformations, as shown by the comparison of rms distance differences, the superimposition of crystallographic structural models [3], and the secondary structure assignments from their solution structures [10]. The construction of a pair of chimeric proteins by swapping the whole α helical domain between I- and LFABP demonstrated the importance of this domain in the determination of FA transfer mechanism in both proteins. The region examined, however contains both αI and αII helixes as well as the βA strand. In IFABP, the α-I helix polar face contains two basic lysine residues at positions 16 and 20, and two negatively charged glutamate residues at positions 15 and 19 [41]. On the other hand, LFABP has only one Lys residue at position 19 and one Glu at position 15 in its α-I helix. It is hypothesized that the charged face of the α-I helix is critical for membrane interactions which lead to the dramatic increase in AOFA transfer to anionic membranes in IFABP but not LFABP [9]. In order to test this hypothesis, in the present studies we engineered a pair of chimeric proteins by exchanging only the α-I helixes from I- and LFABP.
The chimeric protein α(I)LβIFABP showed no major structural differences compared to the wild-type proteins as indicated by the CD spectra and the hydrodynamic analysis by SEC. The molar ellipticity and CD spectral minimum for the chimeric form are very close to those of IFABP, reflecting the large contribution of the β-barrel binding pocket of the parent protein. The almost unmodified Stokes’ radius and monomeric behavior supported its well folded state. The apparent lack of modification of the Qf value of wild-type IFABP by the incorporation of LFABP αI helix, indicates that the binding domain has not suffered a substantial change. Comparing this with the larger change in Qf observed when the entire α-helical domain was exchanged [14], it could be deduced that the αII-helix of LFABP is responsible for the increment in hydrophobicity of the IFABP ligand binding site observed for αLβIFABP [14]. Taken together, the control studies indicate that the relative binding capacity as well as the integrity of the binding pocket have been maintained in the α(I)LβIFABP, without major structural or physical/chemical alterations.
The transfer rates of AOFA from α(I)LβIFABP to zwitterionic SUVs were significantly slower than those obtained from IFABP, suggesting that the αI region contributes to the determination of the absolute transfer rate of the fatty acid. The effect of increasing EPC-SUV concentration on the FA transfer rate from α(I)LβIFABP was notably dampened in comparison with IFABP. This effect could probably be related to the loss of the polar face of the amphipatic helix I, specifically, Lys 16 and Lys 20. Neutralization of Lys 16 and Lys 20 to isoleucine using site directed mutagenesis, also showed a decrease in the AOFA transfer rates without modification of the collisional mechanism [25]. Indeed, a slight but significant increase in the AOFA transfer rate was observed in response to the increase in SUV concentration, suggesting that the collisional FA transfer mechanism of IFABP was not completely abolished in α(I)LβIFABP. The characteristics of the αII domain of IFABP may be responsible for this behavior, presumably owing to the presence of two positively charged residues, Lys 27 and Lys 29. Recent work has demonstrated the importance of these residues in the modulation of FA transfer mechanism from IFABP [25]. It is also noteworthy that a hydrophobic patch exposed to the aqueous milieu close to the portal region which is highly conserved among collisional FABPs has been observed [42]. In IFABP this region contained in or near αII (Val25, Val26, e Ile30) could be driving the collisional interaction towards zwitterionic SUVs. The fact that the exchange of the whole α-helical domain induced a drastic change from the diffusional LFABP to a collisional αIβLFABP [14], is probably pointing towards the αII region of LFABP as the responsible element for the diffusional FA transfer mechanism.
The effect of acidic vesicles on the mechanism of transfer from the chimeric protein was similar to the effects found for zwitterionic vesicles. The absolute transfer rates were decreased when compared with IFABP, and α(I)LβIFABP lost its sensitivity to charge density in comparison to IFABP. Nonetheless a significant increase in transfer rate was observed for vesicles containing 25 % of CL, suggesting once again that the typical collisional FA transfer mechanism of IFABP was not completely abolished.
The exchange of the whole α-helical domain induced a drastic change from the collisional IFABP to a diffusional αLβIFABP [14] while in the present work a more modest modification is observed when αI is exchanged. The direct comparison between these results is probably indicating that the α–helical domain as a whole is determinant for the global process, while each segment could be responsible for different steps contributing to the final fatty acid transfer rate and mechanism.
The loss of sensitivity to SUV charge density could be due to the absence of the αI region of IFABP, which might be acting as a charge sensor in a first step of IFABP-membrane interaction, followed by the FA transfer step, where the αII region could be involved (Fig. 4). It has been observed by crystallography that the ω-terminal methyl group of the fatty acid is interacting with the “portal domain” (αII, βC-βD and βE-βF turns) [15]. The destabilization of αII after releasing the FA could be preceded by the interaction of αI with membranes. An exchange of the αI region of IFABP for that of LFABP causes a net decrease in the number of effective charges in that region, considering that the αI helix of LFABP has only two charged residues (Glu15 and Lys19) while αI helix of IFABP has four charged residues (Glu15, Lys16, Glu19 and Lys20). This considerable decrease in the number of charged residues is most likely affecting the positive electrostatic potential normally found in the α-helical domain of IFABP [37].
Fig. 4.
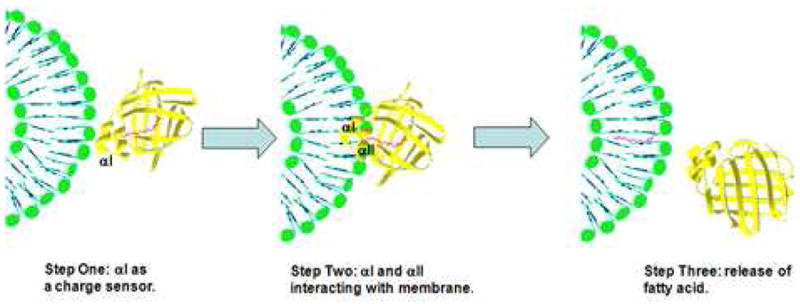
Model to illustrate the possible multistep interaction process of IFABP with model membranes. Initial binding of FABP to vesicles via electrostatic interactions of αI (step 1), then the formation of a stable complex is reached when αII is involved (step 2) to end with the release of fatty acid. This model was made with IFABP PDB sequence 2IFB.
The photocrosslinking experiments, which are useful for assessing direct protein-membrane interactions [27], showed a diminished interaction of α(I)LβIFABP with membranes relative to the native IFABP. This supports the importance of the αI region in the direct interaction of IFABP with membranes. As the interaction was not completely abolished, however, this suggests further that the αII helix is capable of maintaining a lesser degree of physical interaction with membranes. Thus, the crosslinking data are in agreement with the AOFA transfer kinetic results. Nevertheless, it is important to keep in mind that in these experiments the substitution of the α-I region could be modifying the primary charge interaction step, consequently affecting the second step in which α-II is proposed to participate and thereby giving the appearance that the α-II was little involved when it could still be crucial to the ligand transfer process. Overall, it is likely that both the αI and the αII helixes participate, perhaps sequentially, in the interaction of IFABP with membranes, resulting in efficient transfer of the fatty acid ligand to a target membrane.
Acknowledgments
We gratefully acknowledge Dr. M. R. Ermácora and his group for their insightful comments regarding this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bass NM. The cellular fatty acid binding proteins: Aspects of structure, regulation, and function. Int Rev Cytol. 1988;3:143–184. doi: 10.1016/s0074-7696(08)61733-7. [DOI] [PubMed] [Google Scholar]
- 2.Sacchettini JC, Gordon JI, Banaszak LJ. Refined apoprotein structure of rat intestinal fatty acid binding protein produced in Escherichia coli. Proc Natl Acad Sci U S A. 1989;86:7736–7740. doi: 10.1073/pnas.86.20.7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson J, Winter N, Terwey D, Bratt J, Banaszak L. The crystal structure of the liver fatty acid-binding protein. A complex with two bound oleates. J Biol Chem. 1997;272:7140–7150. doi: 10.1074/jbc.272.11.7140. [DOI] [PubMed] [Google Scholar]
- 4.Richieri GV, Ogata RT, Kleinfeld AM. Kinetics of fatty acid interactions with fatty acid binding proteins from adipocyte, heart, and intestine. J Biol Chem. 1996;271:11291–11300. doi: 10.1074/jbc.271.19.11291. [DOI] [PubMed] [Google Scholar]
- 5.Wilkinson TCI, Wilton DC. Studies on fatty acid-binding proteins: The binding properties of rat liver fatty acid-binding protein. Biochem J. 1987;247:485–488. doi: 10.1042/bj2470485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burrier RE, Manson CR, Brecher P. Binding of acyl-CoA to liver fatty acid-binding protein: Effect on acyl-CoA synthesis. Biochim Biophys Acta. 1987;919:221–230. doi: 10.1016/0005-2760(87)90261-x. [DOI] [PubMed] [Google Scholar]
- 7.Thumser AEA, Voysey JE, Wilton DC. The binding of lysiphosphospholipids to rat liver fatty acid-binding protein and albumin. Biochem J. 1994;301:801–806. doi: 10.1042/bj3010801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowe JB, Sacchettini JC, Laposata M, McQuillan JJ, Gordon JI. Expression of rat intestinal fatty acidbinding protein in Escherichia coli: Purification and comparison of ligand binding characteristics with that of Escherichia coliderived rat liver fatty acid-binding protein. J Biol Chem. 1987;262:5931–5937. [PubMed] [Google Scholar]
- 9.Hsu KT, Storch J. Fatty acid transfer from liver and intestinal fatty acid-binding proteins to membranes occurs by different mechanisms. J Biol Chem. 1996;271:13317–13323. doi: 10.1074/jbc.271.23.13317. [DOI] [PubMed] [Google Scholar]
- 10.Hodsdon ME, Cistola DP. Ligand binding alters the backbone mobility of intestinal fatty acid-binding protein as monitored by 15N NMR relaxation and 1H exchange. Biochemistry. 1997;36:2278–2290. doi: 10.1021/bi962018l. [DOI] [PubMed] [Google Scholar]
- 11.Corsico B, Cistola DP, Frieden C, Storch J. The helical domain of intestinal fatty acid binding protein is critical for collisional transfer of fatty acids to phospholipid membranes. Proc Natl Acad Sci U S A. 1998;95:12174–12178. doi: 10.1073/pnas.95.21.12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim K, Cistola DP, Frieden C. Intestinal fatty acid-binding protein: The structure and stability of a helix-less variant. Biochemistry. 1996;35:7553–7558. doi: 10.1021/bi9529115. [DOI] [PubMed] [Google Scholar]
- 13.Wu F, Corsico B, Flach CR, Cistola D, Storch J, Mendelsohn R. Deletion of the helical motif in the intestinal fatty acid-binding protein reduces its interactions with membrane monolayers: Brewster angle microscopy, IR reflection-absorption spectroscopy and surface pressure studies. Biochemistry. 2001;40:1976–1983. doi: 10.1021/bi002252i. [DOI] [PubMed] [Google Scholar]
- 14.Córsico B, Liou HL, Storch J. The α-helical domain of liver fatty acid binding protein is responsible for the diffusionmediated transfer of fatty acids to phospholipid membranes. Biochemistry. 2004;43:3600–3607. doi: 10.1021/bi0357356. [DOI] [PubMed] [Google Scholar]
- 15.Scapin GJ, Gordon I, Sacchettini JC. Refinement of the structure of recombinant rat intestinal fatty acid-binding apoprotein at 1.2 Å resolution. J Biol Chem. 1992;267:4253–4269. doi: 10.2210/pdb1ifc/pdb. [DOI] [PubMed] [Google Scholar]
- 16.Anantharamaiah GM, Jones MK, Segrest JP. In: The Amphipathic Helix. Epand RM, editor. CRC Press; Boston: 1993. pp. 109–141. [Google Scholar]
- 17.Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7667. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clerico EM, Peisajovich SG, Ceolín M, Ghiringhelli PD, Ermácora MR. Engineering a compact non-native state of intestinal fatty acid-binding protein. Biochim Biophys Acta. 2000;1476:203–218. doi: 10.1016/s0167-4838(99)00247-2. [DOI] [PubMed] [Google Scholar]
- 19.Uversky VN. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry. 1993;32(48):13288–13298. doi: 10.1021/bi00211a042. [DOI] [PubMed] [Google Scholar]
- 20.Massey JB, Bick DH, Pownall HJ. Spontaneous transfer of monoacyl amphiphiles between lipid and protein surfaces. Biophys J. 1997;72(4):1732–1743. doi: 10.1016/S0006-3495(97)78819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Storch J. Mechanism for binding of fatty acids to hepatocyte plasma membranes: different interpretation. Hepatology. 1990;12(6):1447–9. doi: 10.1002/hep.1840120632. [DOI] [PubMed] [Google Scholar]
- 22.Roseman MA, Thompson TE. Mechanism of the spontaneous transfer of phospholipids between bilayers. Biochemistry. 1980;19(3):439–44. doi: 10.1021/bi00544a006. [DOI] [PubMed] [Google Scholar]
- 23.Huang C, Thompson TE. Preparation of homogeneous, single-walled phosphatidylcholine vesicles. Methods Enzymol. 1974;32:485–489. doi: 10.1016/0076-6879(74)32048-4. [DOI] [PubMed] [Google Scholar]
- 24.Storch J, Kleinfeld AM. Transfer of long-chain fluorescent free fatty acids between unilamellar vesicles. Biochemistry. 1986;25:1717–1726. doi: 10.1021/bi00355a041. [DOI] [PubMed] [Google Scholar]
- 25.Falomir-Lockhart L, Laborde L, Kahn PC, Storch J, Córsico B. Protein-Membrane Interaction and Fatty Acid Transfer from Intestinal Fatty Acid Binding Protein: Support for a multi step process. J Biol Chem. 2006;281:14232–14240. doi: 10.1074/jbc.M511943200. [DOI] [PubMed] [Google Scholar]
- 26.Weber T, Paesold G, Galli C, Mischler R, Semenza G, Brunner J. Evidence for H(+)-induced insertion of influenza hemagglutinin HA2 N-terminal segment into viral membrane. J Biol Chem. 1994;269(28):18353–8. [PubMed] [Google Scholar]
- 27.Córsico B, Toledo JD, Garda HA. Evidence for a central apolipoprotein A-I domain loosely bound to lipids in discoidal lipoproteins that is capable of penetrating the bilayer of phospholipid vesicles. J Biol Chem. 2001;276:16978–16985. doi: 10.1074/jbc.M011533200. [DOI] [PubMed] [Google Scholar]
- 28.Freskgard PO, Martensson LG, Jonasson P, Jonsson BH, Carlsson U. Assignment of the contribution of the tryptophan residues to the circular dichroism spectrum of human carbonic anhydrase II. Biochemistry. 1994;33(47):14281–8. doi: 10.1021/bi00251a041. [DOI] [PubMed] [Google Scholar]
- 29.Hagan RM, Worner-Gibbs J, Wilton DC. Tryptophan insertion mutagenesis of liver fatty acid-binding protein: L28W mutant provides important insights into ligand binding and physiological function. J Biol Chem. 2005;280(3):1782–9. doi: 10.1074/jbc.M407131200. [DOI] [PubMed] [Google Scholar]
- 30.Storch J, Bass NM, Kleinfeld AM. Studies of the fatty acid-binding site of rat liver fatty acid-binding protein using fluorescent fatty acids. J Biol Chem. 1989;264:8708–8713. [PubMed] [Google Scholar]
- 31.Thumser EA, Storch J. Liver and intestinal fatty acid-binding proteins obtain fatty acids from phospholipid membranes by different mechanisms. J Lipid Res. 2000;41:647–656. [PubMed] [Google Scholar]
- 32.Córsico B, Franchini GR, Hsu KT, Storch J. Electrostatic and hydrophobic interactions contribute to the collisional mechanism of fatty acid transfer from intestinal fatty acid binding protein to phospholipid membranes. J Lipid Res. 2005;46:1765–72. doi: 10.1194/jlr.M500140-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Kim HK, Storch J. Free fatty acid transfer from rat liver fatty acid-binding protein to phospholipid vesicles: Effect of ligand and solution properties. J Biol Chem. 1992;267:77–82. [PubMed] [Google Scholar]
- 34.Wootan MG, Storch J. Regulation of fluorescent fatty acid transfer from adipocyte and heart fatty acid binding proteins by acceptor membrane lipid composition and structure. J Biol Chem. 1994;269:10517–10523. [PubMed] [Google Scholar]
- 35.Shaughnessy S, Smith ER, Kodukula S, Storch J, Fried SK. Adipocyte metabolism in adipocyte fatty acid binding protein knockout mice (aP2-/-) after short-term high-fat feeding: functional compensation by the keratinocyte fatty acid binding protein. Diabetes. 2000;49:904–911. doi: 10.2337/diabetes.49.6.904. [DOI] [PubMed] [Google Scholar]
- 36.Herr FM, Aronson J, Storch J. Role of portal region lysine residues in electrostatic interactions between heart fatty acidbinding protein and phospholipid membranes. Biochemistry. 1996;35:1296–1303. doi: 10.1021/bi952204b. [DOI] [PubMed] [Google Scholar]
- 37.LiCata VJ, Bernlohr DA. Surface properties of adipocyte lipid-binding protein: Response to lipid binding, and comparison with homologous proteins. Proteins: Struct Funct Genet. 1998;33:577–589. doi: 10.1002/(sici)1097-0134(19981201)33:4<577::aid-prot10>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 38.Di Pietro SM, Corsico B, Perduca M, Monaco HL, Santome JA. Structural and biochemical characterization of toad liver fatty acid binding protein. Biochemistry. 2003;42:8192–8203. doi: 10.1021/bi034213n. [DOI] [PubMed] [Google Scholar]
- 39.Yin D, Gavi S, Wang HY, Malbon CC. Probing receptor structure/function with chimeric G-protein-coupled receptors. Mol Pharmacol. 2004 Jun;65(6):1323–32. doi: 10.1124/mol.65.6.1323. [DOI] [PubMed] [Google Scholar]
- 40.Liou HL, Kahn PC, Storch J. Role of the helical domain in fatty acid transfer from adipocyte and heart fatty acid-binding proteins to membranes: analysis of chimeric proteins. J Biol Chem. 2002;277(3):1806–15. doi: 10.1074/jbc.M107987200. [DOI] [PubMed] [Google Scholar]
- 41.Scapin GJ, Gordon I, Sacchettini JC. Refinement of the structure of recombinant rat intestinal fatty acid-binding apoprotein at 1.2 Å resolution. J Biol Chem. 1992;267:4253–4269. doi: 10.2210/pdb1ifc/pdb. [DOI] [PubMed] [Google Scholar]
- 42.Kennedy MW, Beauchamp J. Sticky-finger interaction sites on cytosolic lipid-binding proteins? Cell Mol Life Sci. 2000;57:1379–1387. doi: 10.1007/PL00000623. [DOI] [PMC free article] [PubMed] [Google Scholar]