

Abstract
The metabolic syndrome is a constellation of metabolic disorders including obesity, hypertension, and insulin resistance, components which are risk factors for the development of diabetes, hypertension, cardiovascular, and renal disease. Pathophysiological abnormalities that contribute to the development of the metabolic syndrome include impaired mitochondrial oxidative phosphorylation and mitochondrial biogenesis, dampened insulin metabolic signaling, endothelial dysfunction, and associated myocardial functional abnormalities. Recent evidence suggests that impaired myocardial mitochondrial biogenesis, fatty acid metabolism, and antioxidant defense mechanisms lead to diminished cardiac substrate flexibility, decreased cardiac energetic efficiency, and diastolic dysfunction. In addition, enhanced activation of the renin–angiotensin–aldosterone system and associated increases in oxidative stress can lead to mitochondrial apoptosis and degradation, altered bioenergetics, and accumulation of lipids in the heart. In addition to impairments in metabolic signaling and oxidative stress, genetic and environmental factors, aging, and hyperglycemia all contribute to reduced mitochondrial biogenesis and mitochondrial dysfunction. These mitochondrial abnormalities can predispose a metabolic cardiomyopathy characterized by diastolic dysfunction. Mitochondrial dysfunction and resulting lipid accumulation in skeletal muscle, liver, and pancreas also impede insulin metabolic signaling and glucose metabolism, ultimately leading to a further increase in mitochondrial dysfunction. Interventions to improve mitochondrial function have been shown to correct insulin metabolic signaling and other metabolic and cardiovascular abnormalities. This review explores mechanisms of mitochondrial dysfunction with a focus on impaired oxidative phosphorylation and mitochondrial biogenesis in the pathophysiology of metabolic heart disease.
Keywords: Metabolic impairment, Oxidative phosphorylation, Mitochondrial biogenesis
Introduction
Despite advances in preventive and treatment strategies directed at cardiovascular disease (CVD) risk factors, there remains a high prevalence of obesity, hypertension, diabetes, and cardiovascular and renal disease [1–5]. Indeed, the growing epidemic of obesity and type 2 diabetes continues to hamper the improvements in the prevention and management of CVD. The presence of insulin resistance and excess body weight gain may result in a significant increase in CVD and chronic kidney disease (CKD) morbidity and mortality [2–5]. Cardiovascular risk factors often cluster, most notably metabolic disorders including obesity, insulin resistance, dyslipidemia, and hypertension, collectively referred to as the metabolic syndrome [1]. With clustering of these risk factors, there is an associated increase in incidence and prevalence of CVD and CKD [3–5]. The metabolic syndrome is often associated with inappropriate activation of the renin–angiotensin–aldosterone system (RAAS) [6]. Indeed, elevations in angiotensin II (Ang II) and aldosterone have been shown to promote an impairment in systemic insulin metabolic signaling that leads to endothelial dysfunction and myocardial functional abnormalities [6–8] (Fig. 1). These abnormalities have been observed in insulin-resistant animal models and persons that are at higher risk of developing type 2 diabetes mellitus, hypertension, CVD, and CKD [8, 9].
Fig. 1.
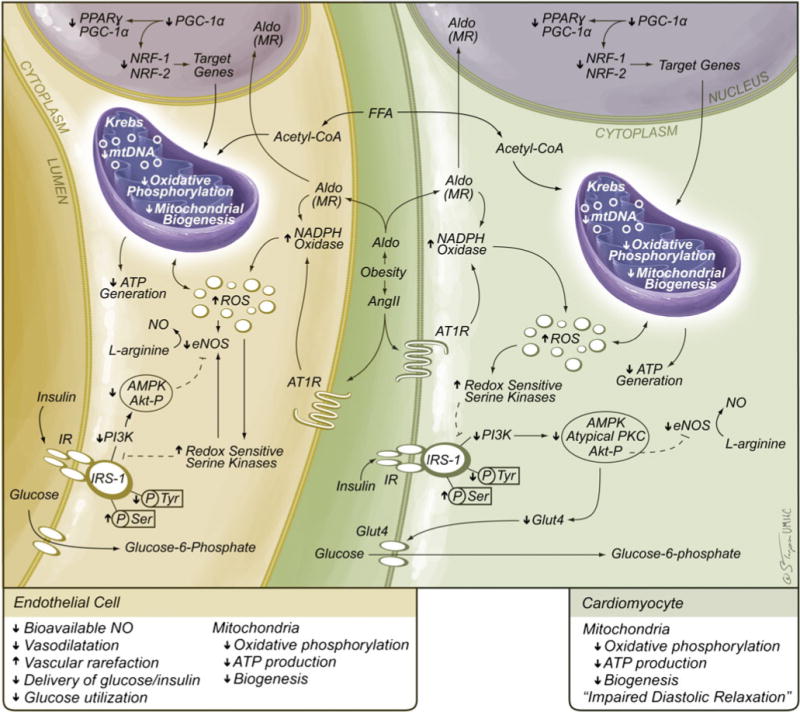
Role of the RAAS in mitochondrial dysfunction. Ang II and aldosterone inhibit insulin metabolic signaling that results in impairments in glucose transport/utilization in cardiovascular tissue as well as other insulin-sensitive tissue. The ensuing increases in oxidative stress reduce glucose transport/utilization as well as mitochondrial ATP generation. Ultimately, these changes will contribute to long-term endothelial (reduced NO) and cardiomyocyte diastolic dysfunction. AP-1 activator protein 1, AT1R Ang type 1 receptor, Ang II angiotensin II, ERK extracellular signal-regulated kinases, FFA free fatty acid, HIF1 hypoxia-inducible factor 1, IRS-1 insulin resistance substrate 1, JNK JUN NH2-terminal kinases, NF-kB nuclear factor-kB, MR mineralocorticoid receptor, PI3-K phosphoinositol 3-kinase, ROS reactive oxygen species
Given the complexity of the pathophysiology and complications of the metabolic syndrome, we will focus on recent information on mitochondrial dysfunction (decreased oxidative phosphorylation) and diminished mitochondrial biogenesis as it relates to myocardial dysfunction and progression of cardiomyopathy associated with the metabolic syndrome.
Mechanisms contributing to myocardial dysfunction in the metabolic syndrome
Impaired myocardial diastolic relaxation (e.g., diastolic dysfunction) is the earliest myocardial contractility observed in metabolic conditions such as obesity, insulin resistance, and hypertension [10–14] (Fig. 1). Diastolic dysfunction manifests as a reduction in velocity of myocardial relaxation, as well as decreasing myocardial compliance [13, 14]. Mechanisms that contribute to this selective cardiac dysfunction include decreases in energy production due to reductions in mitochondrial respiration, increased oxidative stress, and defective contractile and intracellular Ca2+ regulatory proteins [15, 16]. Abnormalities in Ca2+ signaling/flux and myofilament function contribute to the cardiomyopathic alterations observed in the metabolic syndrome [17–19]. Recent data suggests that early stages of myopathic alterations in the metabolic syndrome may be associated with defects in myofilament function independent of alterations in Ca2+ signaling [20, 21]. This is evident in a type 1 diabetes model where there are increases in myocardial action potential duration and collagen expression [21]. Further, in a lipotoxic diabetes model displaying increases in fatty acid uptake, there is slower diastolic muscle relaxation kinetics despite increases in myofilament Ca2+ sensitivity [20].
There is a compelling body of evidence for mitochondrial dysfunction in association with insulin resistance and obesity [7, 22]. This is especially relevant considering mitochondrial dysfunction compromises glucose-stimulated pancreatic insulin secretion as well as insulin-stimulated skeletal muscle glucose utilization [7]. The contribution of mitochondrial dysfunction to impairments in insulin metabolic signaling is suggested by gene array analysis showing reductions in expression of genes regulating mitochondrial adenosine triphosphate (ATP) production associated with insulin resistance [23] and overt type 2 diabetes mellitus [24]. Moreover, reductions in the oxidative capacity of the mitochondrial electron transport chain are manifested in obese, insulin-resistant persons as well as diabetic patients [7, 16]. Mitochondria in endothelial cells are thought to play an important role in cellular signaling as sensors for local oxygen concentration and regulations of nitric oxide (NO) production [25]. RAAS-mediated increases in NADPH oxidase activity and generation of reactive oxygen species (ROS) may result in mitochondrial damage and associated decreases in oxidative phosphorylation, ATP production, and bioavailable NO [26]. Thus, abnormalities in mitochondrial oxidative phosphorylation may predispose decreased coronary arteriolar blood flow to myocardial tissue (Fig. 1). The increasing prevalence of metabolic cardiomyopathy requires a better understanding of the involvement of alterations in mitochondrial integrity, fundamental to the development of optimal strategies of endothelial and myocardial dysfunction in the context of altered mitochondrial function and mitochondrial biogenesis.
Mitochondrial function and mitochondrial biogenesis
Mitochondria play a fundamental role in the survival and function of cardiomyocytes and are critical for the high demand of energy in the myocardium. Mitochondria occupy 20–30% of the cell volume of cardiomyocytes, but the numbers can increase with enhanced myocardial energy requirements. Mitochondria are dynamic organelles that normally respond to the energy needs of tissues [7, 22, 24]. Myocardial energy production occurs primarily in the form of ATP generated by mitochondria to bridge the metabolism of nutrients and oxidative respiration (Fig. 2). The heart consumes the equivalent of 6 kg of ATP per day, the majority of which is generated through mitochondrial oxidative phosphorylation that is fueled by catabolism of lipids and carbohydrates and used for various biological events.
Fig. 2.
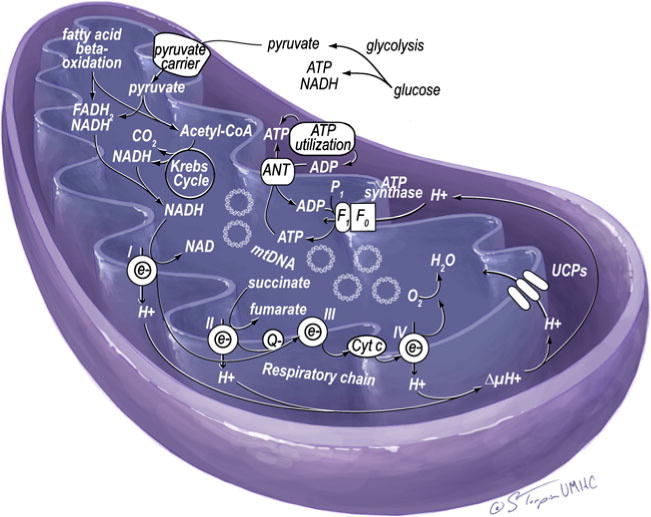
Normal mitochondrial respiratory chain and nutrient metabolism. Reducing agents (NADH or FADH2) are generated from glycolysis and Krebs cycle of glucose metabolism and β-oxidation of fatty acids. While NADH or FADH2 are oxidized to NAD+ or FAD, the electrons are carried to complex I (NADH-ubiquinone reductase), complex II (succinated ubiquinone reductase), complex III (ubiquinone-cytochrome c reductase), complex IV (cytochrome oxidase), and finally to O2, which produces H2O. Oxidation of NADH or FADH2 generates protons that are pumped to intermembrane space through complex I, III, and IV. The pumped protons increase electrochemical gradient across the membrane. This proton gradient is the driving force for F0F1-ATPase (ATP synthase) to produce ATP
There are considerable data supporting the importance of mitochondria in cardiac function; however, there is less information regarding the dynamic regulation of mitochondrial biogenesis in cardiomyocytes. There exists a fine balance between nuclear and mitochondrial gene expression that governs assembly of the mitochondrial respiratory complex. This is especially true under conditions of exercise, wherein mitochondrial biogenesis is triggered through modulation of the ATP/ADP ratio, activation of adenosine monophosphate-activated protein kinase (AMPK), and consequent expression of the transcriptional factor peroxisomal proliferator activator receptor γ co-activator 1α (PGC-1α) and nuclear respiratory factor-1 (NRF1) [7, 27]. In turn, these increases in cardiac energy demands regulate gene expression of nuclear and mitochondrial DNA and maximize the capacity of mitochondria to perform oxidative phosphorylation. In the presence of obesity and insulin resistance, there is an excess of nutrients, coupled with reductions in energy demand, that result in reductions in mitochondrial DNA gene expression that contribute to reduced oxidative phosphorylation capacity [7].
Mitochondrial dysfunction in metabolic syndrome
Mitochondria are not only a major source of energy for the cell but also act as a mediator for biological responses including growth and death [7, 22]. Changes in mitochondrial biogenesis and function have been documented in the metabolic syndrome and diabetes [27, 28]. Both clinical and experimental findings have suggested a 30–70% reduction in phosphocreatine levels and total creatine levels in heart failure. The activity of myofibrillar creatine kinase in heart failure drops to about 50% of the normal level. In general, heart dysfunction is accompanied with defects in all three components of energy metabolism in cardiomyocyte mitochondria, namely, substrate utilization, oxidative phosphorylation, and energy transfer and utilization. These interactions can be even more complicated in the metabolic syndrome with worsening metabolic dysregulation and mitochondrial dysfunction, each reinforcing the other abnormality. This is illustrated in metabolic/diabetic cardiomyopathy wherein the initial manifestations of diastolic dysfunction are more apparent in the presence of endothelial dysfunction [4, 11, 16] (Fig. 1).
The notion that various components of the metabolic syndrome, including that of insulin resistance, contribute to mitochondrial dysfunction is well validated [22]. However, not much is known regarding the extent of mitochondrial dysfunction within the constellation of metabolic risk associated with the metabolic syndrome and the relationship to metabolic cardiomyopathy. Under routine physiological conditions, myocardial function is dependent upon continual recycling of ATP from mitochondrial oxidative phosphorylation. When mitochondria become maladaptive, due to loss and/or reduced efficiency of mitochondria, a cascade of events develops including reduced ATP synthesis, abnormal accumulation of metabolic intermediates, and ROS production, all of which may contribute to impaired mechanical function of the heart [7, 22, 28]. Findings from a wide array of studies support the pivotal role of mitochondrial dysfunction, in particular, reduced mitochondrial biogenesis in the pathogenesis of metabolic cardiomyopathy. For example, diabetic cardiomyopathy is associated with an intracellular accumulation of toxic intermediates, such as long chain acyl-CoA and acylcarnitine, that affect mitochondrial ATP/ADP ratio and lead to diminished mitochondrial metabolic function [29]. Reduced gene expression of mitochondrial regulatory protein PGC-1α has been found to be associated with human heart dysfunction [30]. Finally, pharmacological and genetic intervention data provide compelling support for the critical role of mitochondrial dysfunction in the pathogenesis of cardiomyopathy as improved and reduced mitochondrial function may attenuate and exacerbate heart failure, respectively [30].
Mitochondrial alterations may underlie several aspects of the observed metabolic cardiac phenotypes such as altered Ca2+ handling, interstitial fibrosis, cellular and subcellular remodeling, and cardiomyocyte loss. Reduced mitochondrial biogenesis has been demonstrated in humans and animals with the metabolic syndrome, coinciding with the reduced ATP level and dysfunctional mitochondrial electron transport. The role of an overactive RAAS in the progression of mitochondrial and myocardial dysfunction has been indicated by a number of seminal observations [6, 31–35]. Using rodent models of insulin resistance that display inappropriate activation of the RAAS, such as the transgenic Ren2 rat, we have observed metabolic abnormalities consistent with the metabolic syndrome such as hypertension, insulin resistance, and metabolic cardiomyopathy [34, 35]. Importantly, myocardial mitochondrial biogenesis evaluated by ultrastructural analysis of myocardium utilizing transmission electron microscopy reveals striking alterations in mitochondria including increased numbers of smaller morphologically abnormal mitochondria interspersed between intercalated discs (Fig. 3). These findings are accompanied by increases in mitochondrial complex IV (cytochrome c oxidase) reflecting adaptive increases in mitochondrial activity [34, 35]. The heart in this model has increased tissue Ang II and ROS, both of which are known to exert detrimental effects on mitochondrial integrity. For example, oxidative stress can adversely affect mitochondrial DNA damage [7]. Altered mitochondrial DNA, such as mutant mitochondrial DNA with large deletions, has been shown to enhance the biogenesis of cellular mitochondria. Indeed, mitochondria in a highly oxidative environment proliferate more rapidly and thus tend to be smaller and more plentiful. Observations from these studies indicate that increased mitochondrial biogenesis is an adaptive process to establish a transmembrane proton differential in order to generate ATP, thereby reflecting enhancements in myocardial energy requirements in these insulin-resistant models of RAAS activation. However, this leads to increased numbers of small morphologically abnormal, less efficient mitochondria (Fig. 3). More importantly, the mitochondrial abnormalities were attenuated following inhibition of the RAAS with either Ang II receptor and/or mineralocorticoid receptor blockade [34, 35]. Thereby, these findings support an important role for the RAAS, and associated increases in oxidative stress, in the pathogenesis of mitochondrial abnormalities in metabolic-related cardiomyopathies in the metabolic syndrome (Figs. 1 and 3).
Fig. 3.

Myocardial mitochondrial biogenesis. a Normal cardiac ultrastructure of mitochondria and intercalated disc on ultrastructural analysis utilizing transmission electron microscopy. Note the orderly and linearly arranged sarcomeres and subsarcolemmal (sarcoplasmic reticulum) mitochondria. b Mitochondrial changes in the insulin-resistant Ren2 rat which displays diastolic dysfunction. c Biogenesis of small morphologically abnormal mitochondria depicted in stylized version
Reduced myocardial mitochondrial antioxidant capacity and impaired mitochondrial biogenesis are crucial in the pathogenesis of metabolic-related myocardial dysfunction [7, 10, 13]. This is illustrated in mice lacking endogenous antioxidant capacity that are more vulnerable to dysfunctions in mitochondrial biogenesis and myocardial contractility associated with the metabolic syndrome. Alternatively, increases in antioxidant capacity are capable of slowing the progression of ventricular remodeling and myocardial systolic and diastolic dysfunction [18]. There are several specific defects in antioxidant capacity, biogenesis, and nuclear genes encoding mitochondrial proteins involved in ventricular remodeling and hypertrophy derived from a high-fat diet and associated insulin resistance [18]. Further, data suggest that the mitochondria-mediated cell death through apoptosis, necrosis, and autophagy may contribute to the progression of cardiac geometric and functional changes [7, 32]. Therefore, onset and progression of cardiac structural and functional defects are closely associated with even subtle changes in the mitochondrial antioxidant capacity, energy metabolism, and dysfunction.
Mitochondrial numbers and size are closely associated with mitochondrial oxidative capacity [7]. Reductions in mitochondria number and size in myocardium and skeletal muscle have been observed in those with insulin resistance and obesity [18, 36–38]. Metabolic abnormalities that contribute to reductions in mitochondrial oxidative capacity are often accompanied by a loss of proteins that are encoded by the mitochondrial genome (e.g., cytochrome c oxidase 1) [7]. Mitochondrial biogenesis is driven, in part, through the PGC-1α, which was first discovered as a transcriptional regulator of uncoupling proteins (Figs. 2 and 3). PGC-1α is an integrator of the transcriptional network regulating mitochondrial biogenesis. Indeed, PGC-1α-driven mitochondrial biogenesis is integral to routine cardiac function, as models that have PGC-1α knockout or deficiency have significant alterations in cardiac function [39]. PGC-1α has been shown to mediate downstream transcriptional regulatory circuits such as nuclear respiratory factor-1 and 2 (NRF-1 and NRF-2) which, in turn, govern downstream genes including mitochondrial transcription factor A (MTF-A). NRF-1 regulates a number of mitochondrial genes such as oxidative phosphorylation genes and mitochondrial transcription factor A (TFAM). In addition, PGC-1α regulates genes involved in the cellular uptake of fatty acids and fatty acid oxidation through coactivation of peroxisome proliferator-activated receptors and other fatty acid metabolism genes. Accumulative data support the notion that regulation of mitochondrial biogenesis is facilitated through a number of signaling cascades such as endothelial nitric oxide synthase (eNOS) and AMPK signaling [25, 40].
Expression of PGC-1α may be upregulated by cellular ATP demand [23], coinciding with reductions in mitochondrial biogenesis cofactor PGC-1α [18, 41]. Individuals with insulin resistance display fewer, and sometimes smaller, skeletal muscle mitochondria, which could be attributed to reduced PGC-1α and PGC-1β expression [42, 43]. Preclinical data provides further evidence for the importance of PGC-1α and mitochondrial biogenesis in routine myocardial function. Data from PGC-1α null mice suggest these mice display defects in both skeletal and cardiac muscle contractility [44, 45]. Indeed, DNA microarray analysis shows that expression of PGC-1α and associated increases in mitochondrial biogenesis contribute to skeletal muscle insulin resistance [23, 24]. Thus, PGC-1α-driven mitochondrial biogenesis is integral for normal cardiac and skeletal muscle contractility and relaxation.
Collectively, these data support the notion that abnormal mitochondrial function is mainly due to reduced mitochondrial density. Moreover, the loss of subsarcolemmal mitochondrial and associated diminution of electron transport activity has been demonstrated in insulin resistance and obesity, further substantiating the relationship between mitochondrial electron transport activity and mitochondrial density [38]. It should be noted, there is conflicting data indicating unchanged mitochondrial function normalized to DNA content in those with type 2 diabetes [46]. Additionally, there is normal mRNA expression of PGC-1α, PGC-1β, NRFs, and TFAM in the offspring of type 2 diabetic individuals despite reductions in insulin metabolic signaling [37]. This discrepancy suggests that in some cases, mitochondrial dysfunction cannot be fully attributed to reduced mitochondrial biogenesis.
eNOS and development of mitochondrial biogenesis
PGC-1α is regulated by a number of signaling cascades including the eNOS/NO/cyclic guanosine monophosphate (cGMP) system. Indeed, eNOS plays an important role in mitochondria biogenesis and mitochondrial function [25, 26, 28]. Increases in myocardial NO or cGMP have been shown to stimulate mitochondrial biogenesis [47]. Reductions in mitochondria content and associated defects in fatty acid metabolism are evident in eNOS-deficient mice that manifest insulin resistance and hypertension [28]. More recent observations from our laboratory indicate that eNOS uncoupling with diminished levels of the eNOS cofactor BH4 synthetic enzyme guanosine triphosphate cyclohydrolase I directly leads to myocardial dysfunction accompanied with reduced expression of PGC-1α, its downstream nuclear factors, and attenuation of mitochondrial biogenesis [48]. These data collectively underscore the important functional role of eNOS in the maintenance of myocardial mitochondrial biogenesis and mitochondrial function. However, the mechanism by which eNOS deficiency leads to a decrease in mitochondrial biogenesis is unknown at this time, although eNOS-induced regulation of PGC-1α is believed to play a major role [7]. PGC-1α may be regulated by a number of signaling molecules and transcriptional factors. For example, expression of PGC-1α may be downregulated in mice lacking transcriptional factor myocyte enhancer factor-2 (MEF-2) associated with dysregulated cardiac mitochondrial biogenesis [49]. Inducible overexpression of PGC-1α may itself lead to dilated cardiomyopathy [50], indicating the importance of finely tuned homeostatic regulation of the mitochondrial biogenesis for normal heart structure and cardiac contractile function.
AMPK and mitochondrial biogenesis
AMPK has emerged as a key regulator of energy metabolism and mitochondrial biogenesis in the heart. Pharmacological activators of AMPK [β-guanidinopropionic acid (βGPA) and 5′-D-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR)] stimulate mitochondrial biogenesis through activation of PGC-1α and downstream nuclear factors [51]. In contrast, recent findings from our laboratory failed to detect any change in the expression of PGC-1α in myocardium from murine hearts with overexpression of mutant α2 subunit compared with wild-type mice [52]. Although the precise mechanism behind such discrepancy is unknown, difference in the age and the specific model studied may play a role. AMPK appears to be responsible for exercise-induced phosphorylation/activation of PGC-1α. In this regard, phosphorylation of the α subunit at Thr172 by the AMPK upstream kinase, AMPK kinase (AMPKK), or allosteric modulation by AMP directly regulates AMPK activity and mitochondrial biogenesis [53]. There are two AMPKKs, tumor suppressor kinase LKB1 and calmodulin-dependent protein kinase kinase (CamKK), which have been identified in the heart [7]. Activation of AMPK enhances ATP synthesis by stimulating glucose uptake, fatty acid oxidation, and glycolysis and inhibits energy-consuming anabolic pathways such as protein synthesis to conserve intracellular ATP. Myocardial tissues from individuals with metabolic syndrome display dampened AMPK activation, resulting in downregulation of glucose uptake and glycolysis, as well as decrease in fatty acid oxidation processes leading to cardiac dysfunction. Nonetheless, further investigation is warranted to elucidate if AMPK activation is protective in the metabolic syndrome. It is worth mentioning that AMPK may indirectly participate in the regulation of mitochondrial function via regulation of Bax translocation to mitochondria [54] or through regulation of p53 and cell proliferation [55].
Oxidative stress and mitochondrial biogenesis
Mitochondria are a major source of ROS generation in skeletal muscle and myocardial tissue. This is primarily due to inhibition of complexes I and III leading to accumulation of superoxide anion. However, it should be noted there is extra-mitochondrial oxygen consumption that occurs through enzymatic and non-enzymatic pathways that include xanthine oxidase, NADPH oxidase, uncoupling of NO synthase, D-amino-oxidase, p450 cytochromes, and proline hydroxylases. Mitochondrial production of ROS occurs at complex I (NADH CoQ reductase) and/or complex III (bc1 complex). The presence of excess electrons is then donated to oxygen, which are then converted to superoxide and hydrogen peroxide either spontaneously or by superoxide dismutase (SOD); remaining excess electrons are transferred to oxygen without ATP production resulting in “oxidative stress” [56]. Further, there is emerging evidence that NADPH oxidase-derived ROS can promote mitochondrial oxidative stress [57, 58]. This excess of ROS contributes to detrimental cellular effects despite numerous protective mechanisms including SOD, reduced glutathione, and catalase. In particular, mitochondrial generation of excess ROS contributes to protein, DNA, and lipid injury and, ultimately, reductions in mitochondrial biogenesis and mitochondrial dysfunction [7, 56]. Data from our laboratory has shown that the heavy metal scavenger metallothionein attenuates eNOS uncoupling-induced myocardial dysfunction derived from mitochondrial damage as a result of excess superoxide anion production, loss of mitochondrial DNA, as well as downregulation of mitochondrial biogenesis factor PGC-1α and downstream nuclear factors [18, 48]. Thereby, these data support the notion that oxidative stress is, in part, responsible for alterations in mitochondrial biogenesis as well as downregulation of genes required for mitochondrial oxidative phosphorylation in the metabolic syndrome [7, 18, 28]. Further, mitochondrial antioxidant molecules such as thioredoxin 2 reduce Ang II and mineralocorticoid-induced NADPH oxidase expression and generation of ROS [59]. Thus, these treatments would be expected to correct abnormalities in mitochondrial biogenesis associated with the metabolic syndrome and diabetes [60].
Concluding remarks
Alterations in mitochondrial biogenesis as well as mitochondrial content and function provoke a heterogeneous group of CVD and CKD risk factors that constitute the metabolic syndrome. However, the molecular mechanisms that link defects in mitochondrial DNA to myocardial tissue remodeling and contractile dysfunction are not well delineated. The high demand for energy and excess mitochondrial-derived ROS (e.g., oxidative stress) are both crucial to the development of myocardial dysfunction associated with the metabolic syndrome. Additional changes, such as those derived from increased mitochondrial DNA and alterations in genes critical in mitochondrial biogenesis, in mitochondrial configuration and structure, impede sarcomere alignment and contraction. Mounting evidence suggests that mitochondrial biogenesis in response to energy (ATP) deficiency maybe a compensatory maladaptive response that contributes to metabolic cardiac dysfunction derived from PGC-1α overexpression [50]. It is increasingly recognized that important aspects of mitochondrial dysfunction that contribute to CVDs are induction of apoptosis and changes in mitochondrial morphology under the influence of oxidative stress [61–63]. Finally, inefficient mitochondrial oxidative phosphorylation/biogenesis and increases in oxidative stress appear to be overarching abnormalities contributing to cardiac diastolic function, the hallmark of metabolic cardiomyopathy.
Acknowledgments
Research included in this review is supported by NIH R01HL073101, VA Merit (JRS), and CDA-2 Dept of Veterans Affairs (AWC). The authors thank Brenda Hunter for her assistance in preparing this manuscript.
Footnotes
Conflict of interest statement The authors declare no conflict of interests related to this study.
Contributor Information
Jun Ren, Center for Cardiovascular Research and Alternative Medicine, University of Wyoming College of Health Sciences, Laramie, WY 82071, USA.
Lakshmi Pulakat, Diabetes and Cardiovascular Center, University of Missouri School of Medicine, VA Medical Center, Columbia, MO 65212, USA.
Adam Whaley-Connell, Diabetes and Cardiovascular Center, University of Missouri School of Medicine, VA Medical Center, Columbia, MO 65212, USA.
James R. Sowers, Email: sowersj@health.missouri.edu, Diabetes and Cardiovascular Center, University of Missouri School of Medicine, VA Medical Center, Columbia, MO 65212, USA D109 HSC Diabetes Center, One Hospital Dr, Columbia, MO 65212, USA.
References
- 1.Sullivan PW, Ghushchyan V, Wyatt HR, Wu EQ, Hill JO. Impact of cardiometabolic risk factor clusters on health-related quality of life in the U.S. Obesity (Silver Spring) 2007;15:511–521. doi: 10.1038/oby.2007.580. [DOI] [PubMed] [Google Scholar]
- 2.Cannon CP, Kumar A. Treatment of overweight and obesity: lifestyle, pharmacologic, and surgical options. Clin Cornerstone. 2009;9:55–68. doi: 10.1016/s1098-3597(09)80005-7. discussion 69–71. [DOI] [PubMed] [Google Scholar]
- 3.Sowers JR. Metabolic risk factors and renal disease. Kidney Int. 2007;71:719–720. doi: 10.1038/sj.ki.5002006. [DOI] [PubMed] [Google Scholar]
- 4.Whaley-Connell A, Sowers JR. Hypertension and insulin resistance. Hypertension. 2009;54:462–464. doi: 10.1161/HYPERTENSIONAHA.109.134460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith SC., Jr Multiple risk factors for cardiovascular disease and diabetes mellitus. Am J Med. 2007;120:S3–S11. doi: 10.1016/j.amjmed.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Cooper SA, Whaley-Connell A, Habibi J, et al. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am J Physiol Heart Circ Physiol. 2007;293:H2009–H2023. doi: 10.1152/ajpheart.00522.2007. [DOI] [PubMed] [Google Scholar]
- 7.Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sowers JR, Whaley-Connell A, Epstein M. The emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150:776–785. doi: 10.7326/0003-4819-150-11-200906020-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bassuk SS, Manson JE. Epidemiological evidence for the role of physical activity in reducing risk of type 2 diabetes and cardiovascular disease. J Appl Physiol. 2005;99:1193–1204. doi: 10.1152/japplphysiol.00160.2005. [DOI] [PubMed] [Google Scholar]
- 10.Zhou X, Ma L, Habibi J, Whaley-Connell A, Hayden MR, Tilmon RD, Brown AN, Kim JA, Demarco VG, Sowers JR. Nebivolol improves diastolic dysfunction and myocardial remodeling through reductions in oxidative stress in the Zucker obese rat. Hypertension. 2010;55(4):880–888. doi: 10.1161/HYPERTENSIONAHA.109.145136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J. AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular moycytes: role of the AT1 receptor and NADPH oxidase. Hypertension. 2003;42:206–212. doi: 10.1161/01.HYP.0000082814.62655.85. [DOI] [PubMed] [Google Scholar]
- 12.Ren J, Kelley RO. Cardiac health in women with metabolic syndrome: clinical aspects and pathophysiology. Obesity (Silver Spring) 2009;17:1114–1123. doi: 10.1038/oby.2009.8. [DOI] [PubMed] [Google Scholar]
- 13.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115(19):2540–2548. doi: 10.1161/CIRCULATIONAHA.107.670588. [DOI] [PubMed] [Google Scholar]
- 14.Ren J, Bode AM. Altered cardiac excitation-contraction coupling in ventricular myocytes from spontaneously diabetic BB rats. Am J Physiol Heart Circ Physiol. 2000;279:H238–H244. doi: 10.1152/ajpheart.2000.279.1.H238. [DOI] [PubMed] [Google Scholar]
- 15.Wold LE, Ceylan-Isik AF, Ren J. Oxidative stress and stress signaling: menace of diabetic cardiomyopathy. Acta Pharmacol Sin. 2005;26:908–917. doi: 10.1111/j.1745-7254.2005.00146.x. [DOI] [PubMed] [Google Scholar]
- 16.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 17.Choi KM, Zhong Y, Hoit BD, Grupp IL, Hahn H, et al. Defective intracellular Ca(2+) signaling contributes to cardiomyopathy in type 1 diabetic rats. Am J Physiol Heart Circ Physiol. 2002;283:H1398–H1408. doi: 10.1152/ajpheart.00313.2002. [DOI] [PubMed] [Google Scholar]
- 18.Dong F, Li Q, Sreejayan N, Nunn JM, Ren J. Metal-lothionein prevents high-fat diet induced cardiac contractile dysfunction: role of peroxisome proliferator activated receptor gamma coactivator 1alpha and mitochondrial biogenesis. Diabetes. 2007;56:2201–2212. doi: 10.2337/db06-1596. [DOI] [PubMed] [Google Scholar]
- 19.Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, et al. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. 2006;55:608–615. doi: 10.2337/diabetes.55.03.06.db05-1284. [DOI] [PubMed] [Google Scholar]
- 20.Flagg TP, Cazorla O, Remedi MS, Haim TE, Tones MA, et al. Ca2+-independent alterations in diastolic sarcomere length and relaxation kinetics in a mouse model of lipotoxic diabetic cardiomyopathy. Circ Res. 2009;104:95–103. doi: 10.1161/CIRCRESAHA.108.186809. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Cannell MB, Phillips AR, Cooper GJ, Ward ML. Altered calcium homeostasis does not explain the contractile deficit of diabetic cardiomyopathy. Diabetes. 2008;57:2158–2166. doi: 10.2337/db08-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 23.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 25.Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res. 2007;100:1128–1141. doi: 10.1161/01.RES.0000261970.18328.1d. [DOI] [PubMed] [Google Scholar]
- 26.Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H2023–H2031. doi: 10.1152/ajpheart.01283.2006. [DOI] [PubMed] [Google Scholar]
- 27.Nishio Y, Kanazawa A, Nagai Y, Inagaki H, Kashiwagi A. Regulation and role of the mitochondrial transcription factors in the diabetic heart. Ann NY Acad Sci. 2004;1011:78–85. doi: 10.1007/978-3-662-41088-2_9. [DOI] [PubMed] [Google Scholar]
- 28.Nisoli E, Clementi E, Carruba MO, Moncada S. Defective mitochondrial biogenesis: a hallmark of the high cardiovascular risk in the metabolic syndrome? Circ Res. 2007;100:795–806. doi: 10.1161/01.RES.0000259591.97107.6c. [DOI] [PubMed] [Google Scholar]
- 29.Lopaschuk GD, Spafford M. Response of isolated working hearts to fatty acids and carnitine palmitoyltransferase I inhibition during reduction of coronary flow in acutely and chronically diabetic rats. Circ Res. 1989;65:378–387. doi: 10.1161/01.res.65.2.378. [DOI] [PubMed] [Google Scholar]
- 30.Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol. 2009;46:201–212. doi: 10.1016/j.yjmcc.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Cavanagh EM, Toblli JE, Ferder L, Piotrkowski B, Stella I, Inserra F. Renal mitochondrial dysfunction in spontaneously hypertensive rats is attenuated by losartan but not by amlodipine. Am J Physiol Regul Integr Comp Physiol. 2006;290(6):R1616–R1625. doi: 10.1152/ajpregu.00615.2005. [DOI] [PubMed] [Google Scholar]
- 32.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 33.Mitsuishi M, Miyashita K, Muraki A, Itoh H. Angiotensin II reduces mitochondrial content in skeletal muscle and affects glycemic control. Diabetes. 2009;58:710–717. doi: 10.2337/db08-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stas S, Whaley-Connell A, Habibi J, Appesh L, Hayden MR, et al. Mineralocorticoid receptor blockade attenuates chronic overexpression of the renin-angiotensin-aldosterone system stimulation of reduced nicotinamide adenine dinucleotide phosphate oxidase and cardiac remodeling. Endocrinology. 2007;148:3773–3780. doi: 10.1210/en.2006-1691. [DOI] [PubMed] [Google Scholar]
- 35.Whaley-Connell A, Habibi J, Cooper SA, Demarco VG, Hayden MR, et al. Effect of renin inhibition and AT1R blockade on myocardial remodeling in the transgenic Ren2 rat. Am J Physiol Endocrinol Metab. 2008;295:E103–E109. doi: 10.1152/ajpendo.00752.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 37.Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest. 2005;115:3587–3593. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, et al. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 39.Handschin C, Spiegelman BM. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature. 2008;454:463–469. doi: 10.1038/nature07206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reznick RM, Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol. 2006;574:33–39. doi: 10.1113/jphysiol.2006.109512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ling C, Poulsen P, Carlsson E, Ridderstrale M, Almgren P, et al. Multiple environmental and genetic factors influence skeletal muscle PGC-1alpha and PGC-1beta gene expression in twins. J Clin Invest. 2004;114:1518–1526. doi: 10.1172/JCI21889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, et al. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J Biol Chem. 2003;278:26597–26603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- 43.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 44.Arany Z, He H, Lin J, Hoyer K, Handschin C, et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 45.Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation. 2007;115:909–917. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsoe R, et al. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 2007;50:790–796. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci USA. 2004;101:16507–16512. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ceylan-Isik AF, Guo KK, Carlson EC, Privratsky JR, Liao SJ, et al. Metallothionein abrogates GTP cyclohydrolase I inhibition-induced cardiac contractile and morphological defects: role of mitochondrial biogenesis. Hypertension. 2009;53:1023–1031. doi: 10.1161/HYPERTENSIONAHA.108.123422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci USA. 2003;100:1711–1716. doi: 10.1073/pnas.0337639100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Russell LK, Finck BN, Kelly DP. Mouse models of mitochondrial dysfunction and heart failure. J Mol Cell Cardiol. 2005;38:81–91. doi: 10.1016/j.yjmcc.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 51.Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–E1346. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- 52.Turdi S, Fan X, Li J, Zhao J, Huff AF, et al. AMP-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell. 2010;9:592–606. doi: 10.1111/j.1474-9726.2010.00586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baron SJ, Li J, Russell RR, 3rd, Neumann D, Miller EJ, et al. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res. 2005;96:337–345. doi: 10.1161/01.RES.0000155723.53868.d2. [DOI] [PubMed] [Google Scholar]
- 54.Capano M, Crompton M. Bax translocates to mitochondria of heart cells during simulated ischaemia: involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem J. 2006;395:57–64. doi: 10.1042/BJ20051654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Igata M, Motoshima H, Tsuruzoe K, Kojima K, Matsumura T, et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res. 2005;97:837–844. doi: 10.1161/01.RES.0000185823.73556.06. [DOI] [PubMed] [Google Scholar]
- 56.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brandes RP. Triggering mitochondrial radical release: a new function for NADPH oxidases. Hypertension. 2005;45(5):847–848. doi: 10.1161/01.HYP.0000165019.32059.b2. [DOI] [PubMed] [Google Scholar]
- 58.Wei Y, Whaley-Connell A, Sowers JR, et al. Mineralocorticoid receptor antagonism attenuates vascular apoptosis and injury via rescuing Akt activation. Hypertension. 2009;53(2):158–165. doi: 10.1161/HYPERTENSIONAHA.108.121954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fukai T. Mitochondrial thioredoxin: novel regulator for NADPH oxidase and angiotensin II-induced hypertension. Hypertension. 2009;54(2):224–225. doi: 10.1161/HYPERTENSIONAHA.109.134403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sebastiani M, Giordano C, Nediani C, Travaglini C, Borchi E, et al. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J Am Coll Cardiol. 2007;50:1362–1369. doi: 10.1016/j.jacc.2007.06.035. [DOI] [PubMed] [Google Scholar]
- 61.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010;53:1783–1794. doi: 10.1007/s00125-010-1770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Williamson CL, Dabkowski ER, Baseler WA, Croston TL, Alway SE, Hollander JM. Enhanced apoptotic propensity in diabetic cardiac mitochondria: influence of subcellular spatial location. Am J Physiol Heart Circ Physiol. 2010;298:H633–H642. doi: 10.1152/ajpheart.00668.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]