

Abstract
The initiation of an inflammatory response is critical to the survival of an organism. However, when inflammation fails to reach resolution, a chronic inflammatory state may occur, potentially leading to bystander tissue damage. Accumulating evidence suggests that chronic inflammation contributes to the progression of Alzheimer’s disease (AD), and identifying mechanisms to resolve the pro-inflammatory environment stimulated by AD pathology remains an area of active investigation. Previously, we found that treatment with the pro-resolving mediator aspirin-triggered lipoxin A4 (ATL), improved cognition, reduced Aβ levels, and enhanced microglia phagocytic activity in Tg2576 transgenic AD mice. Here, we evaluated the effect of aging on brain lipoxin A4 (LXA4) levels using non-transgenic and 3xTg-AD mice. Additionally, we investigated the effect of ATL treatment on tau pathology in 3xTg-AD mice. We found that LXA4 levels are reduced with age, a pattern significantly more impacted in 3xTg-AD mice. Moreover, ATL delivery enhanced the cognitive performance of 3xTg-AD mice, reduced Aβ levels, as well as decreased the levels of phosphorylated-tau (p-tau). The decrease in p-tau was due in part to an inhibition of the tau kinases GSK-3β and p38 MAPK. In addition, microglial and astrocyte reactivity was inhibited by ATL treatment. Our results suggest that the inability to resolve the immune response during aging might be an important feature that contributes to AD pathology and cognitive deficits. Furthermore, we demonstrate that activation of LXA4 signaling could serve as a potential therapeutic target for AD related inflammation and cognitive dysfunction.
Keywords: Aspirin-Triggered Lipoxin A4, Lipoxin, Aging, Alzheimer’s disease, Inflammation, 3xTg-AD, resolution, lipoxygenase
Introduction
Alzheimer’s disease (AD) is an age dependent neurodegenerative disorder that impairs memory and causes cognitive deficits [1]. The AD brain is marked by the accumulation of extracellular plaques, primarily composed of aggregated amyloid-β (Aβ) peptide, and intracellular neurofibrillary tangles (NFTs), consisting of filamentous aggregates of hyperphosphorylated-tau protein. Prevailing experimental evidence shows that the accumulation of Aβ and tau in the brain can lead to several downstream events; including, inflammation, synaptic dysfunction, and neuronal cell death [1]. Inflammation in particular can greatly exacerbate the neurodegenerative process of AD if not properly regulated [2, 3]. Immune mediators such as lipids, cytokines, and reactive-oxygen species have been shown to increase the generation of Aβ, and exacerbate tau phosphorylation [4–6].
Under normal circumstances, inflammation is terminated by a resolution response. One group of molecules that function to initiate the resolution of an inflammatory response are the lipid-based lipoxins generated by the activation of the lipoxygenase pathway [7]. The lipoxygenase (LOX) pathway is involved in the activation and amplification, as well as the termination and resolution of cardinal signals of inflammation through the synthesis of leukotrienes and lipoxins [8]. Leukotrienes are primarily involved in the initiation of a pro-inflammatory response through the activation of various immune cells, while lipoxins are molecules with potent anti-inflammatory properties [9]. In addition to endogenous generation of lipoxins, aspirin can also trigger the biosynthesis of a group of lipoxins known as aspirin-triggered lipoxin A4 (ATL), which mimic the anti-inflammatory actions of lipoxins but are more resistant to degradation [10, 11]. Increased expression of multiple LOX isoforms, and levels of the pro-inflammatory leukotriene B4 (LTB4), have been reported in the aged brain; as well as in the AD brain [12–15]. In contrast, experimental evidence indicates that levels of the anti-inflammatory/pro-resolution lipoxin A4 (LXA4) are significantly reduced in the healthy aged brain [16, 17]. Moreover, it has been recently demonstrated that levels of LXA4 are reduced in AD, both in the cerebrospinal fluid (CSF) and hippocampus [18].
We recently demonstrated that LXA4 signaling stimulates alternative activation of microglia and decreases Aβ pathology in the Tg2576 APP transgenic mouse model [19]. These results raise the possibility that aging may trigger the imbalance of endogenous pro-resolution pathways, causing a dysregulated and sustained inflammation in the elderly, and may contribute or exacerbate the pathogenesis of AD. To date, there have been no studies conducted to investigate the in vivo effect of LXA4 treatment on tau pathology. Hence, we sought to test the hypothesized that an impairment of lipoxin-mediated inflammatory homeostasis contributes to the progression of AD-like pathology. We first sought to assess the age dependent change in LXA4 synthesis in non-transgenic (nTg) and 3xTg-AD mice. To test our hypothesis, we also treated aged 3xTg-AD mice, which contain advanced tau and Aβ pathology, with the ATL, and investigated for changes in neuropathology and behavior. Our data suggest that activation of LXA4 signaling may serve as a potential therapy for the management of AD and other age-related disorders.
Methods and Materials
Animals
3xTg-AD mice harboring the presenilin1 mutation (PS1M146V), the APP Swedish double mutation (APPKM670/671ML), and a frontotemporal dementia mutation in tau (tauP301L), all on a mixed 129SvJ/C57BL/6 background, were used for all experiments [20]. Strain-matched non-transgenic (nTg) mice were used as controls. Animals were maintained at controlled room temperature (22°C ± 2°C) and humidity (60% to 80%) under a 12:12-hour light-dark cycle (lights on at 6 AM). All procedures used in the present study followed the Principles of Laboratory Animal Care from the NIH (Bethesda, MD), publication 85-23, and were approved by the University of California, Irvine, Institutional Animal Care and Use Committee.
LTB4 and LXA4 ELISA
Brain lipid extraction was carried out as a modification of the protocol described beforehand [21]. Frozen brains were pulverized in liquid nitrogen. One milliliter of 15% (vol/vol) ethanol, in distilled water (pH 3), was added to each tissue sample. The tissue homogenates were left at 4°C for 10 min and then spun at 375 × g for 10 min at 4°C. The columns (C-18 Sep-Pak cartridges) were conditioned with 4 ml of ethanol followed by 4 ml of distilled water at a flow rate of 2 ml/min. The supernatant from homogenates was then applied to the columns at a flow rate of 0.5 ml/min. The columns were then washed in 2 ml of distilled water followed by 2 ml of petroleum ether. The samples were then eluted with 2 ml of methyl formate at a flow rate of 1 ml/min. The final samples were stored at −80°C until use.
Quantitative analysis of brain LTB4 and LXA4 levels were measured using a commercially available enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s instructions (Neogen Corporation, Lexington, KY, USA).
Treatment with Aspirin-Triggered LXA4
Animals were treated subcutaneously (s.c.) with 15 μg/kg ATL (5S,6R,15R-trihydroxy-7,9,13-trans-11-cis-eicosatetraenoic acid; Cayman Chemical, Ann Arbor, MI), twice daily for 8 weeks [19]. A separate group of animals was treated with 5% polyethylene glycol 200 and 5% Tween 20 in saline (vehicle). Injections were performed from 14 to 16 months of age and completed on the day of euthanasia.
Novel Object Recognition
Each mouse was habituated to an empty arena for 3 consecutive days. On the first day of testing, mice were exposed to two identical objects placed at opposite ends of the arena for 5 minutes. Twenty-four hours later, the mouse was returned to the test box, this time with one familiar object and one novel object. Time spent exploring the objects was recorded for 5 minutes. The recognition index is defined as the ratio of time spent exploring the novel object over the total time spent exploring both familiar and novel objects. Objects used in this task were carefully selected to prevent preference or phobic behavior.
Open field
To discard the possible effect of ATL on locomotor activity, the animals were tested in the open-field task. The apparatus, made of wood covered with impermeable Formica, had a black floor of 30 × 30 cm (divided by white lines into nine squares of 10 × 10 cm) and transparent walls, 15 cm high. Each mouse was placed in the center of the open field, and the total number of squares crossed with the four paws and the rearing behavior were registered for 5 min.
Tissue Preparation
Mice were deeply anesthetized with sodium pentobarbital and sacrificed by perfusion transcardially with 0.1 mol/L PBS (pH 7.4) solution. The brain was removed and one hemisphere was fixed for 48 hours in 4% paraformaldehyde and cryoprotected in 30% sucrose for immunohistochemical (IHC) analysis. Frozen brains were subsequently cut into serial coronal sections (40 μm thick) using a Leica SM2010R freezing microtome (Leica Microsystems, Bannockburn, IL), collected in cold 0.02% sodium azide, and stored at 4°C. The other hemisphere was snap frozen on dry ice; after removal of cerebellum, brainstem, and olfactory bulb, and subjected to protein extraction sequentially: first using T-PER tissue protein extraction reagent (Thermo Scientific, Rockford, IL), then with 70% formic acid. The resulting supernatant from both extractions reagents was stored at −80°C. Protein concentration in the supernatant was determined using the Bradford assay.
Immunoblotting
Equal protein amounts were first separated using 4% to 12% gradient SDS-PAGE gels, and subsequently transferred to nitrocellulose membranes and incubated overnight at 4°C with primary antibody. The following primary antibodies were used in this study: Tau (HT7), AT8, AT100, AT180, AT270 (Thermo Scientific, Rockford, IL), PHF-1 (Dr. Peter Davies, Albert Einstein College of Medicine, Manhasset, NY, USA), CDK5, p35, GSK3β, pSer9-GSK3β, p-p38 MAPK, p-ERK1/2 (Cell Signaling, Danvers, MA), PPA2, YM1 (Stem Cell Technologies, Vancouver, BC, Canada) and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA). Following incubation with the primary antibody, the membranes were incubated with adjusted secondary antibodies coupled to horseradish peroxidase. The immunocomplexes were visualized using the SuperSignal West Pico Kit (Thermo Scientific). Band density measurements were made using ImageJ imaging software version 1.36b (NIH).
Enzyme-Linked Immunosorbent Assay (ELISA)
For determination of the Aβ levels, MaxiSorp immunoplates (Nunc, Rochester, NY) were coated with mAb20.1 antibody (Dr. William E. Van Nostrand, Stony Brook University, Stony Brook, NY) at a concentration of 25 μg/mL in coating buffer (0.1 mol/L Na2CO3, pH 9.6) and blocked with 3% bovine serum albumin. Standard solutions for both Aβ40 and Aβ42 were made in the antigen capture buffer (20 mmol/L NaH2PO4, 2 mmol/L EDTA, 0.4 mol/L NaCl, 0.05% 3-[(3-cholamidopropyl) dimethylammonio]propanesulfonate, and 1% bovine serum albumin, pH 7.0) and loaded onto ELISA plates in duplicate. T-PER soluble fractions were loaded directly onto plates, whereas the formic acid supernatants (insoluble fractions) were diluted 1:20 in a neutralization buffer (1 mol/L Tris base and 0.5 mol/L NaH2PO4) before loading. All samples were loaded in duplicate and incubated overnight at 4°C. Plates were then washed and probed with either horseradish peroxidase-conjugated anti-Aβ40 or anti-Aβ42 (Drs. Vitaly Vasilevko and David H. Cribbs, University of California, Irvine) overnight at 4°C. To develop the plate, the chromogen 3,3′,5,5′-tetramethylbenzidine was added followed, by 30% phosphoric acid to stop the reaction. The plates were read at 450 nm using a plate reader (Molecular Dynamics, Sunnyvale, CA). The readings were then normalized to protein concentrations of the samples.
Immunohistochemistry
Coronal sections (40 μm thick) were incubated overnight at 4°C with anti-Aβ42 (Dr. Vitaly Vasilevko and Dr. David H. Cribbs, University of California, Irvine), anti-GFAP (Millipore, Billerica, MA) or anti-CD45 (AbD Serotec, Raleigh, NC) with 5% normal serum in Tris-buffered solution. The sections were subsequently treated with the appropriate biotinylated secondary antibody; then processed using the Vectastain Elite ABC reagent and 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA), according to the manufacturer’s instructions. Sections from vehicle and ATL treated mice were processed under the same conditions.
The immunostaining was assessed over a region representing a distance of approximately 160 μm obtained between 1.34 and 2.54 mm posterior to the bregma. Images of stained hippocampus, entorhinal cortex, subiculum, and amygdala were acquired using an Axiocam digital camera and AxioVision software version 4.6 connected to an Axioskop 50 microscope (Carl Zeiss MicroImaging, Thornwood, NY). Settings for image acquisition were identical for vehicle and ATL treated tissues.
Staining analyses were calculated as the percentage of labeled area captured (positive pixels)/the full area captured (total pixels) using ImageJ complying with strict standards [22].
Immunofluorescence
Sections were first incubated overnight at 4°C with one of either of the following primary antibodies: GFAP (Dako, Carpentaria, CA), CD45 (AbD Serotec) or Aβ1-16 (6E10) (Covance Research Products, Denver, PA). Sections were then rinsed and incubated for 1 hour with secondary Alexa Fluor-conjugated antibodies (Invitrogen, Carlsbad, CA) at room temperature. Finally, sections were mounted onto gelatin-coated slides in Fluoromount-G (Southern Biotech, Birmingham, AL) and examined under a Leica DM2500 confocal laser microscope using the Leica Application Suite Advanced Fluorescence software version 2.6.0 (Leica Microsystems). The immunofluorescence was also assessed at a distance of approximately 160 μm obtained between 1.34 and 2.54 mm posterior to the bregma.
Thioflavin S Staining
Sections were incubated in 0.5% thioflavin S in 50% ethanol for 10 minutes, differentiated twice in 50% ethanol, and washed in PBS solution. Confocal images were acquired by sequential scanning using a z-separation of 0.5 μm using the Leica Application Suite Advanced Fluorescence software (Leica Microsystems). Volumetric image measurements were made in the subiculum using Imaris software version 7.5.2 (Bitplane Inc., South Windsor, CT).
Statistical Analysis
The statistical evaluation of the results was performed using one-way analysis of variance. After significant analyses of variance, multiple post hoc comparisons were performed using the Bonferroni’s test. Some data were analyzed using the unpaired t-test. The accepted level of significance for the tests was P < 0.05. All tests were performed using the Statistica software version 5.1 (StatSoft Inc., Tulsa, OK). All final data were presented as percentage of control (vehicle-treated samples). All data are expressed as means ± SEM.
Results
Aging and AD Disrupt Inflammatory Resolution
The improper regulation of an inflammatory response has been hypothesized to be the underlining cause of chronic inflammatory conditions that may contribute to frailty and age-related diseases, including AD [23]. To determine if a decline in resolution mediators contributes to the chronic inflammatory condition found in AD, we assessed age-related changes in the levels of LTB4 and LXA4 in the brains of nTg and 3xTg-AD mice. We found that aging resulted in a rise in the levels of LTB4 in nTg mice, an effect that was exacerbated by AD-like pathology in 3xTg-AD mice (Fig. 1A). In contrast, we observed an age-dependent decrease in the levels of LXA4 in nTg and 3xTg-AD mice (Fig. 1B).
Fig. 1. Inflammatory resolution declines in aging and AD.

Age-related changes in the levels of (A) pro-inflammatory LTB4 and (B) anti-inflammatory/pro-resolution LXA4 in the brains of nTg and 3xTg-AD mice. The values represent the mean ± SEM. #P < 0.05 and ##P < 0.01 versus vehicle-treated nTg mice, and *P < 0.05 versus vehicle-treated 3xTg-AD mice.
Aspirin-Triggered LXA4 Improves Cognition
Because LXA4 production was significantly disrupted in aged 3xTg-AD mice, we next sought to determine whether increasing LXA4 signaling activation, through the administration of ATL, would effectively restore the anti-inflammatory responses in AD mouse model pathology; and modulate the progression of the disease. To determine the effect of ATL administration on the cognitive dysfunction associated with AD, 3xTg-AD mice were evaluated in a novel object recognition task. As expected, 3xTg-AD mice performed significantly worse when compared with nTg mice. Notably, ATL-treated 3xTg-AD mice exhibited a significant increase in the amount of time spent exploring the non-familiar object, indicating a rescue of cognition (Fig. 2A). In addition, tests in the open field demonstrate no differences in rearing or number of squares crossed, indicating a lack of anxious-like behavior or impairment of mobility (Fig. 2B,C).
Fig. 2. Aspirin-triggered LXA4 reduces cognitive impairment in 3xTg-AD mice.
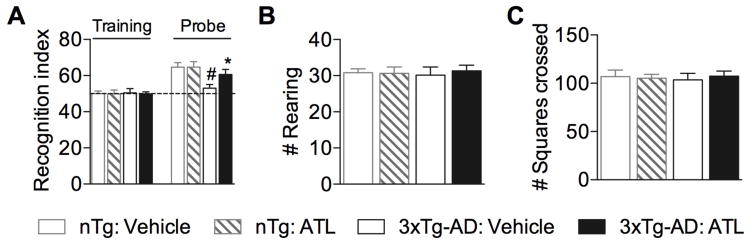
(A) 3xTg-AD mice treated with ATL exhibited a significant increase in the recognition index when compared with vehicle-treated animals. (B,C) No differences shown between ATL- and vehicle-treated mice in the number of rearing and squares crossed in the open field. The values represent means ± SEM. #P < 0.05 versus vehicle-treated nTg mice and *P < 0.05 versus vehicle-treated 3xTg-AD mice.
Aspirin-Triggered LXA4 Reduces Aβ Levels
We next examined for changes in Aβ pathology, as improvements in cognition are often associated with reductions in pathology. We found that ATL administration significantly reduced levels of Aβ40 and Aβ42, in both detergent soluble and insoluble fractions (Fig. 3A). Moreover, we found significantly less amyloid deposition in the ATL-treated animals versus vehicle-treated animals, as indicated by the immunohistochemistry studies (Fig. 3B,C). In addition, since aged 3xTg-AD mice have extensive plaque deposition, we sought to examine the effect of ATL administration on plaque composition, using Thioflavin S staining. We observed significant decrease in Thioflavin S positive plaques greater than 1,000 mm3 and a significant decrease in total plaque number (Fig. 3D,E).
Fig. 3. Aspirin-triggered LXA4 reduces brain Aβ levels in 3xTg-AD mice.

(A) Mice treated with ATL have lower levels of Aβ40 and Aβ42 peptides in both soluble- and insoluble-detergent fractions measured by ELISA. (B,C) ATL-treated mice have less Aβ42 staining in different brain regions when compared with vehicle-treated mice. (D,E) ATL-treated mice have less Thioflavin S positive fibrillar Aβ deposits. Significant reduction in total plaque load, as well as in medium (1,000–5,000) and large (5,000–10,000) size plaques are seen in ATL-treated animals. Representative photomicrographs were taken from the subiculum. The values represent mean ± SEM. *P < 0.05 and **P < 0.01 versus vehicle-treated 3xTg-AD mice.
Aspirin-Triggered LXA4 Reduces Tau Pathology
To examine the effect of ATL administration on tau pathology, we first tested for changes in total tau levels. No changes in total tau levels were observed in response to ATL administration. We next evaluated for potential changes in p-tau. Biochemical analysis of p-tau reviled significant decreases in the p-tau epitopes AT8, AT100, AT180, AT270, and PHF-1 (Fig. 4A,B). To determine if the reductions in p-tau levels were due to modulations in tau kinase function, we measured for changes in several kinases involved in tau phosphorylation. We found that ATL administration significantly increased phosphorylation in GSK3β, at the inhibitory Serine-9 residue, as well as observed significantly decreased p38 MAPK phosphorylation (Fig. 4C,D). ATL administration did not affect the steady state levels of CDK5, p35, p-ERK, PP2A, or GSK3β. Overall, our data indicates that reductions in p-tau were due to the modulation of GSK3β and p38 MAPK.
Fig. 4. Aspirin-triggered LXA4 reduces brain tau pathology in 3xTg-AD mice.

(A,B) Western blots of tau and tau phosphorylation, showing significant reduction in phosphorylated-tau in ATL-treated mice. (C,D) Western blots of tau kinases, showing a significant increase in p-GSK3β (Ser9) and reduction in p-p38 MAPK. The values represent mean ± SEM. *P < 0.05 and **P < 0.01 versus vehicle-treated 3xTg-AD mice.
Aspirin-Triggered LXA4 Reduces Inflammatory Cells
The inflammatory milieu of the AD brain consists of increase numbers of microglia and astrocytes. This characteristic has been recapitulated in aged 3xTg-AD mice. Upon investigation, we found significantly less reactive astrocytes and activated microglia in ATL-treated animals, as indicated by the lower detection of GFAP and CD45, respectively (Fig. 5A,B). The effect was most prominent in the immediate vicinity of plaques (Fig. 5C,D). The immunofluorescence studies indicated lower amyloid load and inflammatory cell numbers.
Fig. 5. Aspirin-triggered LXA4 reduces inflammation.

(A,B) Significant reduction in astrocytes and microglia in ATL-treated animals. (C,D) Colocalization of 6E10 and (C) microglia or (D) astrocytes showing reduction in plaque load and cell intensity in ATL-treated animals. The values represent mean ± SEM. *P < 0.05 and **P < 0.01 versus vehicle-treated 3xTg-AD mice.
Discussion
An exacerbated inflammatory response is a feature of aging, which may trigger loss of function in cells of the central nervous system and increase susceptibility to age-related diseases including AD [2, 3, 23–25]. For this reason, many studies have focused on uncovering the underlying regulatory mechanisms and strategies to down-regulate pro-inflammatory responses. However, the discoveries from recent studies showing that a blockade of inflammatory responses aggravate the progression of AD raise the question about how to best manipulate the immune response to succeed in the management of neurodegenerative disorders [26, 27]. The discoveries that the resolution of inflammation is a highly coordinated and active process controlled by endogenous pro-resolving mediators, and that inflammatory cells undergo classical and alternative activation, highlight new potential molecular targets to modulate inflammation and treat chronic inflammatory diseases [28–30]. Accordingly, we provide critical functional and molecular evidence pointing to the endogenous pro-resolution LXA4 pathway as a potential candidate to treat AD and other age-related inflammatory disorders. In the present study, we report the finding that LXA4 production declines with age, and markedly in AD mouse model. Remarkably, restoring LXA4 signaling with ATL treatment reduced the severity of AD-like neuropathology, as indicated by the decrease in amyloid plaques, tau phosphorylation and inflammation, as well as the improvement in the cognitive performance.
Previous findings identified high expression of 5-LOX and 12/15-LOX as well as increased production of their metabolites 5(S)-HETE, and 12/15(S)-HETE, during aging [12–15]. Most notably, activation of these enzymes has been linked to the progression of AD neuropathology. The expression of LOX enzymes is increased in human AD brains, and genetic or pharmacological approaches to block 5-LOX, 12/15-LOX, and other components of LOX cascade in AD transgenic mice significantly reduce Aβ and tau pathology, and restore cognition [31–36]. Together, these data suggest that an inhibition of LOX activity is beneficial in the treatment and prevention of AD. Our findings showing an age-related imbalance of LTB4 and LXA4 productions in the 3xTg-AD mice support the activity of LOX enzymes as a key step in the coordination of neuroinflammation. These findings also suggest that a deregulation of the inflammatory resolution may represent a critical mechanism to the prolonged inflammation and progressive decline in immune function in aged and AD brains. Although additional studies are necessary to identify the molecular changes taking place in LOX enzymes that favor the pro-inflammatory component of the cascade, our data suggest a novel venue that can be explored to restore and/or increase levels of lipoxins during aging and AD. In support of this hypothesis, we have previously demonstrated that activation of LXA4 pathway by ATL administration reduces Aβ pathology, inflammation, and improves cognition in the Tg2576 mouse model [19]. In this study, we further explored the impact of activating LXA4 in the 3xTg-AD mice, which not only replicate the features of human AD, namely plaques, tangles, inflammation and memory impairment, but also display reduced LXA4 levels [16–18]. Importantly, we provide data in the 3xTg-AD mice that support the notion that restoration of the LXA4 cascade is capable of reducing Aβ and inflammation, and improving cognitive function.
An important characteristic in AD is the progression of tau pathology. Our findings demonstrate that stimulation of LXA4 signaling also results in reduced activation of tau kinases p38 and GSK3β [37]. More importantly, ATL-treatment significantly lowered pathological tau in 3xTg-AD mice, as indicated by the attenuation in the phosphorylation of multiple tau epitopes. Mechanistically, ATL-treatment might have inhibited tau pathology by distinctive processes, specifically through changes in Aβ, inflammation, or direct neuronal modulation. Several studies have shown that Aβ promotes tau hyperphosphorylation and aggregation [38, 39]. Moreover, Aβ pathology precedes tau pathology, and immunotherapy targeting the Aβ can ameliorate both Aβ and soluble tau pathology in the 3xTg-AD mice [20, 40]. More recently, it has been demonstrated that Aβ causes the acceleration of wild-type human tau pathology, which is a critical component of the lasting changes to dendritic spines and cognitive impairment found in AD [41]. Regarding inflammation, several studies have implicated the immune response as a critical component to the progression of tau pathology [42–46]. Lastly, we recently demonstrated that neurons express the LXA4 receptor ALX, which has been directly implicated in the inhibition of numerous protein kinases associated with tau phosphorylation, including p38, ERK, JNK [19, 47–49]. Therefore, it is possible suggest that activation of neuronal ALX by ATL administration may result in inhibition of tau pathology. However, additional studies are necessary to confirm this hypothesis.
In this study, we demonstrated that the resolution phase of the inflammatory response is impaired during aging, and even more so in an AD mouse model. Notably, restoration of this phase, through the administration of ATL, was found to protect against the progression of AD-like neuropathology. A better understanding of what causes the disruption in the LOX cascade will be important to better treat disease progression in humans. Namely, the distinction of whether AD pathology causes the imbalance or the imbalance drives AD pathology, is an important objective for future experimentation. We believe that by further exploring these pathways, we may find a novel process that contributes to the progression of AD. Developing drugs that restore the pro-resolution LXA4 cascade may be a novel therapeutic approach to prevent the progression of AD.
Acknowledgments
This study was supported by grants from the UCI Undergraduate Research Opportunities Program (HCD), UCI Alzheimer’s Disease Research Center through funding from an NIH/NIA grant: P50 AG16573 (RM), Program Project Grant AG00538 (RM & DHC), and an Alzheimer’s Association grant IIRG-11-204835 (DHC).
References
- 1.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 2.McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126:479–497. doi: 10.1007/s00401-013-1177-7. [DOI] [PubMed] [Google Scholar]
- 3.Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 2010;120:277–286. doi: 10.1007/s00401-010-0722-x. [DOI] [PubMed] [Google Scholar]
- 4.Griffin WS, Liu L, Li Y, Mrak RE, Barger SW. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation. 2006;3:5. doi: 10.1186/1742-2094-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Liu L, Barger SW, Griffin WS. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003;23:1605–1611. doi: 10.1523/JNEUROSCI.23-05-01605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279:49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- 7.Chiang N, Arita M, Serhan CN. Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot Essent Fatty Acids. 2005;73:163–177. doi: 10.1016/j.plefa.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Petasis NA, Akritopoulou-Zanze I, Fokin VV, Bernasconi G, Keledjian R, Yang R, Uddin J, Nagulapalli KC, Serhan CN. Design, synthesis and bioactions of novel stable mimetics of lipoxins and aspirin-triggered lipoxins. Prostaglandins Leukot Essent Fatty Acids. 2005;73:301–321. doi: 10.1016/j.plefa.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 9.Henderson WR., Jr The role of leukotrienes in inflammation. Ann Intern Med. 1994;121:684–697. doi: 10.7326/0003-4819-121-9-199411010-00010. [DOI] [PubMed] [Google Scholar]
- 10.McMahon B, Mitchell S, Brady HR, Godson C. Lipoxins: revelations on resolution. Trends Pharmacol Sci. 2001;22:391–395. doi: 10.1016/s0165-6147(00)01771-5. [DOI] [PubMed] [Google Scholar]
- 11.Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci U S A. 1995;92:9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chinnici CM, Yao Y, Pratico D. The 5-lipoxygenase enzymatic pathway in the mouse brain: young versus old. Neurobiol Aging. 2007;28:1457–1462. doi: 10.1016/j.neurobiolaging.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 13.Manev H, Uz T, Sugaya K, Qu T. Putative role of neuronal 5-lipoxygenase in an aging brain. FASEB J. 2000;14:1464–1469. doi: 10.1096/fj.14.10.1464. [DOI] [PubMed] [Google Scholar]
- 14.Pratico D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, Lee VM. 12/15-lipoxygenase is increased in Alzheimer’s disease: possible involvement in brain oxidative stress. Am J Pathol. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao Y, Clark CM, Trojanowski JQ, Lee VM, Pratico D. Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment. Ann Neurol. 2005;58:623–626. doi: 10.1002/ana.20558. [DOI] [PubMed] [Google Scholar]
- 16.Gangemi S, Pescara L, D’Urbano E, Basile G, Nicita-Mauro V, Davi G, Romano M. Aging is characterized by a profound reduction in anti-inflammatory lipoxin A4 levels. Exp Gerontol. 2005;40:612–614. doi: 10.1016/j.exger.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Leo LM, Almeida-Correa S, Canetti CA, Amaral OB, Bozza FA, Pamplona FA. Age-dependent relevance of endogenous 5-lipoxygenase derivatives in anxiety-like behavior in mice. PLoS One. 2014;9:e85009. doi: 10.1371/journal.pone.0085009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Zhu M, Hjorth E, Cortes-Toro V, Eyjolfsdottir H, Graff C, Nennesmo I, Palmblad J, Eriksdotter M, Sambamurti K, Fitzgerald JM, Serhan CN, Granholm AC, Schultzberg M. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement. 2014 doi: 10.1016/j.jalz.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Medeiros R, Kitazawa M, Passos GF, Baglietto-Vargas D, Cheng D, Cribbs DH, LaFerla FM. Aspirin-triggered lipoxin A4 stimulates alternative activation of microglia and reduces Alzheimer disease-like pathology in mice. Am J Pathol. 2013;182:1780–1789. doi: 10.1016/j.ajpath.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 21.Powell WS. Rapid extraction of oxygenated metabolites of arachidonic acid from biological samples using octadecylsilyl silica. Prostaglandins. 1980;20:947–957. doi: 10.1016/0090-6980(80)90144-6. [DOI] [PubMed] [Google Scholar]
- 22.Rossner M, Yamada KM. What’s in a picture? The temptation of image manipulation. J Cell Biol. 2004;166:11–15. doi: 10.1083/jcb.200406019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–169. doi: 10.1111/j.1365-2567.2009.03225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solana R, Pawelec G, Tarazona R. Aging and innate immunity. Immunity. 2006;24:491–494. doi: 10.1016/j.immuni.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Licastro F, Candore G, Lio D, Porcellini E, Colonna-Romano G, Franceschi C, Caruso C. Innate immunity and inflammation in ageing: a key for understanding age-related diseases. Immun Ageing. 2005;2:8. doi: 10.1186/1742-4933-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 27.Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 29.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Firuzi O, Zhuo J, Chinnici CM, Wisniewski T, Pratico D. 5-Lipoxygenase gene disruption reduces amyloid-beta pathology in a mouse model of Alzheimer’s disease. FASEB J. 2008;22:1169–1178. doi: 10.1096/fj.07-9131.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu J, Pratico D. Pharmacologic blockade of 5-lipoxygenase improves the amyloidotic phenotype of an Alzheimer’s disease transgenic mouse model involvement of gamma-secretase. Am J Pathol. 2011;178:1762–1769. doi: 10.1016/j.ajpath.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Yang H, Zhuo JM, Chu J, Chinnici C, Pratico D. Amelioration of the Alzheimer’s disease phenotype by absence of 12/15-lipoxygenase. Biol Psychiatry. 2010;68:922–929. doi: 10.1016/j.biopsych.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 34.Chu J, Lauretti E, Di Meco A, Pratico D. FLAP pharmacological blockade modulates metabolism of endogenous tau in vivo. Transl Psychiatry. 2013;3:e333. doi: 10.1038/tp.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joshi YB, Di Meco A, Pratico D. Modulation of amyloid-beta production by leukotriene B4 via the gamma-secretase pathway. J Alzheimers Dis. 2014;38:503–506. doi: 10.3233/JAD-131223. [DOI] [PubMed] [Google Scholar]
- 36.Giannopoulos PF, Joshi YB, Chu J, Pratico D. The 12-15-lipoxygenase is a modulator of Alzheimer’s-related tau pathology in vivo. Aging Cell. 2013;12:1082–1090. doi: 10.1111/acel.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Medeiros R, Baglietto-Vargas D, LaFerla FM. The role of tau in Alzheimer’s disease and related disorders. CNS Neurosci Ther. 2011;17:514–524. doi: 10.1111/j.1755-5949.2010.00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 39.Gotz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001;276:529–534. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- 40.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 41.Chabrier MA, Cheng D, Castello NA, Green KN, Laferla FM. Synergistic effects of amyloid-beta and wild-type human tau on dendritic spine loss in a floxed double transgenic model of Alzheimer’s disease. Neurobiol Dis. 2014;64:107–117. doi: 10.1016/j.nbd.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O’Banion MK. Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013;33:5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol. 2011;187:6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE, Laferla FM. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol. 2011;178:2811–2822. doi: 10.1016/j.ajpath.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitazawa M, Trinh DN, LaFerla FM. Inflammation induces tau pathology in inclusion body myositis model via glycogen synthase kinase-3beta. Ann Neurol. 2008;64:15–24. doi: 10.1002/ana.21325. [DOI] [PubMed] [Google Scholar]
- 46.Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo CL, Li QQ, Chen XP, Zhang XM, Li LL, Li BX, Zhao ZQ, Tao LY. Lipoxin A4 attenuates brain damage and downregulates the production of pro-inflammatory cytokines and phosphorylated mitogen-activated protein kinases in a mouse model of traumatic brain injury. Brain Res. 2013;1502:1–10. doi: 10.1016/j.brainres.2013.01.037. [DOI] [PubMed] [Google Scholar]
- 48.Wu L, Miao S, Zou LB, Wu P, Hao H, Tang K, Zeng P, Xiong J, Li HH, Wu Q, Cai L, Ye DY. Lipoxin A4 inhibits 5-lipoxygenase translocation and leukotrienes biosynthesis to exert a neuroprotective effect in cerebral ischemia/reperfusion injury. J Mol Neurosci. 2012;48:185–200. doi: 10.1007/s12031-012-9807-4. [DOI] [PubMed] [Google Scholar]
- 49.Wang YP, Wu Y, Li LY, Zheng J, Liu RG, Zhou JP, Yuan SY, Shang Y, Yao SL. Aspirin-triggered lipoxin A4 attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-kappaB and MAPKs in BV-2 microglial cells. J Neuroinflammation. 2011;8:95. doi: 10.1186/1742-2094-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]