

Abstract

Protein arginine methyltransferases (PRMTs) play an important role in diverse biological processes. Among the nine known human PRMTs, PRMT3 has been implicated in ribosomal biosynthesis via asymmetric dimethylation of the 40S ribosomal protein S2 and in cancer via interaction with the DAL-1 tumor suppressor protein. However, few selective inhibitors of PRMTs have been discovered. We recently disclosed the first selective PRMT3 inhibitor, which occupies a novel allosteric binding site and is noncompetitive with both the peptide substrate and cofactor. Here we report comprehensive structure–activity relationship studies of this series, which resulted in the discovery of multiple PRMT3 inhibitors with submicromolar potencies. An X-ray crystal structure of compound 14u in complex with PRMT3 confirmed that this inhibitor occupied the same allosteric binding site as our initial lead compound. These studies provide the first experimental evidence that potent and selective inhibitors can be created by exploiting the allosteric binding site of PRMT3.
INTRODUCTION
Among epigenetic writers, histone methyltransferases (HMTs, also known as protein methyltransferases (PMTs)) are divided into two categories: protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs), which catalyze lysine and arginine methylation, respectively.1,2 Nine human PRMTs have been identified to date and are further divided into three subtypes.3–5 Type I PRMTs (PRMT1–4,6,8) catalyze arginine monomethylation and asymmetric dimethylation, while type II PRMTs such as PRMT5 catalyze arginine monomethylation and symmetric dimethylation. Recently, PRMT7 was characterized as a type III PRMT which catalyzes only arginine monomethylation.6 In addition to histone substrates, PRMTs also methylate many nonhistone proteins.3,7 PRMTs usually methylate GAR (glycine- and arginine-rich) motifs in their substrates,8,9 except for PRMT4, which instead methylates PGM (proline-, glycine-, methionine-, and arginine-rich) motifs.10,11 PRMT5 methylates both GAR and PGM motifs.10,12 Protein arginine methylation catalyzed by PRMTs plays a key role in various biological processes, including gene expression, transcriptional regulation, signal transduction, protein and RNA subcellular localization, RNA splicing, and DNA damage repair.3–5 Dysregulation of PRMTs has been implicated in a number of human conditions, including cancer.3–5
PRMT3 (protein arginine methyltransferase 3), a type I PRMT, was first reported in 199813 and subsequently shown to catalyze asymmetric dimethylation of GAR motifs in its main substrate: the 40S ribosomal protein S2 (rpS2).14,15 The dimethylation of rpS2 by PRMT3 results in stabilization of rpS2 and influences ribosomal biosynthesis.4,14–16 PRMT3 has also been reported to methylate the recombinant mammalian nuclear poly(A)-binding protein (PABPN1)17–19 and a histone peptide (H4 1–24) in vitro.20 In addition, the protein complex consisting of PRMT3, the von Hippel–Lindau (VHL) tumor suppressor protein, and ARF (alternative reading frame) methylates the tumor suppressor p53.21 Importantly, the tumor suppressor DAL-1 (differentially expressed in adeno-carcinoma of the lung-1) inhibits the methyltransferase activity of PRMT3 via its interaction with PRMT3, suggesting that DAL-1 may affect tumor growth by regulating PRMT3 function.22 Therefore, pharmacologic inhibition of PRMT3 may offer a potentially viable option for treating the tumors that display epigenetic down-regulation of DAL-1. In addition, PRMT3 expression levels are elevated in myocardial tissue from patients with atherosclerosis,23 potentially implicating PRMT3 in this and related diseases. Lastly, PRMT3 function has been reported to be essential for dendritic spine maturation in rats.24
Selective small-molecule inhibitors of PRMTs and PKMTs are important tools for investigating the biology of this emerging target class and testing disease and therapeutic hypotheses regarding these enzymes.5,25–28 However, only a limited number of selective inhibitors of PRMTs29–42,44,46 and PKMTs43–56 have been reported. In particular, selective small-molecule inhibitors of PRMT3 were lacking. We recently disclosed the first selective, allosteric inhibitor (compound 1 in Figure 1A) of PRMT3.57 This inhibitor occupies neither the substrate binding groove nor the cofactor binding site. Instead, it occupies a novel allosteric binding site revealed by the X-ray crystal structure of compound 1 in complex with PRMT3 (PDB code 3SMQ). Subsequent mechanism of action (MOA) studies confirmed that this inhibitor is noncompetitive with both the peptide substrate and the cofactor.57 Here we report structure–activity relationship (SAR) studies that focused on extensively exploring three regions of the scaffold represented by compound 1. We describe the design, synthesis, and biochemical evaluation of novel compounds, which resulted in the discovery of potent and selective PRMT3 inhibitors. We obtained an X-ray crystal structure of the compound 14u–PRMT3 complex, which confirmed that this inhibitor occupied the same allosteric binding site previously described. These studies establish that the allosteric binding site of PRMT3 is druggable and can be exploited to generate potent and selective inhibitors.
Figure 1.
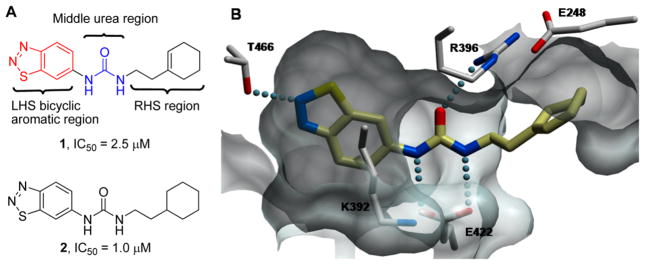
(A) Structure of PRMT3 inhibitors 1 and 2 and three regions explored for SAR. (B) X-ray crystal structure of the inhibitor 1–PRMT3 complex (PDB code 3SMQ). Key interactions include (1) a hydrogen bond between the 2-nitrogen of the benzothiadiazole and T466, (2) two hydrogen bonds between the two amino groups of the middle urea moiety and E422, (3) a hydrogen bond between the oxygen of the urea moiety and R396, and (4) hydrophobic interactions between the cyclohexenylethyl group and a nonpolar surface.
RESULTS AND DISCUSSION
Design and Synthesis
To improve the potency of compound 1 (PRMT3, IC50 = 2.5 μM)57 and demonstrate that potent and selective inhibitors can be generated by exploiting the allosteric binding site of PRMT3, we investigated the following three regions of the chemical series represented by compound 1: (1) the left-hand side (LHS) bicyclic aromatic moiety, (2) the middle urea moiety, and (3) the right-hand side (RHS) moiety (Figure 1A). We previously reported that the uncommon cyclohexenylethyl group of compound 1 could be replaced by a more common and potentially stable group, the cyclohexylethyl group of compound 2 (PRMT3, IC50 = 1.0 μM), without any potency loss, and showed that the alkene functionality was unnecessary (Figure 1A).57
Because the X-ray crystal structure of compound 1 in complex with PRMT3 (PDB code 3SMQ) reveals that the LHS benzothiadiazole moiety fits tightly in the allosteric pocket and forms a hydrogen bond with T466 (threonine 466) (Figure 1B),57 we designed the compounds outlined in Scheme 1 and Table 1 to determine whether the benzothiadiazole ring can be replaced with other fused bicyclic aromatic groups. A representative synthesis of these compounds (3–7) is shown in Scheme 1. The desired compounds were prepared via a standard urea formation reaction from commercially available anilines and 2-cyclohexylethylamine using 1,1′-carbonyldiimi-dazole (CDI) as the coupling reagent.
Scheme 1.

Synthesis of Compounds 3–7a
aReagents and conditions: (a) CDI, THF, 2-cyclohexylethylamine, rt, 41–88%.
Table 1.
SAR of the LHS Bicyclic Aromatic Moiety







IC50 determination experiments were performed in triplicate.
The IC50 value was previously reported.57
We previously synthesized several analogues to confirm the hydrogen bonds between the middle urea moiety of compound 1 and the R396 (arginine 396) and E422 (glutamate 422) of PRMT3 (Figure 1B).57 To further investigate the middle urea moiety, we designed and synthesized the urea bioisosteres and more rigid analogues outlined in Scheme 2. The key intermediate 8 was prepared according to published procedures.58 Commercially available 6-nitrobenzothiazole was first ring-opened to yield the disulfide intermediate, which was then converted to the 6-nitrobenzothiadiazole intermediate. Subsequent reduction of the nitro group afforded the intermediate 8. The nucleophilic substitution reaction of the intermediate 8 with 3,4-diethoxycyclobut-3-ene-1,2-dione59 formed the intermediate 9a, which was then converted to the desired products 10 and 11 by a second nucleophilic substitution reaction with 2-cyclohexylethylamine or 2-amino-1-phenylethanone. Similarly, nucleophilic substitution reaction of the intermediate 8 with diphenyl cyanocarbonimidate yielded the intermediate 9b, which was subsequently converted to the desired product 12 by a second substitution reaction. The aminotriazole 13 was formed from 3-cyclohexylpropanehydrazide and the intermediate 9c, which was prepared from the intermediate 8.
Scheme 2.

Synthesis of Compounds for Exploring the Middle Urea Moietya
aReagents and conditions: (a) N2H4·H2O, EtOH, reflux; (b) H2O2, rt, 87% over two steps; (c) KNO2/H2SO4, 0–10 °C, 62%; (d) SnCl2/HCl, 65 °C, 5 min, 61%; (e) 3,4-diethoxycyclobut-3-ene-1,2-dione, Zn(OTf)2, EtOH, rt, 92%; (f) 2-cyclohexylethylamine or 2-amino-1-phenylethanone, i-PrOH, 120 °C, microwave, 78–85%; (g) diphenyl cyanocarbonimidate, i-PrOH, 100 °C, microwave; (h) 2-cyclohexylethylamine, i-PrOH, 100 °C, microwave, 38% over two steps; (i) BrCN, THF, 40 °C; (j) 3-cyclohexylpropanehydrazide, HCl, i-PrOH, 160 °C, microwave, 32% over two steps.
In the X-ray crystal structure of the inhibitor 1–PRMT3 complex, the RHS cyclohexenylethyl moiety makes hydrophobic interactions with a surface formed from the two subunits of the PRMT3 homodimer (Figure 1B). Because it was not clear what was required for these hydrophobic interactions and whether additional interactions could be made, we extensively explored this region by introducing functional groups with varying steric and electronic characteristics to either an aliphatic or an aromatic moiety. Synthesis of compounds 14a–v and 15 a–e (see structures in Table 3) is outlined in Scheme 3. Compounds 14a–v were prepared via a urea formation reaction from the intermediate 8 and various amines. Compounds 15a–e were obtained via a urea formation reaction followed by the removal of the Boc protecting group.
Table 3.
SAR of the RHS Moiety: Part Ia
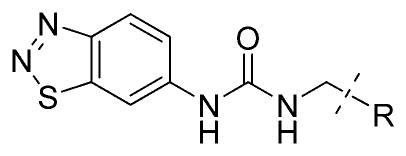
| ||
|---|---|---|
| Compound | R (RHS Moiety) | PRMT3 IC50 (μM) |
| 2 |

|
1.0 ± 0.2 |
| 1 |
|
2.4 ± 0.4 |
| 14a |

|
3.6 ± 0.5 |
| 14b |

|
5.2 ± 1 |
| 14c |

|
>10 |
| 14d |

|
7.2 ± 0.6 |
| 14e |

|
>10 |
| 14f |
|
>50 |
| 14g |

|
>15 |
| 14h |

|
>10 |
| 14i |

|
>25 |
| 14j |

|
>15 |
| 14k |

|
>15 |
| 141 |

|
>10 |
| 14m |

|
>15 |
| 14n |

|
>10 |
| 14o |

|
0.30 ± 0.08 |
| 14p |

|
0.85 ± 0.2 |
| 14q |

|
1.8 ± 0.3 |
| 14r |

|
3.1 ± 0.6 |
| 14s |

|
4.0 ± 1 |
| 14t |
|
>25 |
| 14u |

|
0.48 ± 0.01 |
| 14v |

|
2.2 ± 0.4 |
| 15a |

|
2.7 ± 0.6 |
| 15b |

|
3.6 ± 0.5 |
| 15c |

|
>10 |
| 15d |

|
5.3 ± 0.7 |
| 15e |

|
3.3 ± 0.7 |
IC50 determination experiments were performed in triplicate.
Scheme 3.
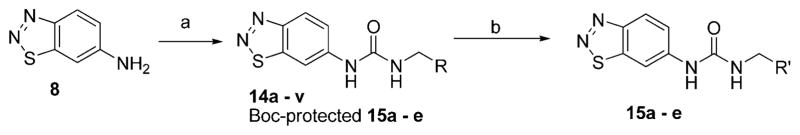
Synthesis of the Compounds Outlined in Table 3a
aReagents and conditions: (a) CDI, DMF, various amines, rt, 35–55%; (b) TFA, DCM, rt, 100%.
To further investigate the benzoyl moiety of compound 14u, the first PRMT3 inhibitor with submicromolar potency (see below and Table 3), we designed and synthesized the compounds outlined in Scheme 4 and Table 4. Compound 16 was prepared via a urea formation reaction followed by the removal of the Boc protecting group, similar to the synthesis of compounds 15a–e. Compounds 17–21 and 24 were synthesized via a urea formation reaction from the intermediate 8 and various amines. Compounds 22 and 23 were obtained via oxidation of respective alcohols 21 and 19 using the sulfur trioxide–pyridine complex. Compounds 16, 17, 19, 20, and 21 were prepared as racemic mixtures.
Scheme 4.

Synthesis of the Compounds Outlined in Table 4a
aReagents and conditions: (a) CDI, DMF, various amines, rt, 35–55%; (b) TFA, DCM, rt, 100%; (c) PySO3, Et3N, DCM/DMSO, rt, 76–78%.
Table 4.
SAR of the RHS Moiety: Part IIa

| ||
|---|---|---|
| Compound | R (RHS Moiety) | PRMT3 IC50 (μM) |
| 14u |

|
0.48 ± 0.01 |
| (±)-16 |

|
5.9 ± 0.5 |
| (±)-17 |

|
1.9 ± 0.2 |
| 18 |

|
5.0 ± 0.2 |
| (±)-19 |

|
4.4 ± 0.4 |
| (±)-20 |
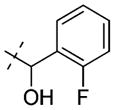
|
3.3 ± 0.4 |
| (±)-21 |

|
0.49 ± 0.08 |
| 22 |

|
0.84 ± 0.1 |
| 23 |

|
0.36 ± 0.07 |
| 24 |

|
0.23 ± 0.03 |
IC50 determination experiments were performed in triplicate.
These synthesized compounds were evaluated in a radioactive biochemical assay which measures the transfer of the tritiated methyl group from the cofactor [3H]-S-adenosylme- thionine (SAM) to a peptide substrate. IC50 values of these compounds in this assay are summarized in Tables 1–4.
SAR of the LHS Bicyclic Aromatic Moiety
We previously reported that the replacement of the benzothiadiazole moiety (compound 2) with the benzothiazole (compound 3), which abolished the hydrogen bond with T466, resulted in complete loss of potency.57 This SAR finding confirmed the key hydrogen bond interaction between the middle nitrogen of the benzothiadiazole moiety and T466, revealed by the X-ray crystal structure of compound 1 in complex with PRMT3. Interestingly, the replacement of the benzothiadiazole moiety (compound 2) with the benzotriazole (compound 4), indazole (compound 5 or 6), or benzoxazolone (compound 7) moiety also led to total loss of potency (Table 1), suggesting that, in addition to making the hydrogen bond interaction with T466, the benzothiadiazole moiety is particularly well accommodated by the PRMT3 allosteric binding pocket due to its unique electronic nature and steric fit. Because these relatively minor modifications to the benzothiadiazole moiety were not tolerated, we did not attempt to introduce more drastic modifications such as substituted monocyclic aromatic or nonaromatic groups and have kept this LHS moiety constant during the subsequent SAR exploration of this hit series.
SAR of the Middle Urea Moiety
The X-ray crystal structure of the inhibitor 1–PRMT3 complex reveals that the carbonyl group of the middle urea moiety forms a hydrogen bond with the guanidinium group of R396 and the two amino groups of the urea moiety form two hydrogen bonds with the carboxylate of E422 (glutamate 422) (Figure 1B).57 We previously synthesized a number of analogues for probing these hydrogen bond interactions and found that the replacement of the oxygen of the urea moiety, which abolished the hydrogen bond with R396, and N-methylation of either nitrogen of the urea moiety, which impaired the hydrogen bonds with E422, led to complete loss of potency.57 These SAR results confirmed the key hydrogen bond interactions between the urea moiety of inhibitor 1 and the allosteric binding site of PRMT3.
We further investigated the middle urea moiety by design, synthesis, and biological evaluation of the urea bioisosteres and more rigid analogues outlined in Table 2. The diaminosquarate 10 displayed >100-fold potency loss compared with its urea analogue (2). Similarly, the diaminosquarate 11 was >200-fold less potent compared with its urea analogue (14u), which contains a preferred RHS moiety (see below and Table 3). In addition, the cyanoguanidine 12 and aminotriazole 13 showed no inhibitory activity against PRMT3 (IC50 values >100 μM). Taken together, these results suggest that the urea moiety is ideal for interacting with the allosteric binding pocket of PRMT3. We therefore held this moiety constant during subsequent SAR exploration.
Table 2.
Bioisosteres of the Middle Urea Moiety
| Compound | Structure | PRMT3 IC50 (μM)a |
|---|---|---|
| 2 |
|
1.0 ± 0.2 |
| 10 |

|
>100 |
| 11 |

|
>100 |
| 12 |
|
>100 |
| 13 |
|
>100 |
IC50 determination experiments were performed in triplicate.
SAR of the RHS Moiety
We previously reported that the uncommon cyclohexenylethyl group of compound 1 could be replaced by a more common and potentially stable group, the cyclohexylethyl group of compound 2, without any potency loss (Table 3).57 To improve potency, we extensively investigated this region by introducing functional groups with varying steric and electronic characteristics to either an aliphatic or an aromatic moiety (Table 3). The replacement of the cyclohexyl ring with a heterocycle such as tetrahydropyran (compound 14a), 1-piperidine (compound 14b), morpholine (compound 14c), 1-pyrrolidine (14e), and 4-piperidine (compound 15a) generally resulted in a decrease in potency. Similarly, compounds 15b–e, which contain a respective terminal amino group to potentially form additional hydrogen bond interaction(s) with backbone amides, were also less potent than compound 2. A longer chain (three-carbon versus two-carbon) was tolerated but did not lead to a significant increase in potency (14d versus 14c, 15d versus 15c, and 15e versus 15a). On the other hand, a shorter chain (one-carbon versus two-carbon) resulted in a complete loss of potency (14f versus 2). The replacement of the cyclohexyl group with a hydrogen (compound 14g) or tert-butyl group (compound 14h) also led to a significant decrease in potency. We next explored the replacement of the cyclohexylethyl group with a motif containing an aromatic ring. Although the unsubstituted benzyl group (compound 14i), three pyridinylmethyl groups (compounds 14j–l), and two benzyl groups substituted with an electron-donating group (compounds 14m,n) led to a significant decrease in potency compared with the cyclo-hexylethyl group (compound 2), we were pleased to find that strong electron-withdrawing groups such as nitro (compound 14o) and methylsulfonyl (compound 14p) groups resulted in similar or higher potency compared with the cyclohexylethyl group (compound 2). Other electron-withdrawing groups such as cyano (compound 14q), trifluoromethyl (compound 14r), and fluoro (compound 14s) groups gave either similar or slightly lower potency compared with the cyclohexylethyl group (compound 2) but significantly better potency compared with the unsubstituted compound (14i) or electron-donating groups (compounds 14m,n). Interestingly, moving the trifluoromethyl group from the para-position (compound 14r) to the ortho-position (compound 14t) resulted in a significant decrease in potency. Further exploration of this aromatic moiety led to the discovery of compound 14u, which contains a benzoyl group, with an IC50 of 0.48 μM and good selectivity for PRMT3 over other methyltransferases (see below). We also found that a longer chain (three-carbon versus one-carbon) maintained good potency (14v versus 14s), which is consistent with the finding in the aliphatic subseries (see above).
Encouraged by the discovery of the first PRMT3 inhibitors with submicromolar potencies, we further investigated the benzoyl moiety of compound 14u (Table 4). The replacement of the carbonyl group (compound 14u) with an α-amino (compound 16), α-hydroxyl (compound 17), or α,α-difluoro (compound 18) group led to a decrease in potency. Adding a p-or o-fluoro group to the phenyl group did not significantly change the potency (compounds 19 and 20 versus compound 17). Interestingly, compound 21, which possesses a cyclohexyl group, was slightly more potent than compound 17, which possesses a phenyl group instead. Although the ketone 22 had potency similar to that of its alcohol analogue (compound 21), the ketone 23 was about 10-fold more potent than its corresponding alcohol (19). Further modifications of the ketone 22 led to the discovery of the amide 24, which is the most potent PRMT3 inhibitor to date with an IC50 of 0.23 μM. Taken together, these results indicate that modifications to the RHS moiety are well tolerated and can lead to a significant increase in potency. These studies resulted in the discovery of multiple PRMT3 inhibitors with submicromolar potencies. Among them, compound 14u was further evaluated in subsequent selectivity, X-ray crystallography, and computational modeling studies.
Selectivity Assessment of Compound 14u
We next determined the selectivity of compound 14u against a number of methyltransferases, including PRMTs, PKMTs, and DNMT1 (DNA methyltransferase 1). As shown in Figure 2, compound 14u was at least 40-fold selective for PRMT3 over G9a, GLP, and SUV39H2 and completely inactive against PRMT5, SETD7, PRC2, SETD8, SETDB1, SUV420H1, SUV420H2, MLL1, SMYD3, SMYD2, DOT1L, and DNMT1. These results are consistent with the selectivity results of compound 1 reported previously,57 suggesting that targeting the allosteric binding site of PRMT3 can yield selective inhibitors.
Figure 2.
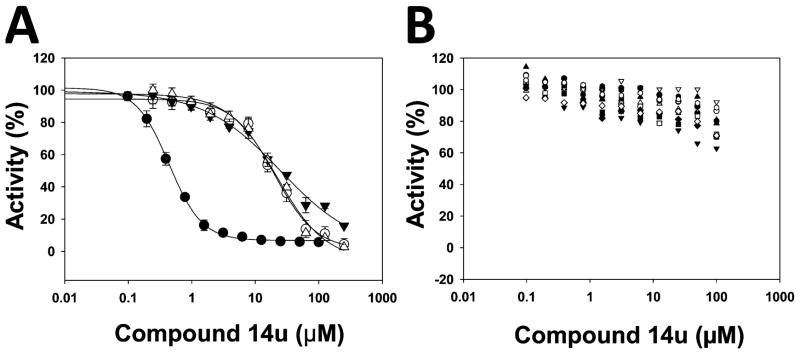
Compound 14u was selective for PRMT3 over other PRMTs, PKMTs, and DNMT1. The effects of compound 14u on the activity of the following methyltransferases were assessed: (A) PRMT3 (●, IC50 = 0.48 ± 0.01 μM), G9a (○, IC50 = 20 ± 1 μM), GLP (▼, IC50 = 25 ± 8 μM), and SUV39H2 (△, IC50 = 22 ± 1 μM); (B) SETD7 (●), PRC2 complex (○), SETD8 (▼), SETDB1 (△), SUV420H1 (■), SUV420H2 (□), MLL1 complex (◇), SMYD3 (▲), SMYD2 (▽), DOT1L (□), PRMT5–MEP50 complex (◆), and DNMT1 (⬢).
X-ray Crystal Structure of the Compound 14u–PRMT3 Complex
To better understand structural determinants for the increased potency of compound 14u, we solved the crystal structure of PRMT3 in complex with compound 14u. Similar to the binding mode of compound 1 reported previously,57 compound 14u binds at the allosteric site located at the interface between the two subunits of the PRMT3 homodimer (Figure 3A). The benzothiadiazole ring and urea moiety engage hydrogen bonds with surrounding side chains (Figure 3B). The benzoyl group is stacked against the guanidinium group of R396, which may explain, in part, the increased binding affinity over that of the parent compound. Nevertheless, we were surprised to observe that the additional carbonyl group is not better exploited in the crystal structure.
Figure 3.

X-ray crystal structure of PRMT3 in complex with the inhibitor 14u (PDB code 4HSG). (A) Compound 14u binds at the allosteric site of PRMT3 and contacts both subunits of the homodimer, as was observed with compound 1.57 (B) Critical interactions include hydrogen bonds with T466, E422, and R396. The letter preceding the residue number indicates the PRMT3 chain (A or B).
Modeling of Compound 14u Binding to PRMT3 Identifies a Potential Hydrogen Bond
Considering the significant increase in potency accompanying the introduction of a carbonyl group to the linker moiety, we were surprised by the absence of a hydrogen bond at this position in the crystal structure. We also noted that alternate rotameric states of K392 could bring its side chain within hydrogen-bonding distance of compound 14u’s carbonyl oxygen. To see whether such a conformation was energetically relevant, we ran a Monte Carlo-based energy minimization of the K392 side chain and found that the second lowest energy conformation was indeed forming a hydrogen bond with compound 14u, within less than 0.5 kcal/mol of the lowest energy state (Figure 4). This suggests that K392 oscillates between two conformational states that are nearly equivalent energetically, one forming a hydrogen bond with compound 14u, the other not.
Figure 4.

Monte Carlo energy minimization identifies a low-energy conformation of K392, which interacts with the carbonyl oxygen of compound 14u via a hydrogen bond. Three independent energy minimization simulations (conformational samplings 1–3) converge toward similar low-energy conformations for K392. Conformational states placing the ε-nitrogen of K392 within hydrogen-bonding distance of 14u are listed in bold. The four lowest energy states from sampling 1 (A–D) and the fourth state from sampling 2 (E) are shown.
CONCLUSION
We designed, synthesized, and evaluated biochemically a series of novel compounds that explore three regions of the scaffold represented by compound 1. The key SAR results revealed by these studies include the following: (1) Modifications to the LHS benzothiadiazole moiety were not tolerated, which is consistent with the key finding revealed by the compound 1–PRMT3 X-ray cocrystal structure that the benzothiadiazole ring fits tightly in the allosteric pocket. (2) Similarly, modifications to the middle urea moiety were not tolerated, again consistent with the findings by the X-ray cocrystal structure. (3) On the other hand, modifications to the RHS cyclohexenylethyl moiety were well tolerated. Our extensive exploration of the RHS cyclohexenylethyl group resulted in the discovery of multiple PRMT3 inhibitors with submicromolar potencies. Among them, compound 14u was selective for PRMT3 over 15 other PRMTs, PKMTs, and DNMT1. The X-ray crystal structure of the compound 14u–PRMT3 complex confirmed that this inhibitor interacts with the same allosteric binding site. Taken together, these studies provide the first experimental evidence that the allosteric binding site of PRMT3 can be exploited to generate potent and selective inhibitors.
EXPERIMENTAL SECTION
Chemistry General Procedures
HPLC spectra for all compounds were acquired using an Agilent 6110 series system with a UV detector set to 254 nm. Samples were injected (5 μL) onto an Agilent Eclipse Plus, 4.6 × 50 mm, 1.8 μM, C18 column at room temperature. Method 1: A linear gradient from 50% to 100% B (MeOH + 0.1% acetic acid) in 5.0 min was followed by pumping 100% B for another 2 min with A being H2O + 0.1% acetic acid. Method 2: A linear gradient from 10% to 100% B (MeOH + 0.1% acetic acid) in 5.0 min was followed by pumping 100% B for another 2 min with A being H2O + 0.1% acetic acid. The flow rate was 1.0 mL/min. Mass spectrometry (MS) data were acquired in positive ion mode using an Agilent 6110 single-quadrupole mass spectrometer with an electrospray ionization (ESI) source. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury spectrometer at 400 MHz for proton (1H NMR) and 100 MHz for carbon (13C NMR); chemical shifts are reported in parts per million (δ). Preparative HPLC was performed on an Agilent Prep 1200 series with a UV detector set to 254 nm. Samples were injected onto a Phenomenex Luna, 75 × 30 mm, 5 μM, C18 column at room temperature. The flow rate was 30 mL/min. A linear gradient was used with 10% (or 50%) MeOH (A) in 0.1% TFA in H2O (B) to 100% MeOH (A). HPLC was used to establish the purity of target compounds; all compounds had >95% purity using the HPLC methods described above. High-resolution (positive ion) mass spectrometry (HRMS) for compounds 14u and 24 was performed using a Thermo LTqFT mass spectrometer under FT control at 100 000 resolution.
Synthesis of compounds 1–3 was reported previously.57
1-(1H-Benzo[d][1,2,3]triazol-6-yl)-3-(2-cyclohexylethyl)urea (4)
To a solution of 6-amino-1,2,3-benzotriazole (72 mg, 0.538 mmol) in DMF (2.0 mL) was added N,N′-carbonyldiimidazole (CDI; 106 mg, 0.654 mmol), and the resulting mixture was stirred for 5 h at rt. Cyclohexylethylamine (109 mg, 0.861 mmol) was then added. After being stirred for 6 h at rt, the resulting mixture was purified by preparative HPLC (50–100% methanol/0.1% TFA in H2O) to afford the title compound 4 as a white solid (65 mg, 42%): 1H NMR (400 MHz, d6-DMSO) δ 15.25 (s, 1H), 8.73 (s, 1H), 8.08 (s, 1H), 7.84 (d, J = 9.2 Hz, 1H), 7.05 (d, J = 8.5 Hz, 1H), 6.16 (s, 1H), 3.17–3.08 (m, 2H), 1.81–1.55 (m, 5H), 1.42–1.05 (m, 6H), 0.97–0.81 (m, 2H); HPLC (method 2) 95%, tR 5.51 min; MS (ESI) m/z 288 [M + H]+.
1-(2-Cyclohexylethyl)-3-(1H-indazol-6-yl)urea (5)
The procedure used for preparation of compound 4 was followed for synthesis of compound 5. The title compound 5 was obtained as a white solid (122 mg, 51%): 1H NMR (400 MHz, d6-DMSO) δ 12.69 (s, 1H), 8.51 (s, 1H), 7.90 (s, 1H), 7.88 (s, 1H), 7.55 (d, J = 8.6 Hz, 1H), 6.81 (dd, J = 8.7, 1.7 Hz, 1H), 6.09 (t, J = 5.5 Hz, 1H), 3.19–3.06 (m, 2H), 1.86–1.50 (m, 5H), 1.44–1.04 (m, 6H), 0.96–0.82 (m, 2H); HPLC (method 2) 96%, tR 5.59 min; MS (ESI) m/z 287 [M + H]+.
1-(2-Cyclohexylethyl)-3-(1H-indazol-5-yl)urea (6)
The procedure used for preparation of compound 4 was followed for synthesis of compound 6. The title compound 6 was obtained as a white solid (78 mg, 47%): 1H NMR (400 MHz, d4-MeOH) δ 7.95 (d, J = 0.8 Hz, 1H), 7.78–7.75 (m, 1H), 7.45 (d, J = 8.9 Hz, 1H), 7.30 (dd, J = 8.9, 2.0 Hz, 1H), 3.26–3.21 (m, 2H), 1.83–1.62 (m, 5H), 1.47–1.13 (m, 6H), 1.02–0.91 (m, 2H); 13C NMR (100 MHz, d4-MeOH) δ 159.0, 138.7, 134.4, 134.2, 124.5, 123.5, 111.6, 111.3, 38.8, 38.6, 36.6, 34.4 (two carbons), 27.7, 27.4 (two carbons); HPLC (method 2) 98%, tR 5.45 min; MS 287 [M + H]+.
1-(2-Cyclohexylethyl)-3-(2-oxo-2,3-dihydrobenzo[d]oxazol-6-yl)urea (7)
The procedure used for preparation of compound 4 was followed for synthesis of compound 7. The title compound 7 was obtained as a white solid (138 mg, 76%): 1H NMR (400 MHz, d6-DMSO) δ 11.36 (s, 1H), 8.40 (s, 1H), 7.53 (d, J = 1.6 Hz, 1H), 7.00–6.85 (m, 2H), 6.04 (t, J = 5.6 Hz, 1H), 3.15–3.03 (m, 2H), 1.75–1.54 (m, 5H), 1.38–1.06 (m, 6H), 0.97–0.80 (m, 2H); 13C NMR (100 MHz, d6-DMSO) δ 155.7, 155.1, 143.89, 136.1, 124.4, 113.5, 109.9, 100.8, 37.7, 37.2, 35.0, 33.1 (two carbons), 26.6, 26.2 (two carbons); HPLC (method 1) 100%, tR 4.59 min; MS (ESI) m/z 304 [M + H]+.
6-Amino-1,2,3-benzothiadiazole (8)
6-Amino-1,2,3-benzothia-diazole (8) was synthesized according to previously reported procedures.57
3 - (Benzo [d ] [1,2,3] thiadiazol - 6- ylamino) - 4 - ((2 -cyclohexylethyl)amino)cyclobut-3-ene-1,2-dione (10)
To a stirred solution of 3,4-diethoxycyclobut-3-ene-1,2-dione (0.26 g, 1.5 mmol, 1.2 equiv) and zinc trifluoromethanesulfonate (59 mg, 0.12 mmol) in ethanol (4.0 mL) at room temperature was added 8 (184 mg, 1.22 mmol). After the solution was stirred for 1 h, a white precipitate was formed, which was centrifuged to remove the ethanol and washed with ethanol (2 mL) twice, yielding the intermediate as a white solid (309 mg, 92%). A mixture of the intermediate (67 mg, 0.244 mmol) and 2-cyclohexylethylamine (32 mg, 0.250 mmol) in 0.2 mL of i-PrOH was heated by microwave irradiation to 120 °C for 10 min in a sealed tube. After concentration in vacuo, the crude product was purified by preparative HPLC with a gradient from 50% MeOH (A) in 0.1% TFA in H2O (B) to 100% MeOH (A) to afford the title compound 10 as a white solid (74 mg, 85%): 1H NMR (400 MHz, d6-DMSO) δ 10.12 (s, 1H), 8.62 (d, J = 9.0 Hz, 1H), 8.25 (s, 1H), 7.93–7.70 (m, 2H), 3.74–3.57 (m, 2H), 1.83–1.55 (m, 5H), 1.49 (q, J = 7.0 Hz, 2H), 1.43–1.28 (m, 1H), 1.29–1.05 (m, 3H), 1.01–0.82 (m, 2H); 13C NMR (100 MHz, d6-DMSO) δ 184.9, 180.1, 169.8, 162.6, 153.9, 142.4, 140.6, 124.3, 119.3, 106.5, 41.6, 38.0, 34.1, 32.5 (two carbons), 26.0, 25.7 (two carbons); HPLC (method 1) 96%, tR 5.39 min; MS (ESI) m/z 357 [M + H]+.
3-(Benzo[d][1,2,3]thiadiazol-6-ylamino)-4-((2-oxo-2-phenylethyl)amino)cyclobut-3-ene-1,2-dione (11)
The procedure used for preparation of compound 10 was followed for synthesis of compound 11. The title compound 11 was obtained as a white solid (80 mg, 78%): 1H NMR (400 MHz, d6-DMSO) δ 10.53 (s, 1H), 8.66 (d, J = 9.0 Hz, 1H), 8.32 (s, 1H), 8.17 (s, 1H), 8.05–7.96 (m, 2H), 7.87 (d, J = 8.9 Hz, 1H), 7.77–7.68 (m, 1H), 7.60 (t, J = 7.7 Hz, 2H), 5.29 (d, J = 5.6 Hz, 2H); HPLC (method 1) 98%, tR 3.66 min; MS (ESI) m/z 365 [M + H]+.
(E)-1-(Benzo[d][1,2,3]thiadiazol-6-yl)-2-cyano-3-(2-cyclohexylethyl)guanidine (12)
A mixture of 8 (65 mg, 0.430 mmol) and diphenyl cyanocarbonimidate (82 mg, 0.344 mmol) in 0.4 mL of i-PrOH was heated by microwave irradiation to 100 °C for 60 min in a sealed tube. To the resulting mixture was added 2-cyclohexylethylamine (109 mg, 0.86 mmol), and then the resulting mixuture was heated by microwave irradiation to 100 °C for another 30 min. The resulting residue was purified by preparative HPLC with a gradient from 50% MeOH (A) in 0.1% TFA in H2O (B) to 100% MeOH (A) to afford the title compound 12 as a white solid (43 mg, 38% in two steps): 1H NMR (400 MHz, d6-DMSO) δ 9.50 (s, 1H), 8.61 (d, J = 9.0 Hz, 1H), 8.22 (s, 1H), 7.65 (t, J = 5.6 Hz, 1H), 7.57 (dd, J = 9.0, 1.9 Hz, 1H), 3.33–3.25 (m, 2H), 1.77–1.56 (m, 5H), 1.48–1.39 (m, 2H), 1.36–1.06 (m, 4H), 0.98–0.82 (m, 2H); HPLC (method 1) 100%, tR 5.12 min; MS (ESI) m/z 329 [M + H]+.
N-(5-(2-Cyclohexylethyl)-4H-1,2,4-triazol-3-yl)benzo[d]-[1,2,3]thiadiazol-6-amine (13)
To a stirred solution of 8 (82 mg, 0.54 mmol) in THF (2 mL) was added cyanogen bromide solution (0.11 mL, 5.0 M in CH3CN) at rt. The resulting mixture was stirred overnight at 40 °C. After removal of the solvents, to the solution of the residue in THF (1 mL) were added 2 drops of HCl solution in i-PrOH (5 N) and 3-cyclohexylpropanehydrazide (184 mg, 1.08 mmol). The resulting mixture was heated by microwave irradiation to 160 °C for 30 min. After being concentrated, the residue was purified by preparative HPLC with a gradient from 50% MeOH (A) in 0.1% TFA in H2O (B) to 100% MeOH (A) to afford the title compound 13 as a white solid (57 mg, 32% in two steps): 1H NMR (400 MHz, d4-MeOH) δ 8.50 (d, J = 2.1 Hz, 1H), 8.44 (d, J = 9.1 Hz, 1H), 7.59 (dd, J = 9.1, 2.1 Hz, 1H), 2.87–2.79 (m, 2H), 1.86–1.71 (m, 4H), 1.71–1.63 (m, 3H), 1.37–1.14 (m, 4H), 1.06–0.92 (m, 2H); 13C NMR (100 MHz, d4-MeOH) δ 157.8, 157.3, 154.8, 144.6, 144.0, 124.6, 119.9, 104.6, 38.4, 36.06, 34.0 (two carbons), 27.6, 27.3 (two carbons), 24.3; HPLC (method 1) 97%, tR 5.76 min; MS (ESI) m/z 329 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-(tetrahydro-2H-pyran-4-yl)ethyl)urea (14a)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14a. The title compound 14a was obtained as a white solid (52 mg, 47%): 1H NMR (400 MHz, d6-DMSO) δ 9.12 (s, 1H), 8.52 (d, J = 1.9 Hz, 1H), 8.50 (d, J = 9.1 Hz, 1H), 7.59 (dd, J = 9.1, 2.0 Hz, 1H), 6.41 (t, J = 5.6 Hz, 1H), 3.82 (dd, J = 11.3, 3.6 Hz, 2H), 3.30–3.21 (m, 2H), 3.21–3.11 (m, 2H), 1.66–1.47 (m, 3H), 1.40 (q, J = 6.8 Hz, 2H), 1.23–1.07 (m, 2H); 13C NMR (100 MHz, d6-DMSO) δ 154.8, 153.2, 142.3, 142.2, 123.4, 119.1, 105.1, 67.0 (two carbons), 36.7, 36.3, 32.6 (two carbons), 31.9; HPLC (method 1) 99%, tR 3.23 min; MS (ESI) m/z 307 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-(piperidin-1-yl)ethyl)-urea (14b)
The procedure used for preparation of compound 4 was followed for synthesis of 14b. The title compound 14b was obtained as a white solid (48 mg, 45%): 1H NMR (400 MHz, d6-acetone) δ 10.04 (s, 1H), 8.65 (d, J = 1.9 Hz, 1H), 8.48 (d, J = 9.1 Hz, 1H), 8.00 (s, 1H), 7.67 (dd, J = 9.1, 2.0 Hz, 1H), 3.92–3.83 (m, 2H), 3.77–3.68 (m, 2H), 3.50–3.42 (m, 2H), 3.20–3.06 (m, 2H), 2.03–1.93 (m, 4H), 1.92–1.83 (m, 1H), 1.66–1.52 (m, 1H); 13C NMR (100 MHz, d6-acetone) δ 154.9, 143.5, 124.4, 120.1, 120.0, 106.2, 106.1, 59.6, 59.5, 54.6, 54.5, 35.3, 23.9, 22.5; HPLC (method 2) 98%, tR 3.08 min; MS (ESI) m/z 306 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-morpholinoethyl)-urea (14c)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14c. The title compound 14c was obtained as a white solid (53 mg, 47%): 1H NMR (400 MHz, d4-MeOH) δ 8.51 (d, J = 1.9 Hz, 1H), 8.45 (d, J = 9.1 Hz, 1H), 7.56 (dd, J = 9.1, 2.0 Hz, 1H), 4.15–4.00 (m, 2H), 3.81 (t, J = 11.6 Hz, 2H), 3.74–3.61 (m, 4H), 3.35 (t, J = 5.6 Hz, 2H), 3.20 (t, J = 10.4 Hz, 2H); 13C NMR (100 MHz, d4-MeOH) δ 158.4, 155.5, 144.0, 142.7, 124.5, 120.9, 107.4, 65.1 (two carbons), 59.4, 53.6 (two carbons), 35.6; HPLC (method 2) 99%, tR 2.63 min; MS (ESI) m/z 308 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(3-morpholinopropyl)-urea (14d)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14d. The title compound 14d was obtained as a white solid (42 mg, 39%): 1H NMR (400 MHz, d4-MeOH) δ 8.48–8.46 (m, 1H), 8.44 (d, J = 9.1 Hz, 1H), 7.55 (dd, J = 9.1, 2.0 Hz, 1H), 4.14–4.02 (m, 2H), 3.79 (t, J = 12.0 Hz, 2H), 3.58–3.47 (m, 2H), 3.37 (t, J = 6.5 Hz, 2H), 3.29–3.23 (m, 2H), 3.16 (td, J = 12.4, 3.3 Hz, 2H), 2.07–1.97 (m, 2H); 13C NMR (100 MHz, d4-MeOH) δ 158.2, 155.4, 144.0, 142.9, 124.5, 120.8, 107.1, 65.1 (two carbons), 56.0, 53.2 (two carbons), 37.6, 25.8; HPLC (method 2) 98%, tR 2.83 min; MS (ESI) m/z 322 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-(pyrrolidin-1-yl)-ethyl)urea (14e)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14e. The title compound 14e was obtained as a white solid (48 mg, 45%): 1H NMR (400 MHz, d4-MeOH) δ 8.50 (d, J = 2.0 Hz, 1H), 8.44 (d, J = 9.1 Hz, 1H), 7.56 (dd, J = 9.1, 2.0 Hz, 1H), 3.90–3.75 (m, 2H), 3.62 (t, J = 5.7 Hz, 2H), 3.39 (t, J = 5.7 Hz, 2H), 3.23–3.06 (m, 2H), 2.25–2.10 (m, 2H), 2.11–1.95 (m, 2H); 13C NMR (100 MHz, d4-MeOH) δ 158.0, 155.4, 143.9, 142.8, 124.5, 120.8, 107.2, 56.8, 55.6 (two carbons), 37.5, 23.9 (two carbons); HPLC (method 2) 100%, tR 2.69 min; MS (ESI) m/z 292 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(cyclohexylmethyl)urea (14f)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14f. The title compound 14f was obtained as a white solid (62 mg, 48%): 1H NMR (400 MHz, d6-DMSO) δ 9.07 (s, 1H), 8.55–8.42 (m, 2H), 7.58 (dd, J = 9.0, 2.1 Hz, 1H), 6.43 (t, J = 5.8 Hz, 1H), 2.98 (t, J = 6.3 Hz, 2H), 1.73–1.64 (m, 4H), 1.64–1.54 (m, 1H), 1.47–1.35 (m, 1H), 1.26–1.07 (m, 3H), 0.97–0.85 (m, 2H); 13C NMR (100 MHz, d6-DMSO) δ 154.8, 153.2, 142.3, 142.2, 123.4, 119.1, 105.1, 45.3, 37.9, 30.3 (two carbons), 26.04, 25.4 (two carbons); HPLC (method 1) 98%, tR 5.28 min; MS (ESI) m/z 291 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-ethylurea (14g)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14g. The title compound 14g was obtained as a white solid (67 mg, 55%): 1H NMR (400 MHz, d6-DMSO) δ 9.13 (s, 1H), 8.52 (d, J = 1.8 Hz, 1H), 8.50 (d, J = 9.1 Hz, 1H), 7.59 (dd, J = 9.1, 2.0 Hz, 1H), 6.40 (t, J = 5.5 Hz, 1H), 3.22–3.09 (m, 2H), 1.08 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, d6-DMSO) δ 154.7, 153.2, 142.3, 142.2, 123.4, 119.1, 105.2, 34.1, 15.3; HPLC (method 1) 100%, tR 2.03 min; MS (ESI) m/z 223 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(3,3-dimethylbutyl)urea (14h)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14h. The title compound 14h was obtained as a white solid (54 mg, 49%): 1H NMR (400 MHz, d6-DMSO) δ 9.13 (s, 1H), 8.52 (d, J = 1.7 Hz, 1H), 8.50 (d, J = 9.0 Hz, 1H), 7.59 (dd, J = 9.1, 2.0 Hz, 1H), 6.33 (t, J = 5.5 Hz, 1H), 3.20–3.07 (m, 2H), 1.45–1.32 (m, 2H), 0.92 (s, 9H); 13C NMR (100 MHz, d6-DMSO) δ 155.1, 153.7, 142.8, 142.7, 123.9, 119.6, 105.6, 43.8, 36.3, 30.0, 29.7 (three carbons); HPLC (method 1) 97%, tR 5.01 min; MS (ESI) m/z 279 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-benzylurea (14i)
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-benzylurea (14i) was synthesized according to previously reported procedures.57
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(pyridin-2-ylmethyl)urea (14j)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14j. The title compound 14j was obtained as a white solid (57 mg, 46%): 1H NMR (400 MHz, d6-DMSO) δ 9.83 (s, 1H), 8.74 (dd, J = 5.4, 0.8 Hz, 1H), 8.57–8.48 (m, 2H), 8.30 (td, J = 7.9, 1.6 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.75–7.70 (m, 1H), 7.64 (dd, J = 9.1, 2.0 Hz, 1H), 7.42 (t, J = 5.5 Hz, 1H), 4.63 (d, J = 5.3 Hz, 2H); 13C NMR (100 MHz, d6-DMSO) δ 156.5, 155.2, 153.5, 143.9, 143.0, 142.3, 141.8, 124.2, 123.8, 123.5, 119.3, 105.7, 42.5; HPLC (method 2) 99%, tR 4.15 min; MS (ESI) m/z 286 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(pyridin-3-ylmethyl)urea (14k)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14k. The title compound 14k was obtained as a white solid (45 mg, 41%): 1H NMR (400 MHz, d6-DMSO) δ 9.72 (s, 1H), 8.84 (d, J = 1.4 Hz, 1H), 8.78 (d, J = 4.8 Hz, 1H), 8.55 (d, J = 1.9 Hz, 1H), 8.52 (d, J = 9.1 Hz, 1H), 8.41 (d, J = 8.1 Hz, 1H), 7.95 (dd, J = 8.0, 5.6 Hz, 1H), 7.63 (dd, J = 9.1, 2.0 Hz, 1H), 7.42 (t, J = 5.9 Hz, 1H), 4.52 (d, J = 5.8 Hz, 2H); 13C NMR (100 MHz, d6-DMSO) δ 155.2, 153.4, 143.0, 142.3, 142.0, 141.9, 141.8, 139.6, 126.3, 123.5, 119.3, 105.6, 40.3; HPLC (method 2) 99%, tR 3.67 min; MS (ESI) m/z 286 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(pyridin-4-ylmethyl)urea (14l)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14l. The title compound 14l was obtained as a white solid (51 mg, 44%): 1H NMR (400 MHz, d6-DMSO) δ 9.87 (s, 1H), 8.83 (d, J = 6.6 Hz, 2H), 8.58–8.49 (m, 2H), 7.91 (d, J = 6.5 Hz, 2H), 7.65 (dd, J = 9.1, 2.0 Hz, 1H), 7.56 (t, J = 5.9 Hz, 1H), 4.61 (d, J = 5.8 Hz, 2H); 13C NMR (100 MHz, d6-DMSO) δ 160.0, 155.2, 153.4, 142.5 (two carbons), 142.3, 141.9, 124.4 (two carbons), 123.5, 119.3, 105.7, 42.6; HPLC (method 2) 97%, tR 3.19 min; MS (ESI) m/z 286 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(4-(dimethylamino)-benzyl)urea (14m)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14m. The title compound 14m was obtained as a white solid (31 mg, 36%): 1H NMR (400 MHz, d6-DMSO) δ 9.39 (s, 1H), 8.54 (d, J = 1.9 Hz, 1H), 8.51 (d, J = 9.1 Hz, 1H), 7.61 (dd, J = 9.1, 2.0 Hz, 1H), 7.33 (d, J = 8.6 Hz, 2H), 7.16 (d, J = 8.1 Hz, 2H), 7.02 (t, J = 5.1 Hz, 1H), 4.29 (d, J = 5.1 Hz, 2H), 3.02 (s, 6H); HPLC (method 2) 97%, tR 4.74 min; MS (ESI) m/z 328 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(4-methoxybenzyl)urea (14n)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14n. The title compound 14n was obtained as a white solid (61 mg, 53%): 1H NMR (400 MHz, d6-DMSO) δ 9.22 (s, 1H), 8.54 (d, J = 1.8 Hz, 1H), 8.51 (d, J = 9.1 Hz, 1H), 7.60 (dd, J = 9.1, 2.0 Hz, 1H), 7.30–7.21 (m, 2H), 6.93–6.86 (m, 2H), 6.83 (t, J = 5.8 Hz, 1H), 4.26 (d, J = 5.8 Hz, 2H), 3.73 (s, 3H); 13C NMR (100 MHz, d6-DMSO) δ 158.2, 154.8, 153.3, 142.3, 142.1, 131.8, 128.6 (two carbons), 123.5, 119.2, 113.7 (two carbons), 105.3, 55.1, 42.3; HPLC (method 1) 99%, tR 3.77 min; MS (ESI) m/z 315 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(4-nitrobenzyl)urea (14o)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14o. The title compound 14o was obtained as a white solid (26 mg, 35%): 1H NMR (400 MHz, d6-DMSO) δ 9.43 (s, 1H), 8.54 (d, J = 1.9 Hz, 1H), 8.52 (d, J = 9.1 Hz, 1H), 8.26–8.16 (m, 2H), 7.63 (dd, J = 9.1, 2.1 Hz, 1H), 7.61–7.54 (m, 2H), 7.08 (t, J = 6.0 Hz, 1H), 4.48 (d, J = 6.0 Hz, 2H); HPLC (method 1) 97%, tR 3.59 min; MS (ESI) m/z 330 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(4-(methylsulfonyl)-benzyl)urea (14p)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14p. The title compound 14p was obtained as a white solid (37 mg, 39%): 1H NMR (400 MHz, d6-DMSO) δ 9.40 (s, 1H), 8.55 (d, J = 2.0 Hz, 1H), 8.52 (d, J = 9.1 Hz, 1H), 7.93–7.87 (m, 2H), 7.63 (dd, J = 9.1, 2.0 Hz, 1H), 7.58 (d, J = 8.3 Hz, 2H), 7.06 (t, J = 6.0 Hz, 1H), 4.46 (d, J = 6.0 Hz, 2H), 3.19 (s, 3H); 13C NMR (100 MHz, d6-DMSO) δ 154.9, 153.4, 146.4, 142.3, 141.9, 139.2, 127.8 (two carbons), 127.1 (two carbons), 123.5, 119.3, 105.6, 43.6, 42.5; HPLC (method 1) 100%, tR 1.92 min; MS (ESI) m/z 363 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(4-cyanobenzyl)urea (14q)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14q. The title compound 14q was obtained as a white solid (34 mg, 41%): 1H NMR (400 MHz, d6-DMSO) δ 9.40 (s, 1H), 8.54 (d, J = 2.0 Hz, 1H), 8.52 (d, J = 9.1 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.62 (dd, J = 9.1, 2.0 Hz, 1H), 7.51 (d, J = 8.1 Hz, 2H), 7.05 (t, J = 6.0 Hz, 1H), 4.43 (d, J = 6.0 Hz, 2H); HPLC (method 1) 97%, tR 4.86 min; MS (ESI) m/z 310 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(4-(trifluoromethyl)-benzyl)urea (14r)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14r. The title compound 14r was obtained as a white solid (52 mg, 47%): 1H NMR (400 MHz, d6-DMSO) δ 9.38 (s, 1H), 8.55 (d, J = 1.9 Hz, 1H), 8.52 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 8.1 Hz, 2H), 7.62 (dd, J = 9.1, 2.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 2H), 7.04 (t, J = 6.0 Hz, 1H), 4.44 (d, J = 6.0 Hz, 2H); 13C NMR (100 MHz, d6-DMSO) δ 155.0, 153.4, 145.1 (q, J = 1.3 Hz), 142.3, 142.0, 127.7, 127.5 (q, J = 31.6 Hz), 125.2 (q, J = 3.8 Hz),124.4 (q, J = 271.9 Hz), 123.5, 119.3, 105.6, 42.5; HPLC (method 1) 98%, tR 4.92 min; MS (ESI) m/z 353 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(4-fluorobenzyl)urea (14s)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14s. The title compound 14s was obtained as a white solid (41 mg, 48%): 1H NMR (400 MHz, d6-DMSO) δ 9.29 (s, 1H), 8.54 (d, J = 1.8 Hz, 1H), 8.51 (d, J = 9.1 Hz, 1H), 7.61 (dd, J = 9.1, 2.0 Hz, 1H), 7.43–7.31 (m, 2H), 7.23–7.10 (m, 2H), 6.93 (t, J = 6.0 Hz, 1H), 4.32 (d, J = 5.9 Hz, 2H); 13C NMR (100 MHz, d6-DMSO) δ 161.15 (d, J = 242.1 Hz), 154.85, 153.33, 142.28, 142.01, 136.22 (d, J = 3.0 Hz), 129.15 (d, J = 8.2 Hz, two carbons), 123.46, 119.23, 115.01 (d, J = 21.2 Hz, two carbons). 105.44, 42.12; HPLC (method 1) 99%, tR 4.20 min; MS (ESI) m/z 303 [M + H]+.
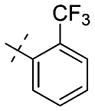
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-(trifluoromethyl)-benzyl)urea (14t)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14t. The title compound 14t was obtained as a white solid (32 mg, 41%): 1H NMR (400 MHz, d6-DMSO) δ 9.43 (s, 1H), 8.55 (d, J = 1.9 Hz, 1H), 8.52 (d, J = 9.1 Hz, 1H), 7.73 (d, J = 7.7 Hz, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.65–7.58 (m, 2H), 7.48 (t, J = 7.5 Hz, 1H), 6.99 (t, J = 6.0 Hz, 1H), 4.54 (d, J = 5.8 Hz, 2H); HPLC (method 1) 99%, tR 4.76 min; MS (ESI) m/z 353 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-oxo-2-phenylethyl)-urea (14u)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14u. The title compound 14u was obtained as a white solid (42 mg, 45%): 1H NMR (400 MHz, d4-MeOH) δ 8.51 (d, J = 1.9 Hz, 1H), 8.46 (d, J = 9.1 Hz, 1H), 8.05 (dt, J = 8.5, 1.7 Hz, 2H), 7.68–7.63 (m, 1H), 7.55 (ddd, J = 9.2, 5.1, 1.9 Hz, 3H), 4.80 (s, 2H); 13C NMR (100 MHz, d4-MeOH) δ 196.7, 157.5, 155.4, 144.1, 143.0, 136.3, 134.9, 130.0 (two carbons), 129.0 (two carbons), 124.6, 120.7, 107.0, 47.9; HPLC (method 1) 97%, tR 3.68 min; MS (ESI) m/z 313 [M + H]+; HRMS calcd. for C15H12N4O2S + H: 313.0759; found: 313.0747 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(3-(4-fluorophenyl)-propyl)urea (14v)
The procedure used for preparation of compound 4 was followed for synthesis of compound 14v. The title compound 14v was obtained as a white solid (42 mg, 45%): 1H NMR (400 MHz, d6-DMSO) δ 9.15 (s, 1H), 8.52 (d, J = 1.9 Hz, 1H), 8.50 (d, J = 9.1 Hz, 1H), 7.60 (dd, J = 9.1, 2.0 Hz, 1H), 7.29–7.22 (m, 2H), 7.14–7.05 (m, 2H), 6.49 (t, J = 5.6 Hz, 1H), 3.18–3.09 (m, 2H), 2.65–2.57 (m, 2H), 1.80–1.69 (m, 2H); 13C NMR (100 MHz, d6-DMSO) δ 160.6 (d, J = 241.0 Hz), 154.8, 153.2, 142.3, 142.2, 137.7 (d, J = 3.1 Hz), 130.0 (d, J = 7.9 Hz, two carbons), 123.4, 119.2, 114.9 (d, J = 20.9 Hz, two carbons), 105.2, 38.6, 31.6, 31.4; HPLC (method 1) 99%, tR 5.02 min; MS (ESI) m/z 331 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-(piperidin-4-yl)ethyl)-urea (15a)
The procedure used for preparation of compound 4 was followed for synthesis of Boc-protected compound 15a, which was purified by flash column chromatography on silica gel (0–10% MeOH/DCM gradient). To a solution of Boc-protected compound 15a (70 mg, 0.172 mmol) in DCM (5 mL) was added TFA (0.5 mL). After being stirred overnight, the resulting mixture was purified by preparative HPLC (10–100% methanol/0.1% TFA in H2O) to afford the title compound 15a as a mono-TFA salt (39 mg, 37% in two steps): 1H NMR (400 MHz, d4-MeOH) δ 8.45 (d, J = 2.0 Hz, 1H), 8.43 (d, J = 9.2 Hz, 1H), 7.53 (dd, J = 9.1, 2.0 Hz, 1H), 3.42–3.34 (m, 2H), 3.33–3.27 (m, 2H), 2.97 (td, J = 12.8, 2.6 Hz, 2H), 2.06–1.97 (m, 2H), 1.79–1.66 (m, 1H), 1.56 (q, J = 6.9 Hz, 2H), 1.48–1.34 (m, 2H); 13C NMR (100 MHz, d4-MeOH) δ 157.6, 155.3, 144.1, 143.2, 124.5, 120.7, 106.8, 45.3 (two carbons), 37.9, 37.4, 32.5, 29.9 (two carbons); HPLC (method 2) 99%, tR 3.13 min; MS (ESI) m/z 306 [M + H]+.
1-(2-(cis-4-Aminocyclohexyl)ethyl)-3-(benzo[d][1,2,3]-thiadiazol-6-yl)urea (15b)
The procedure used for preparation of compound 15a was followed for synthesis of compound 15b. The title compound 15b was obtained as a mono-TFA salt (42 mg, 41% in two steps): 1H NMR (400 MHz, d4-MeOH) δ 8.45 (d, J = 1.9 Hz, 1H), 8.43 (d, J = 9.1 Hz, 1H), 7.53 (dd, J = 9.1, 2.0 Hz, 1H), 3.29–3.23 (m, 3H), 1.87–1.63 (m, 7H), 1.63–1.42 (m, 4H); 13C NMR (100 MHz, d4-MeOH) δ 157.6, 155.3, 144.1, 143.2, 124.5, 120.7, 106.8, 50.1, 38.7, 34.8, 32.6, 27.9 (two carbons), 27.8(two carbons); HPLC (method 2) 98%, tR 3.56 min; MS (ESI) m/z 320 [M + H]+.
1-(2-(trans-4-Aminocyclohexyl)ethyl)-3-(benzo[d][1,2,3]-thiadiazol-6-yl)urea (15c)
The procedure used for preparation of compound 15a was followed for synthesis of compound 15c. The title compound 15c was obtained as a mono-TFA salt (49 mg, 47% in two steps): 1H NMR (400 MHz, d4-MeOH) δ 8.45 (d, J = 1.9 Hz, 1H), 8.43 (d, J = 9.1 Hz, 1H), 7.52 (dd, J = 9.1, 2.0 Hz, 1H), 3.28 (t, J = 7.1 Hz, 2H), 3.09–2.98 (m, 1H), 2.10–2.00 (m, 2H), 1.98–1.90 (m, 2H), 1.53–1.33 (m, 5H), 1.17–1.03 (m, 2H); 13C NMR (100 MHz, d4-MeOH) δ 157.6, 155.3, 144.1, 143.2, 124.5, 120.7, 106.7, 51.6, 38.5, 37.8, 35.1, 31.8 (two carbons), 31.7 (two carbons); HPLC (method 2) 98%, tR 3.43 min; MS (ESI) m/z 320 [M + H]+.
1-(3-(trans-4-Aminocyclohexyl)propyl)-3-(benzo[d][1,2,3]-thiadiazol-6-yl)urea (15d)
The procedure used for preparation of compound 15a was followed for synthesis of compound 15d. The title compound 15d was obtained as a mono-TFA salt (29 mg, 35% in two steps): 1H NMR (400 MHz, d4-MeOH) δ 8.48–8.41 (m, 2H), 7.52 (dd, J = 9.1, 2.0 Hz, 1H), 3.22 (t, J = 7.1 Hz, 2H), 3.02 (tt, J = 11.9, 3.9 Hz, 1H), 2.08–1.97 (m, 2H), 1.97–1.86 (m, 2H), 1.62–1.53 (m, 2H), 1.44–1.25 (m, 5H), 1.13–1.00 (m, 2H); 13C NMR (100 MHz, d4-MeOH) δ 157.6, 155.3, 144.1, 143.2, 124.5, 120.7, 106.8, 51.7, 40.9, 37.3, 34.7, 32.0 (two carbons), 31.8 (two carbons), 28.5; HPLC (method 2) 98%, tR 3.76 min; MS (ESI) m/z 334 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(3-(piperidin-4-yl)-propyl)urea (15e)
The procedure used for preparation of compound 15a was followed for synthesis of compound 15e. The title compound 15e was obtained as a mono-TFA salt (49 mg, 45% in two steps): 1H NMR (400 MHz, d4-MeOH) δ 8.43 (d, J = 2.0 Hz, 1H), 8.40 (d, J = 9.1 Hz, 1H), 7.52 (dd, J = 9.1, 2.0 Hz, 1H), 3.41–3.32 (m, 2H), 3.22 (t, J = 7.0 Hz, 2H), 2.95 (td, J = 12.8, 2.5 Hz, 2H), 1.97–1.89 (m, 2H), 1.66–1.53 (m, 3H), 1.43–1.29 (m, 4H); 13C NMR (100 MHz, d4-MeOH) δ 157.6, 155.2, 144.0, 143.2, 124.4, 120.7, 106.7, 45.3 (two carbons), 40.7, 34.5, 34.1, 30.0 (two carbons), 27.9; HPLC (method 2) 99%, tR 3.32 min; MS (ESI) m/z 320 [M + H]+.
1-(2-Amino-2-phenylethyl)-3-(benzo[d][1,2,3]thiadiazol-6-yl)urea (16)
The procedure used for preparation of compound 15a was followed for synthesis of compound 16. The title compound 16 was obtained as a mono-TFA salt (48 mg, 42% in two steps): 1H NMR (400 MHz, d4-MeOH) δ 8.51–8.47 (m, 1H), 8.45–8.39 (m, 1H), 7.54 (dd, J = 9.1, 2.0 Hz, 1H), 7.52–7.41 (m, 5H), 4.53 (dd, J = 7.9, 5.5 Hz, 1H), 3.79 (dd, J = 14.5, 8.0 Hz, 1H), 3.67 (dd, J = 14.5, 5.5 Hz, 1H); 13C NMR (100 MHz, d4-MeOH) δ 157.8, 155.4, 143.9, 142.8, 136.3, 130.5, 130.4 (two carbons), 128.3 (two carbons), 124.5, 120.8, 107.1, 56.8, 44.7; HPLC (method 2) 99%, tR 3.49 min; MS (ESI) m/z 314 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-hydroxy-2-phenylethyl)urea (17)
The procedure used for preparation of compound 4 was followed for synthesis of compound 17. The title compound 17 was obtained as a white solid (52 mg, 50%): 1H NMR (400 MHz, d4-MeOH) δ 8.44–8.41 (m, 1H), 8.41–8.36 (m, 1H), 7.45 (dd, J = 9.0, 2.0 Hz, 1H), 7.43–7.39 (m, 2H), 7.37–7.31 (m, 2H), 7.29–7.23 (m, 1H), 4.80 (dd, J = 7.9, 4.5 Hz, 1H), 3.56 (dd, J = 13.7, 4.5 Hz, 1H), 3.36 (dd, J = 13.7, 7.9 Hz, 1H); 13C NMR (100 MHz, d4-MeOH) δ 157.5, 155.2, 144.0, 143.9, 143.0, 129.4 (two carbons), 128.6, 127.1 (two carbons), 124.4, 120.6, 106.8, 73.9, 48.3; HPLC (method 1) 99%, tR 3.27 min; MS (ESI) m/z 315 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2,2-difluoro-2-phenylethyl)urea (18)
The procedure used for preparation of compound 4 was followed for synthesis of compound 18. The title compound 18 was obtained as a white solid (42 mg, 38%): 1H NMR (400 MHz, d6-DMSO) δ 9.26 (s, 1H), 8.55–8.48 (m, 2H), 7.63–7.55 (m, 3H), 7.53–7.47 (m, 3H), 6.81 (t, J = 6.3 Hz, 1H), 3.95 (td, J = 15.0, 6.3 Hz, 2H); HPLC (method 1) 98%, tR 4.15 min; MS (ESI) m/z 335 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-(4-fluorophenyl)-2-hydroxyethyl)urea (19)
To a stirred solution of 4-fluorobenzalde-hyde (1.24 g, 10.0 mmol) and nitromethane (2.14 mL, 40.0 mmol) in ethanol (40 mL) was added 4.2 mL of 10% aq NaOH at 0 °C. The resulting mixture was stirred for 2 h at 0 °C and quenched with acetic acid (0.61 mL, 10.5 mmol). After addition of 30 mL of saturated aq NaHCO3 and extraction with ethyl acetate (100 mL × 3), the combined organic phase was dried with sodium sulfate, filtered, and concentrated to afford the desired crude product 1-(4-fluorophenyl)-2-nitroethanol. A stirred suspension of the intermediate and palladium carbon in methanol (100 mL) was treated with hydrogen at 1 atm overnight at rt. The product 2-amino-1-(4-fluorophenyl)ethanol was obtained after filtration and concentration. The procedure used for preparation of compound 4 was followed for synthesis of compound 19 from 2-amino-1-(4-fluorophenyl)ethanol. The title compound 19 was obtained as a white solid (47 mg, 49%): 1H NMR (400 MHz, d4-MeOH) δ 8.46 (dd, J = 2.0, 0.5 Hz, 1H), 8.43 (dd, J = 9.0, 0.5 Hz, 1H), 7.49 (dd, J = 9.0, 2.0 Hz, 1H), 7.46–7.40 (m, 2H), 7.12–7.03 (m, 2H), 4.80 (dd, J = 7.7, 4.5 Hz, 1H), 3.53 (dd, J = 13.7, 4.5 Hz, 1H), 3.35 (dd, J = 13.8, 7.8 Hz, 1H); HPLC (method 1) 98%, tR 3.59 min; MS (ESI) m/z 333 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-(2-fluorophenyl)-2-hydroxyethyl)urea (20)
The procedure used for preparation of compound 19 was followed for synthesis of compound 20. The title compound 20 was obtained as a white solid (57 mg, 54%): 1H NMR (400 MHz, d4-MeOH) δ 8.45–8.37 (m, 2H), 7.58 (td, J = 7.5, 1.6 Hz, 1H), 7.46 (dd, J = 9.0, 2.0 Hz, 1H), 7.29 (tdd, J = 7.3, 5.3, 1.8 Hz, 1H), 7.19 (td, J = 7.5, 0.8 Hz, 1H), 7.06 (ddd, J = 10.5, 8.2, 0.9 Hz, 1H), 5.13 (dd, J = 7.3, 4.5 Hz, 1H), 3.60 (dd, J = 13.7, 4.5 Hz, 1H), 3.44 (dd, J = 13.7, 7.4 Hz, 1H); 13C NMR (100 MHz, d4-MeOH) δ 159.9 (d, J = 244.4 Hz), 156.14, 153.88, 142.65, 141.60, 129.4 (d, J = 13.7 Hz), 128.9 (d, J = 8.3 Hz), 127.5 (d, J = 4.4 Hz), 124.0 (d, J = 3.5 Hz), 123.1, 119.2, 114.7 (d, J = 22.0 Hz), 105.4, 66.4, 45.6; HPLC (method 1) 100%, tR 3.26 min; MS (ESI) m/z 333 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-cyclohexyl-2-hydroxyethyl)urea (21)
The procedure used for preparation of compound 19 was followed for synthesis of compound 21. The title compound 21 was obtained as a white solid (57 mg, 48%): 1H NMR (400 MHz, d4-MeOH) δ 8.46 (dd, J = 2.0, 0.5 Hz, 1H), 8.42 (dd, J = 9.1, 0.5 Hz, 1H), 7.49 (dd, J = 9.1, 2.0 Hz, 1H), 3.51 (dd, J = 13.6, 3.4 Hz, 1H), 3.41 (ddd, J = 8.2, 6.4, 3.4 Hz, 1H), 3.09 (dd, J = 13.6, 8.2 Hz, 1H), 1.97–1.87 (m, 1H), 1.84–1.63 (m, 4H), 1.47–1.35 (m, 1H), 1.35–0.99 (m, 5H); 13C NMR (100 MHz, d4-MeOH) δ 157.7, 155.3, 144.1, 143.1, 124.5, 120.6, 106.7, 76.0, 44.5, 43.2, 30.3, 29.4, 27.6, 27.4, 27.2; HPLC (method 1) 100%, tR 4.51 min; MS (ESI) m/z 321 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-cyclohexyl-2-oxoethyl)urea (22)
To a solution of compound 21 (46 mg, 0.143 mmol) in 2.2 mL of DCM/DMSO (2:1) were added triethylamine (0.10 mL, 0.72 mmol) and Py·SO3 (92 mg, 0.574 mmol) at rt. The resulting mixture was stirred for 1 h at rt, quenched with 1.0 mL of water, concentrated, and purified by preparative HPLC (50–100% methanol/0.1% TFA in H2O) to afford the title compound 23 as a white solid (35 mg, 78%): 1H NMR (400 MHz, d6-DMSO) δ 9.50 (s, 1H), 8.54–8.49 (m, 2H), 7.59 (dd, J = 9.2, 2.0 Hz, 1H), 6.60 (t, J = 5.2 Hz, 1H), 4.12 (d, J = 5.2 Hz, 2H), 2.49–2.42 (m, 1H), 1.86–1.55 (m, 5H), 1.33–1.09 (m, 5H); HPLC (method 1) 98%, tR 4.38 min; MS (ESI) m/z 319 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-(4-fluorophenyl)-2-oxoethyl)urea (23)
The procedure used for preparation of compound 22 was followed for synthesis of compound 23. The title compound 23 was obtained as a white solid (36 mg, 76%): 1H NMR (400 MHz, d6-DMSO) δ 9.60 (s, 1H), 8.56–8.51 (m, 2H), 8.17–8.06 (m, 2H), 7.63 (dd, J = 9.1, 2.0 Hz, 1H), 7.44–7.34 (m, 2H), 6.78 (t, J = 5.2 Hz, 1H), 4.72 (d, J = 5.2 Hz, 2H); 13C NMR (100 MHz, d6-DMSO) δ 194.5, 165.2 (d, J = 252.2 Hz), 154.8, 153.4, 142.3, 141.9, 131.6 (d, J = 2.9 Hz), 130.9 (d, J = 9.6 Hz, two carbons), 123.6, 119.1, 115.9 (d, J = 21.9 Hz, two carbons), 105.4, 46.8; HPLC (method 1) 98%, tR 3.82 min; MS (ESI) m/z 331 [M + H]+.
1-(Benzo[d][1,2,3]thiadiazol-6-yl)-3-(2-oxo-2-(piperidin-1-yl)ethyl)urea (24)
The procedure used for preparation of compound 4 was followed for synthesis of compound 24. The title compound 24 was obtained as a white solid (57 mg, 51%): 1H NMR (400 MHz, d6-DMSO) δ 9.60 (s, 1H), 8.57–8.46 (m, 2H), 7.60 (dd, J = 9.2, 2.0 Hz, 1H), 6.60 (t, J = 4.5 Hz, 1H), 4.01 (d, J = 4.5 Hz, 2H), 3.53–3.45 (m, 2H), 3.38–3.31 (m, 2H), 1.63–1.48 (m, 4H), 1.48–1.39 (m, 2H). 13C NMR (100 MHz, d6-DMSO) δ 166.6, 154.6, 153.3, 142.3, 142.0, 123.6, 119.0, 105.2, 44.8, 42.3, 41.2, 25.8, 25.2, 23.9. HPLC (method 1) 98%, tR 3.26 min; MS (ESI) m/z 320 [M + H]+; HRMS calcd. for C14H17N5O2S + H: 320.1181; found: 320.1181 [M+H]+.
Methyltransferase Activity Assays
Methyltransferase activity assays were performed by monitoring the incorporation of tritium-labeled methyl group to biotinylated peptide substrates using scintillation proximity assay (SPA) for PRMT3, SETD7, G9a, GLP, SETDB1, SETD8, SUV420H1, SUV420H2, SUV39H2, PRC2 trimeric complex (EZH2:EED:SUZ12), MLL1 tetrameric complex (MLL:WDR5:RbBP5:ASH2L), PRMT5–MEP50 complex, and SMYD2. Assay components for all assays are summarized in Table S1 (Supporting Information). The reaction buffer for SMYD2 and SMYD3 was 50 mM Tris, pH 9.0, 5 mM DTT, and 0.01% TritonX-100, that for G9a, GLP, and SUV39H2 was 25 mM potassium phosphate, pH 8.0, 1 mM EDTA, 2 mM MgCl2, and 0.01% Triton X-100, and that for the other HMTs was 20 mM Tris, pH 8.0, 5 mM DTT, and 0.01% TritonX-100. To stop the enzymatic reactions, 10 μL of 7.5 M guanidine hydrochloride was added, followed by 180 μL of buffer, and the resulting reaction mixture was mixed and transferred to a 96-well FlashPlate (catalog no. SMP103, Perkin-Elmer, www.perkinelmer.com). After mixing, the reaction mixtures were incubated, and the counts per minute (cpm) were measured using a Topcount plate reader (Perkin-Elmer). The cpm values in the absence of compound for each data set were defined as 100% activity. In the absence of the enzyme, the cpm values in each data set were defined as background (0%). IC50 values were determined using compound concentrations ranging from 100 nM to 100 μM. The IC50 values were determined using SigmaPlot software.
For DNMT1, the assay was performed as described above using hemimethylated dsDNA as a substrate. The dsDNA substrate was prepared by annealing two complementary strands (biotinylated forward strand, B-GAGCCCGTAAGCCCGTTCAGGTCG; reverse strand, CGACCTGAACGGGCTTACGGGCTC) synthesized by Eurofins MWG Operon. The reaction buffer was 20 mM Tris–HCl, pH 8.0, 5 mM DTT, and 0.01% Triton X-100.
Methyltransferase activity assays for DOT1L and SMYD3 were performed using filter plates (catalog no. MSFBN6B10, Millipore, www.millipore.com). Reaction mixtures in 20 mM Tris–HCl, pH 8.0, 5 mM DTT, 2 mM MgCl2, and 0.01% Triton X-100 were incubated at room temperature for 1 h, 100 μL 10% TCA was added, and the resulting reaction mixture was mixed and transferred to a filter plate. The plates were centrifuged at 2000 rpm for 2 min followed by two additional 10% TCA washes and one ethanol wash (180 μL) followed by centrifugation. The plates were dried, 100 μL of MicroO was added, and the plates were centrifuged. A 70 μL volume of MicroO was added, and the cpm values were measured using a Topcount plate reader.
Cocrystallization Protocols and Structure Refinement
PRMT3 was incubated at 1.1 mg/mL overnight with compound 14u at a 1:30 molar ratio (PRMT3:compound 14u). Following incubation, protein was concentrated to 3 mg/mL and crystallized using the sitting drop diffusion method at 20 °C by mixing 1 μL of the protein solution with 1 μL of the reservoir solution containing 25% PEG 3350, 0.2 M LiSO4, and 0.1 M Hepes, pH 7.5. Prior to freezing, the crystals were soaked for 10 min in the same buffer with 10% glycerol.
Data Collection and Processing
The native data set was collected on APS beamline 19-ID at 100 K. Program HKL2000 was used for data processing and scaling.60
Structure Determination and Refinement
The structure of PRMT3 in complex with compound 14u was determined by molecular replacement using MOLREP.61 PDB entry 2FYT was used as a search template. The graphic program Coot62 was used for manual model refinement and visualization. Refmac563 was used to refine the model. MolProbity64 was used to validate the refined structure; 98.0% of the residues are the favored regions of the Ramachandran plot, and none of them are in the disallowed regions. The structure has been deposited in the RCSB with PDB code 4HSG.
Modeling
Monte Carlo energy minimization simulations were run in triplicate with ICM version 3.7-2c (Molsoft, San Diego) using the “ssearch” command. Briefly, the conformational space accessible to K392 was sampled in the internal coordinate space, with global conformational sampling steps followed by local energy minimization and calculation of the complete energy potential, including surface and advanced electrostatics terms.65 Compound 14u and residues surrounding K392 were kept static.
Supplementary Material
Acknowledgments
We thank the University Cancer Research Fund (UCRF) and the Carolina Partnership from the University of North Carolina at Chapel Hill for financial support. The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from Canadian Institutes of Health Research, Eli Lilly Canada, Genome Canada, GlaxoSmithKline, the Ontario Ministry of Economic Development and Innovation, the Novartis Research Foundation, Pfizer, AbbVie, Takeda, and the Wellcome Trust. C.H.A. holds a Canada Research Chair in Structural Genomics.
ABBREVIATIONS USED
- PRMTs
protein arginine methyltransferases
- PRMT3
protein arginine methyltransferase 3
- rpS2
40S ribosomal protein S2
- SAR
structure–activity relationship
- HMTs
histone methyl-transferases
- PMTs
protein methyltransferases
- PKMTs
protein lysine methyltransferases
- GAR
glycine- and arginine-rich
- PGM
proline-, glycine-, methionine-, and arginine-rich
- PABPN1
nuclear poly(A)-binding protein
- MOA
mechanism of action
- LHS
left-hand side
- RHS
right-hand side
- SAM
S-adenosylmethionine
- DNMT1
DNA methyltransferase 1
Footnotes
The authors declare no competing financial interest.
Methyltransferase assay components and conditions, data collection and refinement statistics of the compound 14u–PRMT3 cocrystal structure, and 1H and 13C NMR spectra of compounds 14u and 24. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discovery. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 2.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discovery. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 3.Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell. 2005;18:263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Di Lorenzo A, Bedford MT. Histone arginine methylation. FEBS Lett. 2011;585:2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yost JM, Korboukh I, Liu F, Gao C, Jin J. Targets in epigenetics: inhibiting the methyl writers of the histone code. Curr Chem Genomics. 2011;5:72–84. doi: 10.2174/1875397301005010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zurita-Lopez CI, Sandberg T, Kelly R, Clarke SG. Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming omega-NG-monomethylated arginine residues. J Biol Chem. 2012;287:7859–7870. doi: 10.1074/jbc.M111.336271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, La Thangue NB. Arginine methylation regulates the p53 response. Nat Cell Biol. 2008;10:1431–1439. doi: 10.1038/ncb1802. [DOI] [PubMed] [Google Scholar]
- 8.Bedford MT. Arginine methylation at a glance. J Cell Sci. 2007;120:4243–4246. doi: 10.1242/jcs.019885. [DOI] [PubMed] [Google Scholar]
- 9.Pahlich S, Bschir K, Chiavi C, Belyanskaya L, Gehring H. Different methylation characteristics of protein arginine methyltransferase 1 and 3 toward the Ewing sarcoma protein and a peptide. Proteins: Struct, Funct Bioinf. 2005;61:164–175. doi: 10.1002/prot.20579. [DOI] [PubMed] [Google Scholar]
- 10.Cheng DH, Cote J, Shaaban S, Bedford MT. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell. 2007;25:71–83. doi: 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 11.Bedford MT, Reed R, Leder P. WW domain-mediated interactions reveal a spliceosome-associated protein that binds a third class of proline-rich motif: the proline glycine and methionine-rich motif. Proc Natl Acad Sci US A. 1998;95:10602–10607. doi: 10.1073/pnas.95.18.10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Branscombe TL, Frankel A, Lee JH, Cook JR, Yang Z, Pestka S, Clarke S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J Biol Chem. 2001;276:32971–32976. doi: 10.1074/jbc.M105412200. [DOI] [PubMed] [Google Scholar]
- 13.Tang J, Gary JD, Clarke S, Herschman HR. PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J Biol Chem. 1998;273:16935–16945. doi: 10.1074/jbc.273.27.16935. [DOI] [PubMed] [Google Scholar]
- 14.Bachand F, Silver PA. PRMT3 is a ribosomal protein methyltransferase that affects the cellular levels of ribosomal subunits. EMBO J. 2004;23:2641–2650. doi: 10.1038/sj.emboj.7600265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swiercz R, Person MD, Bedford MT. Ribosomal protein S2 is a substrate for mammalian PRMT3 (protein arginine methyltransferase 3) Biochem J. 2005;386:85–91. doi: 10.1042/BJ20041466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swiercz R, Cheng D, Kim D, Bedford MT. Ribosomal protein rpS2 is hypomethylated in PRMT3-deficient mice. J Biol Chem. 2007;282:16917–16923. doi: 10.1074/jbc.M609778200. [DOI] [PubMed] [Google Scholar]
- 17.Smith JJ, Rucknagel KP, Schierhorn A, Tang J, Nemeth A, Linder M, Herschman HR, Wahle E. Unusual sites of arginine methylation in poly(A)-binding protein II and in vitro methylation by protein arginine methyltransferases PRMT1 and PRMT3. J Biol Chem. 1999;274:13229–13234. doi: 10.1074/jbc.274.19.13229. [DOI] [PubMed] [Google Scholar]
- 18.Fronz K, Otto S, Kolbel K, Kuhn U, Friedrich H, Schierhorn A, Beck-Sickinger AG, Ostareck-Lederer A, Wahle E. Promiscuous modification of the nuclear poly(A)-binding protein by multiple protein-arginine methyltransferases does not affect the aggregation behavior. J Biol Chem. 2008;283:20408–20420. doi: 10.1074/jbc.M802329200. [DOI] [PubMed] [Google Scholar]
- 19.Tavanez JP, Bengoechea R, Berciano MT, Lafarga M, Carmo-Fonseca M, Enguita FJ. Hsp70 chaperones and type I PRMTs are sequestered at intranuclear inclusions caused by polyalanine expansions in PABPN1. PloS One. 2009;4:e6418. doi: 10.1371/journal.pone.0006418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allali-Hassani A, Wasney GA, Siarheyeva A, Hajian T, Arrowsmith CH, Vedadi M. Fluorescence-based methods for screening writers and readers of histone methyl marks. J Biomol Screening. 2012;17:71–84. doi: 10.1177/1087057111422256. [DOI] [PubMed] [Google Scholar]
- 21.Lai Y, Song M, Hakala K, Weintraub ST, Shiio Y. Proteomic dissection of the von Hippel-Lindau (VHL) interactome. J Proteome Res. 2011;10:5175–5182. doi: 10.1021/pr200642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh V, Miranda TB, Jiang W, Frankel A, Roemer ME, Robb VA, Gutmann DH, Herschman HR, Clarke S, Newsham IF. DAL-1/4. 1B tumor suppressor interacts with protein arginine N-methyltransferase 3 (PRMT3) and inhibits its ability to methylate substrates in vitro and in vivo. Oncogene. 2004;23:7761–7771. doi: 10.1038/sj.onc.1208057. [DOI] [PubMed] [Google Scholar]
- 23.Chen X, Niroomand F, Liu Z, Zankl A, Katus HA, Jahn L, Tiefenbacher CP. Expression of nitric oxide related enzymes in coronary heart disease. Basic Res Cardiol. 2006;101:346–353. doi: 10.1007/s00395-006-0592-5. [DOI] [PubMed] [Google Scholar]
- 24.Miyata S, Mori Y, Tohyama M. PRMT3 is essential for dendritic spine maturation in rat hippocampal neurons. Brain Res. 2010;1352:11–20. doi: 10.1016/j.brainres.2010.07.033. [DOI] [PubMed] [Google Scholar]
- 25.Bissinger EM, Heinke R, Sippl W, Jung M. Targeting epigenetic modifiers: inhibitors of histone methyltransferases. Med Chem Comm. 2010;1:114–124. [Google Scholar]
- 26.He Y, Korboukh I, Jin J, Huang J. Targeting protein lysine methylation and demethylation in cancers. Acta Biochim Biophys Sin. 2012;44:70–79. doi: 10.1093/abbs/gmr109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller S, Brown PJ. Epigenetic chemical probes. Clin Pharmacol Ther. 2012;92:689–693. doi: 10.1038/clpt.2012.154. [DOI] [PubMed] [Google Scholar]
- 28.Blancafort P, Jin J, Frye SV. Writing and re-writing the epigenetic code of cancer cells: from engineered proteins to small molecules. Mol Pharmacol. 2013;83:563–576. doi: 10.1124/mol.112.080697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng DH, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases. J Biol Chem. 2004;279:23892–23899. doi: 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- 30.Bonham K, Hemmers S, Lim YH, Hill DM, Finn MG, Mowen KA. Effects of a novel arginine methyltransferase inhibitor on T-helper cell cytokine production. FEBS J. 2010;277:2096–2108. doi: 10.1111/j.1742-4658.2010.07623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trapp J, Meier R, Hongwiset D, Kassack MU, Sippl W, Jung M. Structure-activity studies on suramin analogues as inhibitors of NAD(+)-dependent histone deacetylases (sirtuins) Chem Med Chem. 2007;2:1419–1431. doi: 10.1002/cmdc.200700003. [DOI] [PubMed] [Google Scholar]
- 32.Ragno R, Simeoni S, Castellano S, Vicidomini C, Mai A, Caroli A, Tramontano A, Bonaccini C, Trojer P, Bauer I, Brosch G, Sbardella G. Small molecule inhibitors of histone arginine methyltransferases: homology modeling, molecular docking, binding mode analysis, and biological evaluations. J Med Chem. 2007;50:1241–1253. doi: 10.1021/jm061213n. [DOI] [PubMed] [Google Scholar]
- 33.Mai A, Cheng D, Bedford MT, Valente S, Nebbioso A, Perrone A, Brosch G, Sbardella G, De Bellis F, Miceli M, Altucci L. Epigenetic multiple ligands: mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors. J Med Chem. 2008;51:2279–2290. doi: 10.1021/jm701595q. [DOI] [PubMed] [Google Scholar]
- 34.Purandare AV, Chen Z, Huynh T, Pang S, Geng J, Vaccaro W, Poss MA, Oconnell J, Nowak K, Jayaraman L. Pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2008;18:4438–4441. doi: 10.1016/j.bmcl.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 35.Huynh T, Chen Z, Pang S, Geng J, Bandiera T, Bindi S, Vianello P, Roletto F, Thieffine S, Galvani A, Vaccaro W, Poss MA, Trainor GL, Lorenzi MV, Gottardis M, Jayaraman L, Purandare AV. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2009;19:2924–2927. doi: 10.1016/j.bmcl.2009.04.075. [DOI] [PubMed] [Google Scholar]
- 36.Allan M, Manku S, Therrien E, Nguyen N, Styhler S, Robert MF, Goulet AC, Petschner AJ, Rahil G, MacLeod AR, Deziel R, Besterman JM, Nguyen H, Wahhab A. N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2009;19:1218–1223. doi: 10.1016/j.bmcl.2008.12.075. [DOI] [PubMed] [Google Scholar]
- 37.Spannhoff A, Heinke R, Bauer I, Trojer P, Metzger E, Gust R, Schule R, Brosch G, Sippl W, Jung M. Target-based approach to inhibitors of histone arginine methyltransferases. J Med Chem. 2007;50:2319–2325. doi: 10.1021/jm061250e. [DOI] [PubMed] [Google Scholar]
- 38.Heinke R, Spannhoff A, Meier R, Trojer P, Bauer I, Jung M, Sippl W. Virtual screening and biological characterization of novel histone arginine methyltransferase PRMT1 inhibitors. Chem Med Chem. 2009;4:69–77. doi: 10.1002/cmdc.200800301. [DOI] [PubMed] [Google Scholar]
- 39.Spannhoff A, Machmur R, Heinke R, Trojer P, Bauer I, Brosch G, Schule R, Hanefeld W, Sippl W, Jung M. A novel arginine methyltransferase inhibitor with cellular activity. Bioorg Med Chem Lett. 2007;17:4150–4153. doi: 10.1016/j.bmcl.2007.05.088. [DOI] [PubMed] [Google Scholar]
- 40.Feng Y, Li MY, Wang BH, Zheng YG. Discovery and mechanistic study of a class of protein arginine methylation inhibitors. J Med Chem. 2010;53:6028–6039. doi: 10.1021/jm100416n. [DOI] [PubMed] [Google Scholar]
- 41.Selvi BR, Batta K, Kishore AH, Mantelingu K, Varier RA, Balasubramanyam K, Pradhan SK, Dasgupta D, Sriram S, Agrawal S, Kundu TK. Identification of a novel inhibitor of coactivator-associated arginine methyltransferase 1 (CARM1)-mediated methylation of histone H3 Arg-17. J Biol Chem. 2010;285:7143–7152. doi: 10.1074/jbc.M109.063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J, Chen L, Sinha SH, Liang Z, Chai H, Muniyan S, Chou YW, Yang C, Yan L, Feng Y, Kathy, Li K, Lin MF, Jiang H, George Zheng Y, Luo C. Pharmacophore-based virtual screening and biological evaluation of small molecule inhibitors for protein arginine methylation. J Med Chem. 2012;55:7978–7987. doi: 10.1021/jm300521m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 44.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wigle TJ, Wasney GA, Dong A, Senisterra G, Chau I, Siarheyeva A, Norris JL, Kireev DB, Jadhav A, Herold JM, Janzen WP, Arrowsmith CH, Frye SV, Brown PJ, Simeonov A, Vedadi M, Jin J. Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J Med Chem. 2010;53:5844–5857. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang Y, Ganesh T, Horton JR, Spannhoff A, Liu J, Sun A, Zhang X, Bedford MT, Shinkai Y, Snyder JP, Cheng X. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J Mol Biol. 2010;400:1–7. doi: 10.1016/j.jmb.2010.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wasney GA, Dong A, Barsyte D, Kozieradzki I, Senisterra G, Chau I, Siarheyeva A, Kireev DB, Jadhav A, Herold JM, Frye SV, Arrowsmith CH, Brown PJ, Simeonov A, Vedadi M, Jin J. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J Med Chem. 2009;52:7950–7953. doi: 10.1021/jm901543m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu F, Barsyte-Lovejoy D, Allali-Hassani A, He Y, Herold JM, Chen X, Yates CM, Frye SV, Brown PJ, Huang J, Vedadi M, Arrowsmith CH, Jin J. Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. J Med Chem. 2011;54:6139–6150. doi: 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, DiMaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang SC, Chen X, Chau I, Mangano T, Huang XP, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, Jin L, Kuntz KW, Chesworth R, Moyer MP, Bernt KM, Tseng JC, Kung AL, Armstrong SA, Copeland RA, Richon VM, Pollock RM. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferguson AD, Larsen NA, Howard T, Pollard H, Green I, Grande C, Cheung T, Garcia-Arenas R, Cowen S, Wu J, Godin R, Chen H, Keen N. Structural basis of substrate methylation and inhibition of SMYD2. Structure. 2011;19:1262–1273. doi: 10.1016/j.str.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 51.Yao Y, Chen P, Diao J, Cheng G, Deng L, Anglin JL, Prasad BVV, Song Y. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis and crystallographic studies. J Am Chem Soc. 2011;133:16746–16749. doi: 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan Y, Wang Q, Paulk J, Kubicek S, Kemp MM, Adams DJ, Shamji AF, Wagner BK, Schreiber SL. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem Biol. 2012;7:1152–1157. doi: 10.1021/cb300139y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, AD, Diaz E, Lafrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 54.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, Richon VM, Copeland RA, Keilhack H, Pollock RM, Kuntz KW. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 55.Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, Graves AP, Scherzer DA, Shu A, Thompson C, Ott HM, Aller GSV, Machutta CA, Diaz E, Jiang Y, Johnson NW, Knight SD, Kruger RG, McCabe MT, Dhanak D, Tummino PJ, Creasy CL, Miller WH. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–1096. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng W, Ibáñez G, Wu H, Blum G, Zeng H, Dong A, Li F, Hajian T, Allali-Hassani A, Amaya MF, Siarheyeva A, Yu W, Brown PJ, Schapira M, Vedadi M, Min J, Luo M. Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J Am Chem Soc. 2012;134:18004–18014. doi: 10.1021/ja307060p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Siarheyeva A, Senisterra G, Allali-Hassani A, Dong A, Dobrovetsky E, Wasney GA, Chau I, Marcellus R, Hajian T, Liu F, Korboukh I, Smil D, Bolshan Y, Min J, Wu H, Zeng H, Loppnau P, Poda G, Griffin C, Aman A, Brown Peter J, Jin J, Al-awar R, Arrowsmith CH, Schapira M, Vedadi M. An allosteric inhibitor of protein arginine methyltransferase 3. Structure. 2012;20:1425–1435. doi: 10.1016/j.str.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 58.Ward E, Poesche W, Higgins D, Heard D. 458. 1, 2, 3-Benzothiadiazole. Part I. Nitro-, amino-, and hydroxy-derivatives. J Chem Soc. 1962:2374–2379. [Google Scholar]
- 59.Rostami A, Colin A, Li XY, Chudzinski MG, Lough AJ, Taylor MS. N,N′-Diarylsquaramides: general, high-yielding synthesis and applications in colorimetric anion sensing. J Org Chem. 2010;75:3983–3992. doi: 10.1021/jo100104g. [DOI] [PubMed] [Google Scholar]
- 60.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution— from diffraction images to an initial model in minutes. Acta Crystallogr, D: Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 61.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 62.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 63.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 64.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr, D: Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.