

Abstract
Waterfowl (Anseriformes) and shorebirds (Charadriiformes) are the most common wild vectors of influenza A viruses. Due to their migratory behavior, some may transmit disease over long distances. Migratory connectivity studies can link breeding and nonbreeding grounds while illustrating potential interactions among populations that may spread diseases. We investigated Dunlin (Calidris alpina), a shorebird with a subspecies (C. a. arcticola) that migrates from nonbreeding areas endemic to avian influenza in eastern Asia to breeding grounds in northern Alaska. Using microsatellites and mitochondrial DNA, we illustrate genetic structure among six subspecies: C. a. arcticola,C. a. pacifica,C. a. hudsonia,C. a. sakhalina,C. a. kistchinski, and C. a. actites. We demonstrate that mitochondrial DNA can help distinguish C. a. arcticola on the Asian nonbreeding grounds with >70% accuracy depending on their relative abundance, indicating that genetics can help determine whether C. a. arcticola occurs where they may be exposed to highly pathogenic avian influenza (HPAI) during outbreaks. Our data reveal asymmetric intercontinental gene flow, with some C. a. arcticola short-stopping migration to breed with C. a. pacifica in western Alaska. Because C. a. pacifica migrates along the Pacific Coast of North America, interactions between these subspecies and other taxa provide route for transmission of HPAI into other parts of North America.
Keywords: Calidris alpina, Dunlin, genetic structure, highly pathogenic avian influenza, human disease, influenza A, migratory connectivity, migratory short-stopping
Introduction
Birds are primary reservoirs for all known influenza A virus subtypes (Webster et al. 1992). In particular, waterfowl (Anseriformes) and shorebirds (Charadriiformes) are the most common wild vectors (Olsen et al. 2006). Infected birds generally harbor low-pathogenic avian influenza (AI) strains; however, outbreaks of highly pathogenic avian influenza strains (HPAI) such as the H5N1 and H7N9 subtypes are becoming more common, especially in South-East Asia (Chen et al. 2004, 2006; Li et al. 2004; Ferguson et al. 2005; Gao et al. 2013; Uyeki and Cox 2013). Concerns surrounding the spread of HPAI exist, particularly as mediated through avian vectors given the long distance seasonal migratory behavior of many virus hosts (Kilpatrick et al. 2006). Although most migratory movements occur within continents, intercontinental migration can also occur. For example, up to three million birds and thousands of infected individuals cross the Bering Strait from Asia into Alaska each year (Winker and Gibson 2010).
The likelihood that an individual bird species may contribute to the intercontinental spread of avian influenza depends in part on the details of its seasonal migratory patterns. Thus, migratory connectivity studies of birds can be used to define important migratory pathways and identify the population of origin of individuals at all stages of the annual cycle (Webster et al. 2002). Such studies take on new importance in the age of widespread disease transfer by birds (e.g., Rappole et al. 2000; Ishiguro et al. 2005; Morshed et al. 2005; Fergus et al. 2006; Gilbert et al. 2006; Dusek et al. 2014). If the identity and origin of avian disease carriers can be determined and if their migratory pathways are understood, it may be possible to predict the next occurrence of a virulent disease near human population centers, implement precautionary measures to limit human–bird contact, and adopt practices to try to minimize the potential for further spread of the disease to other geographic regions.
The Dunlin (Calidris alpina) is a circumpolar migratory shorebird that breeds throughout arctic and subarctic tundra regions and winters in the southern portion of the Northern Hemisphere (Del Hoyo et al. 1996). There are up to 11 described subspecies that show varying degrees of morphological variation (Greenwood 1986; Tomkovich 1986; Nechaev and Tomkovich 1987; Browning 1991; AOU 2013). These purported subspecies are believed to use separate breeding grounds, but their migratory flyways and nonbreeding areas may overlap (Warnock and Gill 1996; Lappo et al. 2012; Gill et al. 2013). Five subspecies of Dunlin breed in the East Asia and Alaska region known as Beringia (Fig.1): Calidris alpina actites,C. a. kistchinski, and C. a. sakhalina breed in the Russian Far East while C. a. arcticola and C. a. pacifica breed in Alaska (Warnock and Gill 1996; AOU 2013; Fig.1). A sixth North American subspecies exists (C. a. hudsonia), but breeds in central and eastern Canada and winters along the Atlantic Coast and Gulf of Mexico (Fernández et al. 2008). Potential interactions among the Beringia subspecies are complex: C. a. pacifica breeds in western Alaska and migrates south along the Pacific Coast of North America to winter in the western United States and Mexico (Fernández et al. 2008; Gill et al. 2013). Calidris alpina arcticola breeds in northern Alaska, but migrates across the Bering Strait to winter along the Pacific Coast of Asia where it potentially intermixes with the three East Asia subspecies (Fernández et al. 2008; Lanctot et al. 2009; Gill et al. 2013).
Figure 1.

Breeding distribution of six subspecies of Dunlin (Calidris alpina) sampled for genetic analysis. Sites codes are congruent with those listed in Table1.
Dunlin were ranked the second-highest of 26 priority taxa to be routinely monitored for HPAI in Alaska when extensive sampling was initiated during the H5N1 HPAI outbreak in 2006 (U.S. Fish and Wildlife Service and U.S. Geological Survey 2007). The rankings were based on each taxon's distribution in Asia, proximity to locations where HPAI has been previously identified, general habitat use patterns, ease of sampling, and population size in Alaska (Alaska Interagency HPAI Bird Surveillance Working Group 2006; Ip et al. 2008). Dunlin ranked high primarily because they winter in areas where outbreaks of HPAI occur in Asia and because so many individuals (300 000–700 000 birds; Andres et al. 2012) migrate from Asia to Alaska each year. Dunlin are also highly susceptible to HPAI H5N1 (Hall et al. 2011). Mortality is likely common among infected juveniles (Hall et al. 2011), but infected adults may survive and transmit viruses. Surveys of wild-caught Dunlin in Alaska between 2006 and 2007 revealed that 0.22% were positive for AI based on RT-PCR analyses of cloacal swabs or fecal samples (Ip et al. 2008), indicating that active shedding of AI viruses was occurring at the time of sampling. This value likely underestimates the true infection rate, as Hall et al. (2011) found that RT-PCR detection of H5N1 in experimental challenges was longer lasting and more consistent from oropharyngeal samples as opposed to cloacal samples. Furthermore, Pearce et al. (2012) found that 2.6% of Dunlin sampled in Alaska during the late summer of 2010 demonstrated evidence for prior AI exposure based on serologic assays. While actual numbers are likely to vary substantially from year to year based on the dynamics of viral outbreaks in Asia, these studies nominally suggest that between 1540 and 18 200 (based on estimated population sizes) infected Dunlin could be in Alaska in any given year. Collectively, this information indicates that C. a. arcticola is an important subspecies to consider when evaluating potential routes and mechanisms by which Asian influenza strains can be transmitted to North America.
Although all Dunlin subspecies show some phenotypic variation (Tomkovich 1986; Nechaev and Tomkovich 1987; Browning 1991), it is difficult to separate them outside of the breeding grounds using commonly employed morphological characters such as plumage or culmen, head, wing, and tarsus measurements (Warnock and Gill 1996; Wennerberg et al. 1999; but see Gates et al. 2013). This is particularly true in eastern Asia where four subspecies are thought to intermix during the nonbreeding season (Lanctot et al. 2009; Gates et al. 2013). In these circumstances, hypervariable molecular markers and DNA sequences may be useful for illuminating patterns of population connectivity and movements of individuals throughout the annual cycle (Haig et al. 2011). We therefore used mitochondrial DNA sequences (mtDNA) from the cytochrome b gene and control region along with eight nuclear microsatellite loci to address multiple questions associated with the differentiation of Dunlin subspecies and the extent of gene flow and interactions among groups from Asia and North America. (i) Do genetic data provide evidence for differentiation among Dunlin subspecies and breeding populations from the region? While prior work has examined phylogeographic patterns in the Northern Hemisphere, most studies were based on small sample sizes and had limited (or no) sampling within Beringia-associated subspecies (e.g., Wenink and Baker 1996; Wenink et al. 1996; Wennerberg et al. 1999; Marthinsen et al. 2007). (ii) Does genetic differentiation among subspecies provide a basis for the probabilistic identification of subspecies where they co-occur (sensu Patten and Unitt 2002)? In particular, we were interested in determining whether genetic data can distinguish C. a. arcticola from the three other Dunlin subspecies that winter in East Asia (C. a. actites, C. a. kistchinski, and C. a. sakhalina). If distinguishable, then nonbreeding populations of C. a. arcticola could be more easily identified, leading to a better understanding of the likelihood of this subspecies becoming infected and transmitting HPAI into North America. (iii) Can genetic data characterize the extent of gene flow and interaction among the three proximate Beringian subspecies (C. a. sakhalina,C. a. arcticola, and C. a. pacifica)? Given the geographic locations of their breeding ranges (Fig.1), opportunities for gene flow among subspecies may occur. Furthermore, a portion of the C. a. arcticola and C. a. pacifica populations intermix during postbreeding staging in western Alaska (Gill et al. 2013), but the extent of gene flow between these groups is not well known. If gene flow is extensive, then the data may point to greater-than-expected interactions between these two subspecies. Because C. a. pacifica winters along the Pacific Coast of North America, interactions with C. a. arcticola during the breeding or postbreeding season may increase the risk of transmission of Asian influenza strains from Alaska into other parts of North America.
Materials and methods
Sample collection and molecular methods
We collected 370 Dunlin blood or tissue samples from 18 breeding areas during the 2003 to 2009 breeding seasons (Fig.1, Table1). Samples included putative representatives from the five subspecies that inhabit eastern Asia and Alaska (C. a. actites,C. a. kistchinski,C. a. sakhalina,C. a. pacifica, and C. a. arcticola) and were our primary focus for this study. However, we also included samples from three C. a. hudsonia breeding populations in eastern North America to help provide greater genetic and spatial context to our analyses. Individual birds were captured with bownets at nest sites (most subspecies) or lethally collected (C. a. kistchinski samples) on breeding territories. Live-captured birds had up to 0.3 mL of blood collected into a heparinized tube via brachial puncture with a 26- to 27.5-gauge needle. Additional breeding season tissues were obtained from the University of Washington Burke Museum to augment the Russian populations (UWBM Accession Numbers 43910, 44120, 44121, 51684, 51687, 51693, 51694, 51695, and 69903). Blood or tissue samples were preserved in Longmire buffer (Longmire et al. 1997) until used for genetic analyses.
Table 1.
Sample sizes and locations of six subspecies of Dunlin (Calidris alpina) sampled for microsatellites (n = 370) and mtDNA (n = 234). Locations are indicated by site code on Fig.1
| Subspecies | Location (site code) | Latitude | Longitude | N (microsats) | N (mtDNA) |
|---|---|---|---|---|---|
| arcticola | Barrow, AK, USA (A) | 71.27 | −156.53 | 87 | 32 |
| Canning, AK, USA (B) | 70.10 | −145.85 | 34 | 15 | |
| Prudhoe Bay, AK, USA (C) | 70.35 | −148.64 | 23 | 13 | |
| actites | Schiavo Bay, Sakhalin, Russia (D) | 52.55 | +143.30 | 23 | 23 |
| hudsonia | Nunavut, NU, Canada (E) | 63.97 | −80.28 | 3 | 3 |
| Churchill, MB, Canada (F) | 58.74 | −94.07 | 10 | 10 | |
| Rasmussen, NU, Canada (G) | 69.02 | −93.85 | 3 | 3 | |
| kistchinski | Kamchatka, Russia (H) | 52.81 | +156.42 | 30 | 25 |
| Magadanskaya Oblast, Russia (I) | 59.38 | +149.07 | 12 | 5 | |
| pacifica | Platinum, AK, USA (J) | 59.02 | −161.82 | 8 | 7 |
| Cold Bay, AK, USA (K) | 55.24 | −162.84 | 25 | 21 | |
| Nome, AK, USA (L) | 64.45 | −164.93 | 5 | 4 | |
| Kanaryarmiut, AK, USA (M) | 61.36 | −165.15 | 8 | 8 | |
| Manokinak, AK, USA (N) | 61.19 | −165.10 | 30 | 11 | |
| sakhalina | Wrangel, Russia (O) | 71.41 | −179.67 | 20 | 16 |
| Meinopylgino, Chukotka, Russia (P) | 62.55 | +177.08 | 11 | 10 | |
| Belyaka Spit, Chukota, Russia (Q) | 67.15 | −174.68 | 22 | 16 | |
| Anadyr, Chukota, Russia (R) | 64.70 | +177.63 | 16 | 12 |
DNA was extracted as described in Haig et al. (2004). We used polymerase chain reaction (PCR) to amplify partial sequences of the mitochondrial cytochrome b gene (cyt b) and control region (D-loop) in 234 samples (Table1). Primer pairs, including L14996-H15646 (http://people.bu.edu/msoren/primers.html, accessed 15 January 2015) and TS96L-TS778H (Wenink et al. 1994), were used to amplify the mitochondrial cyt b and D-loop sequences, respectively. All primer sequences and annealing temperatures are shown in Appendix A. PCR amplifications were performed in 20 μL reactions containing 2.5 mm MgCl2, 1 μm of primers, 100 μm of each dNTP, 1× PCR buffer (Perkin Elmer, Waltham, MA, USA), and 1 U AmpliTaq Gold DNA polymerase (Perkin Elmer). Thermal-cycling parameters included initial denaturation at 94°C followed by 35 cycles of denaturing at 94°C (30 s), the annealing temperature listed in Appendix A (30 s), and extension at 72°C (60 s). PCR products were bidirectionally sequenced with BigDye® Terminator 3.1 Cycle Sequencing chemistry (Life Technologies, Grand Island, NY, USA) and resolved on an ABI 3730 automated DNA sequencer, with resulting chromatograms aligned, edited, and trimmed using the program SeqMan ver. 8.0.2 (DNAStar Inc., Madison, WI, USA). The final 1112-bp alignment contained concatenated sequences from each individual and included 633 bp of cyt b and 479 bp from the D-loop.
Nuclear microsatellite genotypes were obtained at eight loci for 370 individuals (Table1; Appendix A). We obtained primers for loci CALP2 and 4A11 from Wennerberg (2001a), and for loci Cme2, Cme10, and Cme12 from van Treuren et al. (1999), whereas loci D25, D26, and D110 were characterized de novo for this specific investigation during an Illumina GAIIx Genome Analyzer paired-end 80 run (sensu Jennings et al. 2011). Library construction followed recommended Illumina protocols with the exception that index sequencing ‘bar-coded’ adapters (Craig et al. 2008; Cronn et al. 2008) were substituted for standard paired-end adapters. Primer sequences and annealing temperatures for all microsatellites are provided in Appendix A. PCRs were performed in a 10 μL reaction volume with the following reagent concentrations: 1× PCR buffer (Promega Inc., Madison, WI, USA), 0.5 μm of each primer, 2.5 mm MgCl2, 100 μm of each dNTP, and 1 U Taq DNA polymerase (Promega, Inc.). Thermal-cycling parameters included 2 min denaturation at 93°C, followed by 30 cycles of 30 s at 93°C, 30 s at the appropriate annealing temperature, and elongation at 72°C for 1 min. Amplification products were analyzed on an ABI 3100 capillary DNA automated sequencer. ABI GENESCAN software was used to size fragments based on internal lane standard GeneScan 500 [Rox]. ABI GENEMAPPER software was used to score alleles sizes.
Differentiation among subspecies
We characterized the mitochondrial and microsatellite data to provide heuristic indicators of differences among subspecies. For the mtDNA data, we used FaBox (Villesen 2007) to identify unique haplotypes in the data set and create tables reflecting haplotype frequencies and shared haplotypes among groups. ARLEQUIN version 3.1 (Excoffier et al. 2005) was used to quantify gene diversity (H) and nucleotide diversity (π) in mtDNA data within each subspecies. Tables documenting microsatellite allele frequency variation among subspecies were created using CONVERT (Glaubitz 2004). Likewise, program GDA version 1.1 (Lewis and Zaykin 2002) was used to calculate allelic richness and observed and expected heterozygosity (HO and HE, respectively). HP-Rare (Kalinowski 2005) was used to obtain rarefied estimates of allelic richness that accounted for differences in sample size.
We used phylogenetic analyses to examine differentiation of subspecies based on the mtDNA data. The program PhyML 3.0 (Guindon et al. 2010) was used to infer phylogenetic relationships among haplotypes using the maximum-likelihood (ML) criterion. The best-fit nucleotide substitution model was identified using jModeltest2 (Darriba et al. 2012). One thousand bootstrap replicates were used to evaluate clade support. Bayesian phylogenetic analyses were performed using MRBAYES version 3.1.2 (Huelsenbeck and Ronquist 2001), where four concurrent chains were run for 6 × 106 generations. Trees were sampled every 2000 generations and ‘burn in’ included the initial 25% of samples. jModeltest 2 was also used to identify nucleotide substitution models for Bayesian analyses, but was restricted to the subset of models supported by MRBAYES when performing model selection. Resulting phylogenetic trees from both analyses were visualized and annotated using MEGA 5.2 (Tamura et al. 2011).
We used STRUCTURE version 2.2.3 (Pritchard et al. 2000) to analyze the microsatellite data to identify the number of genetic clusters and to probabilistically assign each analyzed individual to one of the identified clusters. Analyses assumed numbers of clusters (K) ranging from one through seven and were based on the uncorrelated allele frequency model and no admixture. Ten replicate analyses were performed for each value of K with each replicate using an initial 106 burn-in steps followed by 107 analysis replicates. We evaluated the outcome of analyses in two different ways: by identifying the value of K that produced the highest average likelihood score over replicates and through the use of the ΔK procedure of Evanno et al. (2005). In both cases, results were summarized over replicates using the program CLUMPP (Jakobsson and Rosenberg 2007). Prior to all microsatellite analyses, we used GDA version 1.1 (Lewis and Zaykin 2002) to identify deviations from Hardy–Weinberg genotypic proportions and test for linkage disequilibrium between pairs of loci within each subspecies. Composite test results for Hardy–Weinberg disequilibrium within each subspecies were obtained by combining P-values from locus-specific analyses using the Z-transform test (Whitlock 2005).
ARLEQUIN was used to perform an analysis of molecular variance (amova; Excoffier et al. 1992) and quantify genetic structure among Dunlin subspecies. In this analysis, Φst (for mtDNA), FST, and RST (both for microsatellite data, the latter assuming a strict stepwise mutation; Slatkin 1995) were calculated to determine the overall and pairwise levels of differentiation among different subspecies. P-values associated with these statistics were obtained using 10 000 randomization replicates.
Distinguishing C. a. arcticola from other subspecies that winter in Asia
Results from STRUCTURE analyses (described above) were further evaluated to determine whether the microsatellite data could be used to probabilistically distinguish among Dunlin subspecies that winter in Asia. If STRUCTURE identified more than one cluster, then assignment values for individuals within each cluster may facilitate accurate subspecific diagnoses of individual birds from mixed groups on the nonbreeding grounds. We also used the individual assignment approach encapsulated in GeneClass2 (Piry et al. 2004), where we determined whether birds could be assigned to one of the predefined Dunlin subspecies with a high degree of confidence. Analyses used the Bayesian computation criterion of Rannala and Mountain (1997) and probability computations as described in Cornuet et al. (1999) using 10 000 simulated individuals. After analyses, we determined the proportion of individuals that were correctly reassigned to their respective subspecies and the average probability associated with correct assignments.
The diagnostic utility of the mtDNA data was also evaluated. Results from the phylogenetic analyses initially suggested that our mtDNA could be used to distinguish C. a. arcticola from other subspecies that winter in Asia (see Results and Discussion). Specifically, haplotypes from C. a. arcticola and C. a. pacifica (hereafter referred to as clade I haplotypes) formed a clade that was largely distinct from haplotypes detected in the Asian subspecies C. a. kistchinski,C. a. sakhalina, and C. a. actites (see Results and Fig.2). The sole exception to this pattern was the detection of seven C. a. sakhalina individuals that possessed clade I haplotypes (Fig.2). Therefore, to more formally quantify the diagnostic potential of the mtDNA, we applied a simple formulation of Bayes' theorem (Sokal and Rohlf 1995) to estimate P(arcticola|I): the probability that an individual sampled on the nonbreeding grounds with a haplotype from clade I is actually C. a. arcticola rather than C. a. sakhalina. The probability is calculated as
| 1 |
and relies on the following quantities: the probability of detecting clade I haplotypes in C. a. arcticola:P(I|arcticola) = 60/60 = 1.0; the probability of detecting clade I haplotypes in C. a. sakhalina:P(I|sakhalina) = 7/54 = 0.13; the probability of selecting a bird that is C. a. arcticola:P(arcticola); and the probability of selecting a bird that is C. a. sakhalina:P(sakhalina) = 1 − P(arcticola). Calidris alpina arcticola and C. a. sakhalina are believed to use similar areas during the winter, primarily Japan, coastal mainland China, Taiwan, and South Korea (Lanctot et al. 2009; Clements et al. 2013; Gill et al. 2013). Because P(arcticola) and P(sakhalina) reflect the probability of randomly selecting an individual from each subspecies, these quantities therefore depend on the abundance of each subspecies on the wintering grounds. Based on population estimates, there are 100 000 to 1 000 000 C. a. sakhalina individuals (Bamford et al. 2008), whereas 300 000 to 700 000 C. a. arcticola winter in East Asia (Andres et al. 2012). We therefore calculated P(arcticola|I) using the upper bound, lower bound, and approximate midpoint of each population size estimate in calculations.
Figure 2.
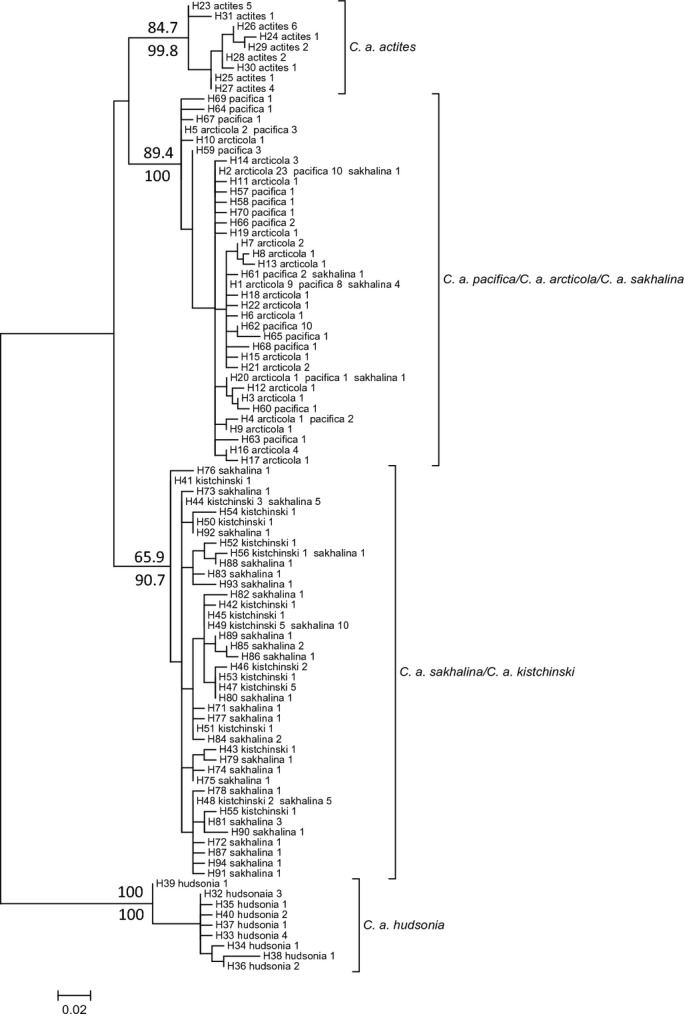
Unrooted maximum-likelihood (ML) tree generated from 94 mitochondrial DNA haplotypes detected in six subspecies of Dunlin (Calidris alpina). Labels at the terminus of each branch provide information on haplotype codes (Appendix B) and the number of individuals from each subspecies that possessed a given haplotype. Branch support values for four major clades of interest are indicated (above branch: bootstrap values from ML analyses; below branch: posterior probabilities from Bayesian analyses).
Quantifying gene flow among Beringian subspecies
We used MIGRATE-N version 3.5.1 (Beerli and Palczewski 2010) to obtain Bayesian estimates of mutation-scaled effective population sizes and asymmetric migration rates among the three proximate Beringian subspecies (C. a. sakhalina, C. a. arcticola, and C. a. pacifica) that were most likely to exhibit gene flow. Limiting our analyses to three subspecies substantially reduced the number of parameters that needed to be simultaneously estimated, thereby providing a more tractable computational problem with a greater likelihood of success relative to analysis of the full data set (analysis required estimation of three as opposed to six effective population size parameters and six rather than thirty gene flow parameters) (Beerli 2009). MIGRATE-N estimates long-term effective population sizes as θ = xNeμ, where μ is the mutation rate and x is an inheritance scaling factor that takes on values of 1 for mtDNA and 4 for codominant nuclear markers such as microsatellites. Long-term migration patterns are estimated over the time scales reflected by the set of sampled gene genealogies using the mutation-scaled quantity M = m/μ, where m is the proportion of immigrants. Note that the product of the parameter estimates divided by the scaling factor (θM/x) provides a basis for estimating Nem, the effective number of immigrants into a population per generation.
Analysis parameter values and settings for MIGRATE-N were selected after preliminary exploratory analyses and with input from the program's developer (P. Beerli, personal communication). mtDNA analyses used the basic DNA sequence model, and priors for θ were specified as a uniform distribution with minimum and maximum values of 0 and 0.03, respectively. Uniform priors with minimum and maximum values of 0 and 10 000 were likewise specified for M. Two independent runs based on random starting trees were performed to ensure convergence and consistency of parameter estimates. Each run was based on 106 recorded steps with a recording interval of 50 steps. Four concurrent chains were implemented during each run, with each chain using a static heating scheme based on temperature values of 1.0, 1.5, 3.0, and 105. Microsatellite analyses were performed using the Brownian motion model. Lower and upper bounds for the uniform prior on θ were specified as 0 and 10.0, whereas uniform priors for M were bound by 0 and 500. Two completely independent runs using starting UPGMA trees were performed, with each run based on 20 concurrent chains with 1000 recording steps made at 100 step intervals. The same heating scheme used for the mtDNA was applied to the microsatellites.
Results
Differentiation among subspecies
We observed 78 variable sites within the concatenated 1112-bp cyt b and D-loop sequence alignment (41 variable sites from cyt b and 37 from D-loop), which resulted in 94 unique haplotypes among the 234 Dunlin specimens examined (Appendix B; GenBank accessions for D-loop: KP205084– KP205177; GenBank accessions for cyt b: KP205178– KP205271). At the subspecies level, lowest values of mitochondrial gene and nucleotide diversities (H and π; Table2) were found in C. a. arcticola (H = 0.830, π = 0.0015), while the highest values were detected in C. a. sakhalina (H = 0.953, π = 0.0058, Table2). Most haplotypes were restricted to a single subspecies (84 of 94; Appendix B).
Table 2.
Genetic diversity in Dunlin (Calidris alpina)
| Subspecies | Microsatellites | mtDNA | |||||
|---|---|---|---|---|---|---|---|
| N | A | HE | HO | N | H | π | |
| arcticola | 144 | 6.00 (4.46) | 0.543 | 0.497 | 60 | 0.830 | 0.0015 |
| actites | 23 | 3.88 (3.71) | 0.47 | 0.462 | 23 | 0.870 | 0.0019 |
| hudsonia | 16 | 4.38 (4.38) | 0.536 | 0.508 | 16 | 0.908 | 0.0023 |
| kistchinski | 42 | 5.50 (4.64) | 0.574 | 0.568 | 30 | 0.931 | 0.0028 |
| pacifica | 76 | 6.13 (4.66) | 0.551 | 0.512 | 51 | 0.900 | 0.0023 |
| sakhalina | 69 | 6.63 (5.15) | 0.604 | 0.545 | 54 | 0.953 | 0.0058 |
N, sample size; A, allelic richness (rarefied estimates accounting for differences in sample size provided in parentheses); HE, expected heterozygosity; HO, observed heterozygosity; H, gene diversity; π, nucleotide diversity.
jModeltest2 identified the TrN+I+G model as most appropriate for ML analyses. The unrooted ML tree grouped the 94 unique haplotypes into four clades that included (1) a C. a. actites group, (2) a C. a. hudsonia group, (3) a C. a. kistchinski/sakhalina group, and (4) a group comprised primarily of C. a. arcticola/pacifica specimens (Fig.2). With the exception of the detection of four C. a. arcticola/pacifica haplotypes in seven C. a. sakhalina specimens, there was no additional evidence of haplotype sharing among groups (Fig.2 and Appendix B). jModeltest2 indicated that the HKY model was most appropriate of those supported by MRBAYES. Trees from Bayesian analyses showed clear signs of convergence across the four runs (scale reduction factor of estimated parameters ranged from 0.99 to 1.01; standard deviation of split frequencies = 0.0093) and were virtually indistinguishable from the ML tree. Consequently, only the ML tree is presented here (Fig.2).
There was highly significant differentiation among subspecies based on the mitochondrial data (ΦST = 0.773, P < 0.001, Table3A). All pairwise comparisons among subspecies were highly significant (Table3A). Consistent with the phylogenetic analysis (Fig.2), the lowest ΦST values were detected in the C. a. pacifica/arcticola contrast and the C. a. sakhalina/kistchinski contrast—the two subspecies pairs that were not phylogenetically distinct in our analyses.
Table 3.
Pairwise and global estimates of FST for Dunlin (Calidris alpina) subspecies. FST values are shown below matrix diagonals while P-values are above matrix diagonals. (A) mtDNA; (B) microsatellite analyses; (C) microsatellite analyses assuming a stepwise mutational model
| A. ΦST = 0.773, P < 0.001 | arcticola | actites | hudsonia | kistchinski | pacifica | sakhalina |
|---|---|---|---|---|---|---|
| C. a. arcticola | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | |
| C. a. actites | 0.858 | <0.001 | <0.001 | <0.001 | <0.001 | |
| C. a. hudsonia | 0.942 | 0.92 | <0.001 | <0.001 | <0.001 | |
| C. a. kistchinski | 0.867 | 0.797 | 0.894 | <0.001 | 0.009 | |
| C. a. pacifica | 0.048 | 0.814 | 0.92 | 0.832 | <0.001 | |
| C. a. sakhalina | 0.713 | 0.622 | 0.807 | 0.071 | 0.676 |
| B. FST = 0.032, P = 0.001 | arcticola | actites | hudsonia | kistchinski | pacifica | sakhalina |
|---|---|---|---|---|---|---|
| C. a. arcticola | <0.001 | <0.001 | 0.005 | 0.001 | <0.001 | |
| C. a. actites | 0.095 | <0.001 | <0.001 | <0.001 | <0.001 | |
| C. a. hudsonia | 0.062 | 0.126 | <0.001 | <0.001 | <0.001 | |
| C. a. kistchinski | 0.010 | 0.097 | 0.065 | 0.019 | 0.129 | |
| C. a. pacifica | 0.009 | 0.130 | 0.081 | 0.008 | <0.001 | |
| C. a. sakhalina | 0.014 | 0.094 | 0.058 | 0.004 | 0.022 |
| C. RST = 0.039, P < 0.001 | arcticola | actites | hudsonia | kistchinski | pacifica | sakhalina |
|---|---|---|---|---|---|---|
| C. a. arcticola | 0.042 | <0.001 | 0.380 | <0.001 | 0.033 | |
| C. a. actites | 0.025 | <0.001 | 0.013 | <0.001 | 0.002 | |
| C. a. hudsonia | 0.163 | 0.264 | 0.001 | <0.001 | 0.003 | |
| C. a. kistchinski | 0.001 | 0.057 | 0.130 | 0.006 | 0.414 | |
| C. a. pacifica | 0.039 | 0.100 | 0.110 | 0.031 | 0.012 | |
| C. a. sakhalina | 0.012 | 0.067 | 0.085 | 0.000 | 0.019 |
As with the mtDNA data, C. a. sakhalina demonstrated the highest microsatellite allelic richness and HE values. However, the lowest microsatellite diversity was detected in C. a. actites (Table2). The microsatellites demonstrated no evidence for significant deviations from Hardy–Weinberg genotypic proportions after sequential Bonferroni corrections. Likewise, the 168 linkage disequilibrium tests performed (28 locus-pair analyses per subspecies * 6 subspecies) revealed only five significant results at the 0.05 level. These significant tests were detected across several subspecies (C. a. actites,C. a. pacifica, and C. a. sakhalina) and could have been observed by chance alone given the large number of individual tests that were performed.
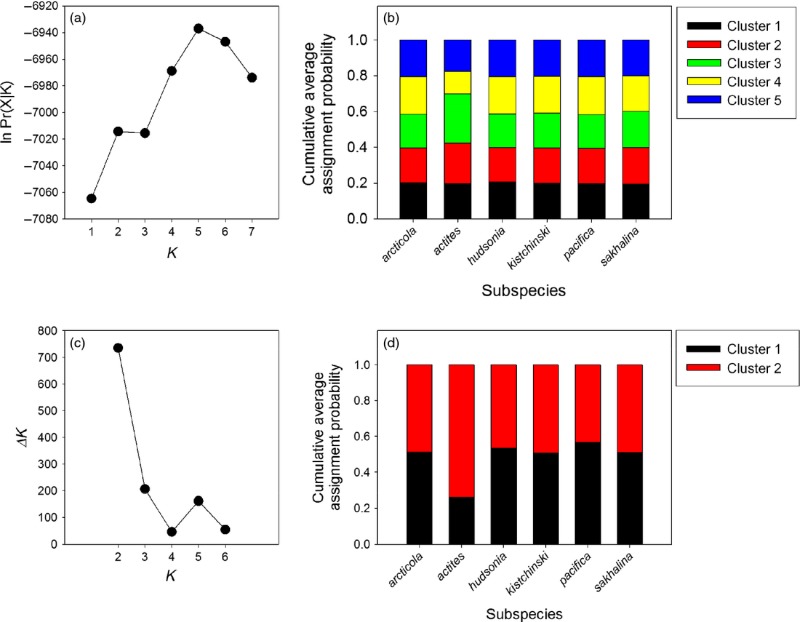
The microsatellite analyses provided varying insights regarding genetic differentiation patterns in Dunlin. STRUCTURE suggested no evidence of differentiation among subspecies. Although the greatest average likelihood score was observed for the K = 5 case and the ΔK procedure suggested that there were K = 2 clusters, individual assignment probabilities to individual clusters were low and nearly uniform across clusters (Appendix C). This outcome indicates that the analysis procedure overestimated the true number of clusters and that subspecies-level subdivisions cannot be resolved with this analytical approach. In contrast, the global estimate of FST from the microsatellite data indicated that significant genetic structure existed (FST = 0.032, P < 0.001) (Table3B). However, in comparison with the mitochondrial analysis, the microsatellite differentiation was generally small and reflected subtle differences in allele frequencies among subspecies (Appendix D). Most pairwise subspecific measures of differentiation were significant, with the exception of the comparison of C. a. sakhalina and C. a. kistchinski (FST = 0.004, P = 0.129) (Table3B). The pairwise RST values and their associated P-values were similar to those of FST estimate, with the added finding of nonsignificant differentiation between C. a. arcticola and C. a. kistchinski (Table3C).
Distinguishing C. a. arcticola from other subspecies that winter in Asia
Our STRUCTURE analyses suggested that the microsatellites possessed little utility for diagnosing subspecies (Appendix C). The GeneClass2 assignment tests provided similar insights. In general, success of the assignment approach was poor, with only 128 (34.6%) of the 370 individuals successfully assigned to the correct subspecies and only 31 of the 144 C. a. arcticola specimens (21.5%) correctly assigned. The average assignment probability of a properly assigned C. a. arcticola was only 0.576, indicating that there was low confidence in the correct assignments that were observed.
By contrast, our application of Bayes' theorem indicated a greater potential for genetic identification of C. a. arcticola if mtDNA data were used. In this case, the probability of a correct identification depends in part on the relative population sizes of C. a. arcticola and C. a. sakhalina (eqn 1; Table4): the two subspecies that winter in Asia and that also can possess a type I haplotype. Using the upper and lower bounds of population size estimates for each subspecies, our calculations suggest that, under the extreme case where the ratio of C. a. sakhalina to C. a. arcticola is 1 000 000:300 000, the probability that a bird possessing a clade I haplotype is a C. a. arcticola individual is 0.698 (Table4). This probability increases to 0.885 when population sizes are assumed to be equal and is as high as 0.982 when the population size of C. a. arcticola is assumed to be the upper extent of its estimated range and C. a. sakhalina is assumed to be at the lower extent of its range (Table4).
Table 4.
Outcomes of calculations to infer P(arcticola|I): the probability that a randomly selected nonbreeding bird in East Asia with a haplotype from the main C. a. arcticola/pacifica group (Fig.2) is actually C. a. arcticola as opposed to C. a. sakhalina. Calculations depend on the relative abundance of C. a. arcticola and C. a. sakhalina and are described in the Materials and methods (eqn 1). This table presents outcomes that evaluated upper, lower, and approximate midpoint population size estimates given by Bamford et al. (2008) and Andres et al. (2012)
| Population estimate | |||||
|---|---|---|---|---|---|
| Total | P(sakhalina) | P(arcticola) | P(arcticola|I) | C. a. sakhalina | C. a. arcticola |
| 100 000 | 300 000 | 400 000 | 0.250 | 0.750 | 0.958 |
| 100 000 | 700 000 | 800 000 | 0.125 | 0.875 | 0.982 |
| 1 000 000 | 300 000 | 1 300 000 | 0.769 | 0.231 | 0.698 |
| 1 000 000 | 700 000 | 1 700 000 | 0.588 | 0.412 | 0.843 |
| 500 000 | 500 000 | 1 000 000 | 0.500 | 0.500 | 0.885 |
Gene flow among Beringian subspecies
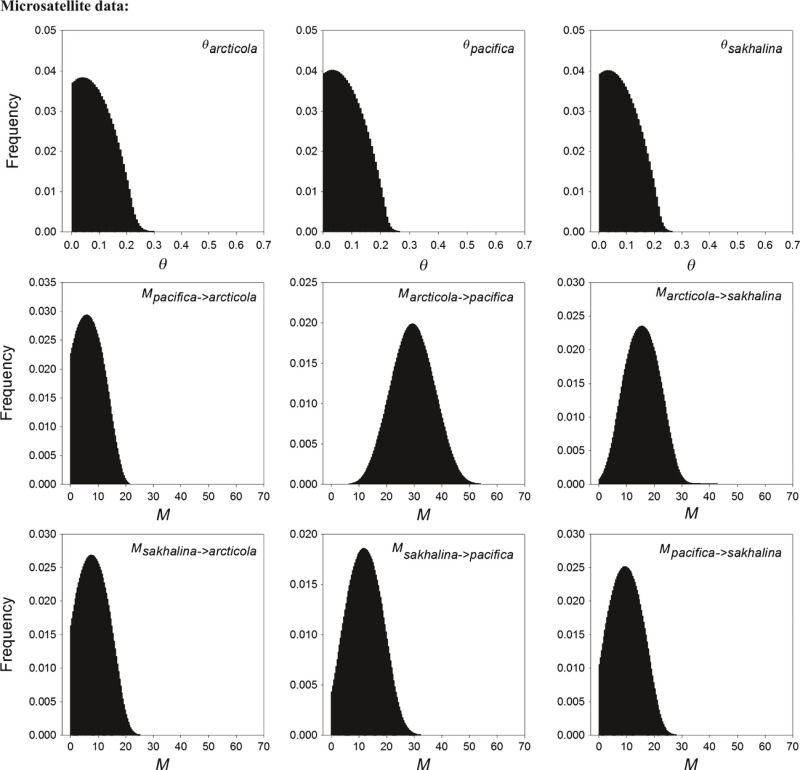
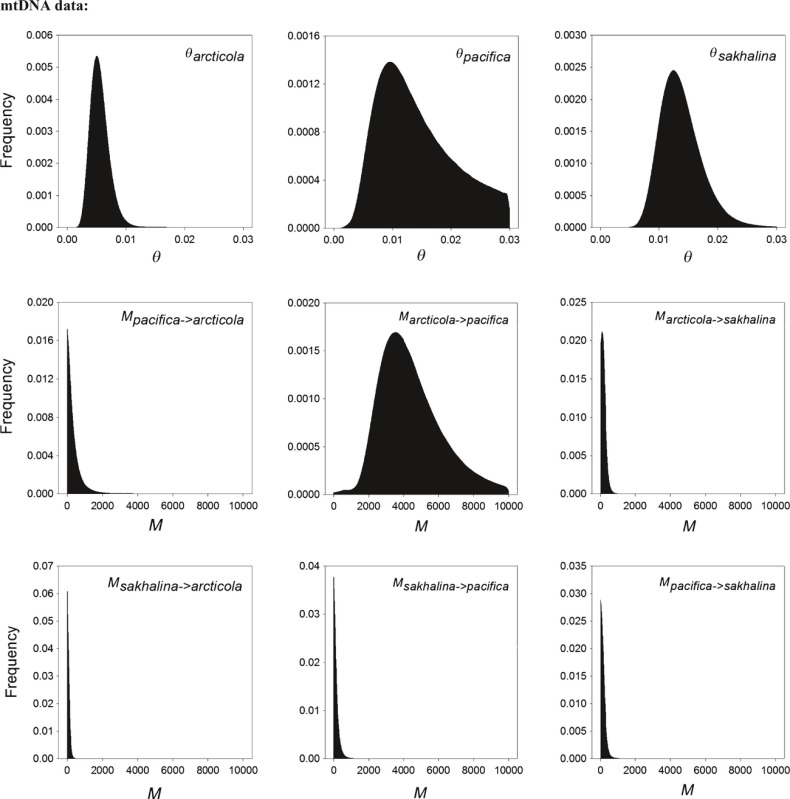
Results of MIGRATE-N analyses were comparable between independent runs for each data set, indicating that convergence had occurred. The posterior distributions for each parameter were also well defined (Appendix E), thus facilitating the generation of point estimates and credibility intervals for each parameter (Table5). In general, gene flow estimates were low. However, the signature of asymmetric gene flow was present in both data sets, where C. a. arcticola was a source of migrants into both C. a. pacifica and C. a. sakhalina, but comparatively little gene flow occurred in the opposite direction. Migration from C. a. arcticola into C. a. pacifica was particularly pronounced, especially based on the results of the mtDNA analysis (Marcticola → pacifica = 3583.3) relative to the microsatellite data (Marcticola → pacifica = 26.83). Migration from C. a. arcticola into C. a. sakhalina (mtDNA: Marcticola → sakhalina = 90.0; microsatellites: Marcticola → sakhalina = 13.5) was also detected, albeit at lower levels than the rate into C. a. pacifica (Table5).
Table 5.
Bayesian estimates of mutation-scaled effective population sizes (θ) and asymmetric migration rates (M) among the Dunlin subspecies C. a. arcticola, C. a. pacifica, and C. a. sakhalina. 95% credibility intervals are reported for each parameter, as is the derived parameter Nem reflecting the effective number of migrants per generation. See text for more details. Posterior distributions of estimated parameters are illustrated in Appendix E
| mtDNA | Microsatellites | |||||
|---|---|---|---|---|---|---|
| 2.5% | Mode | 97.5% | 2.5% | Mode | 97.5% | |
| θarcticola | 0.0026 | 0.0049 | 0.0088 | 0.0000 | 0.0367 | 0.2000 |
| θpacifica | 0.0047 | 0.0096 | 0.0281 | 0.0000 | 0.0300 | 0.1930 |
| θsakhalina | 0.0075 | 0.0125 | 0.0209 | 0.0000 | 0.0300 | 0.1930 |
| Mpacifica → arcticola (Nem) | 0.0 | 3.3 (0.016) | 1313.3 | 0.000 | 8.500 (0.078) | 17.000 |
| Msakhalina → arcticola (Nem) | 0.0 | 3.3 (0.016) | 206.7 | 0.000 | 3.500 (0.032) | 11.667 |
| Marcticola → pacifica (Nem) | 1586.7 | 3583.3 (34.4) | 8320.0 | 10.000 | 26.833 (0.201) | 44.330 |
| Msakhalina → pacifica (Nem) | 0.0 | 3.3 (0.032) | 453.3 | 0.000 | 7.500 (0.056) | 16.333 |
| Marcticola → sakhalina (Nem) | 0.0 | 90.0 (1.125) | 460.0 | 2.667 | 13.500 (0.101) | 24.000 |
| Mpacifica → sakhalina (Nem) | 0.0 | 3.3 (0.041) | 413.3 | 0.000 | 7.167 (0.054) | 15.333 |
Discussion
Migratory birds may facilitate the spread of HPAI from Asia to North America (Winker and Gibson 2010). In this investigation, we used large sample sizes and two genetic data sources (mitochondrial DNA and microsatellites) to determine genetic structure patterns among six Dunlin subspecies that reside in and migrate through eastern Asia and North America. We specifically focused on determining whether the four subspecies of Dunlin that winter in Asia can be differentiated and if genetic evidence for gene flow among Beringian subspecies exists. We suggest that our results may be useful for documenting potential HPAI transmission routes and the pathways that may facilitate the spread of disease across continents.
Birds have reduced genetic structure relative to many other organisms, likely due to their capacity for flight and long distance movement (Greenwood and Harvey 1976; Zink et al. 1997). Many Arctic avian species, particularly migratory species, show lower levels of population genetic structure as a result of these high dispersal tendencies (Crochet 1996). For example, most shorebirds migrate long distances between breeding and nonbreeding areas (Brown et al. 2001), which may result in high gene flow and reduced genetic differentiation (e.g., Baker et al. 1994; Wenink et al. 1994; Haig et al. 1997; Wennerberg 2001b; Draheim et al. 2010; Miller et al. 2012). In contrast to past genetic studies of Dunlin that included limited sampling of Beringia-associated subspecies (e.g., Wenink et al. 1993; Wenink and Baker 1996; Wennerberg et al. 1999, 2008), genetic analyses from our investigation revealed marked genetic differentiation among some Dunlin subspecies based on mtDNA analyses. Phylogenetic analysis revealed four separate phylogroups with high levels of statistical support (Fig.2). Two of these groups consisted of samples from only C. a. hudsonia or C. a. actites, which occur in the most eastern and western regions of our study area. The other two groups contained mixtures of birds from more than one subspecies. The latter groups largely corresponded to birds that breed in relatively close proximity to one another, either in Asia (C. a. sakhalina and C. a. kistchinski) or in Alaska (C. a. arcticola and C. a. pacifica), although a few C. a. sakhalina birds from sites O and Q (Fig.1) possessed haplotypes from the C. a. arcticola/C. a. pacifica group (Fig.2). The lack of clear structure between the C. a. sakhalina/kistchinski and C. a. arcticola/pacifica groups suggests, in part, that the taxonomic status of these subspecies may require revision, although we recognize that other factors are important for defining subspecies (e.g., morphology, behavior, etc.; Haig et al. 2006).
Differentiation among subspecies was less pronounced based on the microsatellites, but significant structure was nonetheless detected between most subspecies pairs (Table3). Male-biased gene flow (Clark et al. 1997; Gibbs et al. 2000) or different evolutionary rates among markers (Brown 1983) are plausible hypotheses that may explain differences between data sets. However, adult male Dunlin usually exhibit higher breeding site fidelity relative to females (Soikkeli 1967, 1970; Jackson 1994; Tomkovich 1994; Hill 2012). Thus, the lower effective population size and greater strength of genetic drift associated with maternally inherited haploid genomes may be the most reasonable explanation for the greater differentiation identified in the mtDNA data. Regardless of data set, the genetic structure patterns that we detected are likely the result of some degree of breeding site fidelity (Warnock and Gill 1996; Hill 2012) and reasonably strong population-specific migratory connectivity exhibited by some subspecies (Fernández et al. 2008; Gill et al. 2013; S. Yezerinac and R. Lanctot, unpublished data).
Assuming that our sample of individuals and subspecies is representative of Dunlin from East Asia, our analysis suggests that we can use our data to obtain rudimentary estimates of the probability of correctly distinguishing Asian- versus Alaskan-breeding birds with mtDNA when sampling takes place in the East Asian nonbreeding areas. With the exception of seven C. a. sakhalina individuals, our representative mtDNA sequences from C. a. sakhalina,C. a. kistchinski, and C. a. actites (n = 107 total) were phylogenetically distinct from the haplotypes identified in Alaskan breeders (C. a. arcticola:n = 60; C. a. pacifica:n = 51; Fig.2). Thus, if an individual possessed a haplotype associated with the C. a. actites or C. a. kistchinski/sakhalina groups, the probability that the individual also breeds in Asia approaches 100% because no Alaska breeders possessed haplotypes from those groups. By contrast, if a bird sampled on the East Asia nonbreeding grounds possesses a haplotype from the main C. a. arcticola/pacifica group, our results suggest that the individual may either be C. a. sakhalina or C. a. arcticola (Fig.2; C. a. pacifica can be excluded from consideration given that this subspecies is entirely restricted to western North America). In this case, our application of Bayes' theorem indicates that there is nominally a ∽70% chance that a randomly selected bird possessing a haplotype from group I is C. a. arcticola (Table4). The probability of a correct inference becomes even larger as the population size ratio of C. a. arcticola to C. a. sakhalina increases (Table4). These probabilities are higher than the 53–60% correct assignment rates found by Gates et al. (2013) when using morphology to differentiate subspecies. Future analyses that combine genetic and morphological data may increase the likelihood of identifying C. a. arcticola in the Asian nonbreeding areas.
An unexpected outcome of our analyses included the detection of asymmetric gene flow from C. a. arcticola into C. a. pacifica and to a lesser extent also into C. a. sakhalina (Table5). After considering potential reasons for this pattern, we highlight the simple fact that C. a. arcticola performs the longest spring migration out of all of the subspecies examined and that its northbound migration pathway crosses over part of the C. a. sakhalina and C. a. pacifica breeding areas (Fig.1). It is feasible that some C. a. arcticola individuals ‘short-stop’ their migration in eastern Russia before crossing the Bering Sea to breed with C. a. sakhalina, and even more stop in western Alaska rather than continuing on to northern Alaska. Most reported cases of migratory short-stopping are associated with fall migrations en route to nonbreeding grounds, with the increased availability of supplemental food from agricultural systems (Wilson 1999; Jefferies et al. 2003) or climate change (Austin and Rehfisch 2005; La Sorte and Thompson 2007; Visser et al. 2009; Charmantier and Gienapp 2013) commonly invoked as possible explanations. In our case, we suggest that the frequency of short-stopping during spring migration may instead be correlated with poor weather conditions, resource limitations encountered during migration, or with the overall health and condition of the short-stopping individuals themselves. Evidence for migratory short-stopping during northbound breeding migrations has also been identified in lesser snow geese (Chen caerulescens caerulescens; Shorey et al. 2011). Given the shallow mitochondrial differentiation of C. a. pacifica and C. a. arcticola (Fig.2), we also cannot rule out the possibility that the signal of asymmetric gene flow is the result of recent divergence of the two subspecies. However, a recent divergence does not preclude the possibility of ongoing gene flow, especially considering the geographic proximity of the breeding ranges of the two subspecies, the long migration flight undertaken by C. a. arcticola, and the fact that the northbound migratory path leads directly over C. a. pacifica's breeding range. In contrast, the signal of asymmetric gene flow from C. a. arcticola into C. a. sakhalina is most likely not the result of a recent divergence. The mtDNA-based phylogenetic tree illustrates that the two subspecies are reasonably well differentiated (Fig.2), thereby leaving gene flow as a more tenable explanation for the analysis outcome.
Our finding of asymmetric gene flow indicates that, in addition to C. a. arcticola's usual northern Alaska breeding grounds, the western Alaska breeding grounds for C. a. pacifica need to be considered as a possible secondary entry point for Dunlin to carry AI into North America. This may be especially relevant if the migratory short-stopping behavior is influenced by an individual's health status, particularly if ill due to a viral disease. Because western Alaska and northern Alaska do not possess the same avian assemblages (Gabrielson and Lincoln 1959; Johnson and Herter 1989), the introduction of AI into western Alaska could lead to outbreaks in an additional and different suite of species than would an outbreak centered in northern Alaska.
The inference of asymmetric gene flow also implies the occurrence of direct interactions between C. a. arcticola and C. a. pacifica that could facilitate virus transmission between subspecies. Prior studies indicated that the two subspecies intermix during the fall after the breeding season (Taylor et al. 2011; Gill et al. 2013). If this were the only period of interaction, then the likelihood of HPAI spreading between subspecies would be low because any C. a. arcticola individuals harboring the virus would have had to (i) be infected on the wintering grounds and then (ii) live for 3–4 months with an active infection prior to intermixing with C. a. pacifica in the fall. However, our results suggest that individuals of the two subspecies sexually reproduce and thus likely share incubation duties for about 20 days (Warnock and Gill 1996). The breeding period occurs not long after migration and may coincide with the time when active shedding of HPAI by infected individuals is occurring.
Although our new findings do not specifically identify strategies for preventing the transmission of HPAI into North America, they nonetheless reveal a mechanism by which Dunlin could facilitate the spread of HPAI into North America and Mexico. This is particularly pertinent given that Dunlin are highly susceptible to infection with the H5N1 HPAI, and that some individuals may live to spread the disease, possibly after undergoing a migration (Hall et al. 2011). Although only a few Dunlin sampled in western North America have been documented with actively shedding AI (Ip et al. 2008; Iverson et al. 2008; USFWS and USGS 2011), the continued emergence of new HPAI strains (e.g., H5N8, H7N9) and the fact that most efforts to date have detected prior exposure (i.e., antibodies, see Pearce et al. 2012; Johnson et al. 2014) indicates that the evolution of new strains remains problematic and that Dunlin are a potential route for HPAI to reach and spread within North America.
Acknowledgments
We are grateful to the many individuals that provided samples for this study, including S. Drovetski, D. Edwards, D. Hope, J. Liebezeit, T. Miller, Y. Red'kin, B. Schwartz, C. Gratto-Trevor, U. Somjee, and V. Sotnikov. Samples were collected under the USFWS IACUC and salvage permits 2009012, MB085371-0, and MB025076-0, and the State of Alaska scientific permit #09-071. Dunlin were collected in Russia under permit 87 # 01/2009, Division for Conservation and Use of Animals, Department of Agricultural Policy and Use of Nature Resources, Chukotskiy Autonomous Area. We thank the Burke Museum of Natural History and Culture for providing tissue samples from their collections and H. Draheim for additional project assistance. P. Beerli and D. Dalthorp provided helpful discussion and guidance on some of the statistical approaches that were employed. S. Saalfeld graciously produced Fig.1. J. Busch provided helpful comments on an earlier manuscript draft. Funding was provided by the U.S. Geological Survey Forest and Rangeland Ecosystem Science Center, USFWS's Avian Health and Disease Program and the Region 7 Migratory Bird Management Division, Arctic Expedition of the Institute of Ecology and Evolution in Moscow, and Amur-Ussuri Centre for Avian Biodiversity. Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the U.S. Government. The findings and conclusions in this article are those of the authors and do not necessarily represent the views of the U.S. Fish and Wildlife Service.
Appendix A
Microsatellite and mitochondrial primer sequencers and PCR annealing temperatures (TA) used in Dunlin (Calidris alpina) analyses
| Primer names | Primer sequences | TA (°C) | |
|---|---|---|---|
| Microsatellites | CALP2R | 5′-CAG AGC TGG AAG GT-3′ | 58 |
| CALP2F | 5′-CAA AGG ATG TGG TT-3′ | ||
| CME2R | 5′-TTA AAA GGG ACC GAG TGT CCT-3′ | 58 | |
| CME2F | 5′-GGC TCT GCA TGA AAG TCT AAA TG-3′ | ||
| CME10R | 5′-TGT TAC CAA AGG CTT AAG CAA AG-3′ | 58 | |
| CME10F | 5′-GAA GGC GAG GAG AAC TTC TGT-3′ | ||
| CME12R | 5′-GTT GGG GGA CTA AAG GAA GAC-3′ | 58 | |
| CME12F | 5′-GAG CGG GAC GAG GAC AGT-3′ | ||
| 4A11R | 5′-GGC ACA AAG CTC ACA CCT CTA TG-3′ | 58 | |
| 4A11F | 5′-TCT AGC CTG AAA ATC TGT CCT TG-3′ | ||
| D25R2 | 5′-CCT TGC TTT AGT CAA AGG TGA-3′ | 54 | |
| D25F2 | 5′-GAG AGG ACC AGG AAA CAC T-3′ | ||
| D26R | 5′-GGA AGG CGT GTT GAT ACT G-3′ | 58 | |
| D26F1 | 5′-CAG CGT GAC ATT AAC TCT CTG-3′ | ||
| D110R1 | 5′-GAA ATT ACA AAG TAT GCT GAG-3′ | 54 | |
| D110F1 | 5′-CAA CTA TAT CAG CAG GAA GCT-3′ | ||
| Cytochrome b | L14996 | 5′-AAY ATY TCW GYH TGA TGA AAY TTY GG-3′ | 55 |
| H15646 | 5′-GGN GTR AAG TTT TCT GGG TCN CC-3′ | ||
| Control region | TS 96L | 5′-GCA TGT AAT TTG GGC ATT TTT TG-3′ | 53 |
| TS 778H | 5′-AAA CAC TTG AAA CCG TCT CAT-3′ |
Appendix B
Absolute and relative (in parentheses) frequencies of 94 combined mitochondrial cytochrome b and D-loop haplotypes within six Dunlin (Calidris alpina) subspecies
| Subspecies | ||||||
|---|---|---|---|---|---|---|
| Haplotype | arcticola | actites | hudsonia | kistchinski | pacifica | sakhalina |
| H1 | – | – | 2 (0.125) | – | – | – |
| H2 | – | – | 1 (0.063) | – | – | – |
| H3 | – | – | 4 (0.250) | – | – | – |
| H4 | – | – | 2 (0.125) | – | – | – |
| H5 | – | – | 1 (0.063) | – | – | – |
| H6 | – | – | 1 (0.063) | – | – | – |
| H7 | – | – | 1 (0.063) | – | – | – |
| H8 | – | – | 3 (0.188) | – | – | – |
| H9 | – | – | 1 (0.063) | – | – | – |
| H10 | 1 (0.017) | – | – | – | – | – |
| H11 | – | – | – | 2 (0.067) | – | – |
| H12 | – | – | – | 5 (0.167) | – | – |
| H13 | – | 1 (0.043) | – | – | – | – |
| H14 | – | 1 (0.043) | – | – | – | – |
| H15 | – | – | – | – | 1 (0.020) | – |
| H16 | – | 2 (0.087) | – | – | – | – |
| H17 | – | – | – | – | – | 1 (0.019) |
| H18 | – | – | – | 6 (0.200) | – | 9 (0167) |
| H19 | – | – | – | – | – | 1 (0.019) |
| H20 | – | – | – | – | 1 (0.020) | – |
| H21 | – | – | – | 1 (0.033) | – | – |
| H22 | – | – | – | – | – | 1 (0.019) |
| H23 | – | – | – | 1 (0.033) | – | – |
| H24 | – | 6 (0.261) | – | – | – | – |
| H25 | – | 1 (0.043) | – | – | – | – |
| H26 | – | 2 (0.087) | – | – | – | – |
| H27 | – | – | – | – | – | 1 (0.019) |
| H28 | – | – | – | – | – | 1 (0.019) |
| H29 | – | 1 (0.043) | – | – | – | – |
| H30 | – | 4 (0.174) | – | – | – | – |
| H31 | – | 5 (0.217) | – | – | – | – |
| H32 | – | – | – | – | – | 3 (0.056) |
| H33 | – | – | – | 1 (0.033) | – | – |
| H34 | – | – | – | – | – | 1 (0.019) |
| H35 | – | – | – | – | – | 1 (0.019) |
| H36 | – | – | – | – | – | 1 (0.019) |
| H37 | – | – | – | – | – | 1 (0.019) |
| H38 | – | – | – | – | 1 (0.020) | – |
| H39 | – | – | – | 1 (0.033) | – | – |
| H40 | – | – | – | 2 (0.067) | – | 5 (0.093) |
| H41 | – | – | – | – | 1 (0.020) | |
| H42 | – | – | – | – | 10 (0.196) | |
| H43 | – | – | – | – | 1 (0.019) | |
| H44 | 23 (0.383) | – | – | – | 10 (0.196) | 1 (0.019) |
| H45 | 1 (0.017) | – | – | – | – | – |
| H46 | – | – | – | 1 (0.020) | – | |
| H47 | 2 (0.033) | – | – | – | 3 (0.059) | – |
| H48 | 1 (0.017) | – | – | – | 2 (0.039) | – |
| H49 | 1 (0.017) | – | – | – | – | – |
| H50 | – | – | – | – | 2 (0.039) | 1 (0.019) |
| H51 | – | – | – | – | 1 (0.020) | – |
| H52 | – | – | – | – | 3 (0.059) | – |
| H53 | – | – | – | 1 (0.033) | – | – |
| H54 | – | – | – | – | – | 1 (0.019) |
| H55 | – | – | – | 1 (0.033) | – | – |
| H56 | – | – | – | 1 (0.033) | – | – |
| H57 | – | – | – | – | – | 1 (0.019) |
| H58 | – | – | – | – | – | 1 (0.019) |
| H59 | 1 (0.017) | – | – | – | – | – |
| H60 | – | – | – | – | 1 (0.020) | – |
| H61 | – | – | – | – | – | 1 (0.019) |
| H62 | – | – | – | 1 (0.033) | – | – |
| H63 | – | – | – | – | – | 1 (0.019) |
| H64 | – | – | – | 1 (0.033) | – | – |
| H65 | – | – | – | 1 (0.033) | – | – |
| H66 | – | – | – | – | – | 2 (0.019) |
| H67 | 1 (0.017) | – | – | – | – | – |
| H68 | – | – | – | – | 2 (0.039) | – |
| H69 | – | – | – | 3 (0.100) | – | 5 (0.093) |
| H70 | – | – | – | – | – | 2 (0.037) |
| H71 | – | – | – | – | – | 1 (0.019) |
| H72 | – | – | – | 1 (0.033) | – | |
| H73 | – | – | – | – | – | 1 (0.019) |
| H74 | – | – | – | – | – | 1 (0.019) |
| H75 | – | – | – | 1 (0.033) | – | 1 (0.019) |
| H76 | – | – | – | – | – | 1 (0.019) |
| H77 | – | – | – | – | – | 1 (0.019) |
| H78 | 9 (0.150) | – | – | – | 8 (0.157) | 4 (0.074) |
| H79 | 2 (0.033) | – | – | – | – | – |
| H80 | 4 (0.067) | – | – | – | – | – |
| H81 | 1 (0.017) | – | – | – | 1 (0.020) | 1 (0.019) |
| H82 | 1 (0.017) | – | – | – | – | – |
| H83 | 1 (0.017) | – | – | – | – | – |
| H84 | 3 (0.050) | – | – | – | – | – |
| H85 | 1 (0.017) | – | – | – | – | – |
| H86 | 1 (0.017) | – | – | – | – | – |
| H87 | 1 (0.017) | – | – | – | – | – |
| H88 | 2 (0.033) | – | – | – | – | – |
| H89 | 1 (0.017) | – | – | – | – | – |
| H90 | 1 (0.017) | – | – | – | – | – |
| H91 | 1 (0.017) | – | – | – | – | – |
| H92 | – | – | – | – | 1 (0.020) | – |
| H93 | – | – | – | – | 1 (0.020) | – |
| H94 | – | – | – | – | 1 (0.020) | – |
| Total | 60 | 23 | 16 | 30 | 51 | 54 |
Appendix C
Results from STRUCTURE analyses using eight microsatellite loci. The highest average likelihood was associated with the K = 5 case (panel A), suggesting the presence of five genetic clusters. However, assignment probabilities of individuals to these clusters were nearly uniform (panel B), indicating that K had been overestimated. Use of the Evanno et al. (2005) ΔK approach (panel C) suggested that there were two clusters; however, average assignment probabilities of individuals to these clusters were also uninformative (panel D). These results suggest that there is no detectable subspecies subdivision based on the microsatellites.
Appendix D
Allele frequencies from eight microsatellite loci within six Dunlin (Calidris alpina) subspecies
| Subspecies | ||||||||
|---|---|---|---|---|---|---|---|---|
| Locus | Allele size | arcticola | actites | hudsonia | kistchinski | pacifica | sakhalina | Overall |
| Calp2 | 120 | 0.0069 | – | – | – | – | – | 0.0027 |
| 122 | 0.0278 | – | – | 0.0595 | 0.0132 | 0.0362 | 0.0270 | |
| 124 | 0.0174 | 0.2609 | – | 0.1190 | 0.0263 | 0.0942 | 0.0595 | |
| 126 | 0.2535 | 0.1957 | 0.1875 | 0.1071 | 0.1579 | 0.1594 | 0.1932 | |
| 128 | 0.1979 | – | 0.0625 | 0.0595 | 0.0921 | 0.0870 | 0.1216 | |
| 130 | 0.0486 | – | 0.0625 | 0.1071 | 0.1053 | 0.0580 | 0.0662 | |
| 132 | 0.0625 | 0.0652 | 0.0938 | 0.1667 | 0.0592 | 0.1087 | 0.0838 | |
| 134 | 0.0208 | 0.0435 | 0.1562 | 0.0595 | 0.0461 | 0.0580 | 0.0446 | |
| 136 | 0.1111 | 0.3696 | 0.1250 | 0.1429 | 0.0855 | 0.1667 | 0.1365 | |
| 138 | 0.2361 | 0.0652 | 0.3125 | 0.1548 | 0.3289 | 0.1377 | 0.2203 | |
| 140 | 0.0174 | – | – | 0.0119 | 0.0789 | 0.0725 | 0.0378 | |
| 142 | – | – | – | 0.0119 | 0.0066 | 0.0145 | 0.0054 | |
| 144 | – | – | – | – | – | 0.0072 | 0.0014 | |
| Cme2 | 137 | 0.0243 | – | 0.0312 | – | 0.0132 | – | 0.0135 |
| 139 | – | 0.0870 | – | – | 0.0066 | 0.0145 | 0.0095 | |
| 141 | 0.0903 | 0.1739 | – | 0.0119 | 0.0395 | 0.0435 | 0.0635 | |
| 143 | – | – | – | 0.0357 | – | – | 0.0041 | |
| 145 | 0.0069 | 0.0652 | – | – | 0.0197 | 0.0290 | 0.0162 | |
| 147 | 0.1562 | 0.0217 | 0.0312 | 0.2024 | 0.0789 | 0.2101 | 0.1419 | |
| 149 | 0.2986 | 0.4783 | 0.0938 | 0.3452 | 0.3618 | 0.2464 | 0.3095 | |
| 151 | 0.2535 | 0.0652 | 0.2188 | 0.1548 | 0.2829 | 0.1739 | 0.2203 | |
| 153 | 0.0938 | 0.0217 | – | 0.1190 | 0.1316 | 0.0942 | 0.0959 | |
| 155 | 0.0590 | – | 0.4375 | 0.1071 | 0.0658 | 0.1014 | 0.0865 | |
| 157 | 0.0174 | 0.0870 | 0.1250 | 0.0238 | – | 0.0870 | 0.0365 | |
| 159 | – | – | 0.0312 | – | – | – | 0.0014 | |
| 163 | – | – | 0.0312 | – | – | – | 0.0014 | |
| Cme10 | 177 | 0.0035 | – | – | – | 0.0066 | 0.0072 | 0.0041 |
| 181 | 0.9826 | 1.0000 | 0.9062 | 0.9881 | 0.9276 | 0.9565 | 0.9649 | |
| 183 | 0.0139 | – | 0.0938 | 0.0119 | 0.0658 | 0.0145 | 0.0270 | |
| 187 | – | – | – | – | – | 0.0217 | 0.0041 | |
| Cme12 | 164 | – | – | – | 0.0119 | 0.0066 | – | 0.0027 |
| 166 | 0.0069 | – | – | – | – | 0.0145 | 0.0054 | |
| 168 | 0.0035 | – | – | – | – | – | 0.0014 | |
| 170 | 0.0278 | 0.0870 | 0.0312 | 0.0119 | 0.0329 | 0.0290 | 0.0311 | |
| 172 | 0.3229 | 0.1304 | 0.1562 | 0.3690 | 0.3816 | 0.3406 | 0.3243 | |
| 174 | 0.5174 | 0.3478 | 0.7500 | 0.4643 | 0.5000 | 0.5290 | 0.5095 | |
| 176 | 0.1215 | 0.4348 | 0.0625 | 0.1429 | 0.0789 | 0.0870 | 0.1257 | |
| 4A11 | 139 | – | – | – | – | 0.0066 | 0.0072 | 0.0027 |
| 141 | 0.2743 | 0.0870 | 0.2188 | 0.3452 | 0.3289 | 0.4130 | 0.3054 | |
| 143 | 0.6840 | 0.8696 | 0.7500 | 0.5833 | 0.6316 | 0.4928 | 0.6405 | |
| 145 | 0.0417 | 0.0435 | 0.0312 | 0.0714 | 0.0329 | 0.0870 | 0.0514 | |
| D25 | 326 | 0.0521 | 0.2391 | 0.0312 | 0.0714 | 0.0592 | 0.1594 | 0.0865 |
| 328 | 0.8229 | 0.7609 | 0.5625 | 0.7381 | 0.8092 | 0.6957 | 0.7716 | |
| 330 | 0.0556 | – | 0.3750 | 0.1310 | 0.0329 | 0.0435 | 0.0676 | |
| 332 | 0.0694 | – | 0.0312 | 0.0476 | 0.0921 | 0.0870 | 0.0689 | |
| 334 | – | – | – | 0.0119 | 0.0066 | 0.0072 | 0.0041 | |
| 336 | – | – | – | – | – | 0.0072 | 0.0014 | |
| D26 | 237 | 0.0382 | – | 0.0312 | 0.0476 | 0.0461 | 0.0942 | 0.0486 |
| 239 | 0.0104 | – | – | – | – | – | 0.0041 | |
| 241 | – | – | – | – | – | 0.0362 | 0.0068 | |
| 243 | 0.3993 | 0.5652 | 0.5938 | 0.3095 | 0.2500 | 0.4348 | 0.3838 | |
| 245 | 0.4167 | 0.1739 | 0.3750 | 0.4881 | 0.5592 | 0.3043 | 0.4162 | |
| 247 | 0.1111 | 0.2174 | – | 0.1429 | 0.0987 | 0.1159 | 0.1149 | |
| 249 | – | 0.0435 | – | 0.0119 | 0.0132 | 0.0072 | 0.0081 | |
| 251 | 0.0139 | – | – | – | 0.0263 | 0.0072 | 0.0122 | |
| 253 | 0.0104 | – | – | – | 0.0066 | – | 0.0054 | |
| D110 | 184 | 0.0729 | – | 0.0312 | 0.0238 | 0.0658 | 0.0290 | 0.0514 |
| 186 | 0.1007 | 0.0217 | 0.1562 | 0.0833 | 0.1250 | 0.1159 | 0.1041 | |
| 188 | 0.3299 | 0.7826 | 0.5625 | 0.3452 | 0.2697 | 0.3188 | 0.3554 | |
| 190 | 0.4826 | 0.1957 | 0.2500 | 0.5119 | 0.5329 | 0.4928 | 0.4703 | |
| 192 | 0.0139 | – | – | 0.0357 | 0.0066 | 0.0362 | 0.0176 | |
| 196 | – | – | – | – | – | 0.0072 | 0.0014 | |
Appendix E
Posterior distributions for Bayesian estimates of mutation-scaled effective population sizes (θ) and migration rates (M) obtained from MIGRATE-N (Beerli and Palczewski 2010)
Point estimates and credibility intervals calculated from the posterior distributions are provided in Table5. See text for more details.
mtDNA data:
Data archiving statement
Data for this study are available at: GenBank Accession Numbers KP205084–KP205177 and KP205178–KP205271. Dryad Digital Repository http://dx.doi.org/10.5061/dryad.4t806.
Literature cited
- Alaska Interagency HPAI Bird Surveillance Working Group. 2006. Anchorage, AK Sampling protocol for highly pathogenic Asian H5N1 avian influenza in migratory birds in Alaska. Interagency planning report, http://www.fws.gov/alaska/mbsp/mbm/ai/AlaskaInteragencyHPAISampleProtocol_FinalAdob5.pdf (accessed on 12 June 2014)
- American Ornithologists' Union. Checklist of North American Birds. Washington, D.C: American Ornithologists' Union; 2013. [Google Scholar]
- Andres BA, Smith PA, Morrison RIG, Gratto-Trevor CL, Brown SC. Friis CA. Population estimates of North American shorebirds, 2012. Wader Study Group Bulletin. 2012;119:178–194. [Google Scholar]
- Austin GE. Rehfisch MM. Shifting nonbreeding distributions of migratory fauna in relation to climatic change. Global Change Biology. 2005;11:31–38. [Google Scholar]
- Baker AJ, Piersma T. Rosenmeier L. Unraveling the intraspecific phylogeography of knots Calidris canutus, a progress report on the search for genetic markers. Journal für Ornithologie. 1994;135:599–608. [Google Scholar]
- Bamford M, Watkins D, Bancroft W, Tischler G. Wahl J. Migratory Shorebirds of the East Asian – Australasian Flyway; Population Estimates and Internationally Important Sites. Canberra, ACT, Australia: Wetlands International – Oceania; 2008. [Google Scholar]
- Beerli P. How to use Migrate or why are Markov chain Monte Carlo programs difficult to use? In: Vernesi C, editor; Bertorelle G, Bruford MW, Haue HC, Rizzoli A, editors. Population Genetics for Animal Conservation. Cambridge: Cambridge University Press; 2009. pp. 42–79. [Google Scholar]
- Beerli P. Palczewski M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics. 2010;185:313–326. doi: 10.1534/genetics.109.112532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WM. Evolution of animal mitochondrial DNA. In: Koehn RK, editor; Nei M, editor. Evolution of Genes and Proteins. Sunderland: Sinauer Associates; 1983. pp. 62–88. [Google Scholar]
- Brown S, Hickey C, Harrington B. Gill R. United States Shorebird Conservation Plan. 2nd edn. Manomet, MA: Manomet Center for Conservation Sciences; 2001. [Google Scholar]
- Browning MR. Taxonomic comments on the Dunlin, Calidris alpine, from northern Alaska and eastern Siberia. Bulletin of the British Ornithologists' Club. 1991;111:140–145. [Google Scholar]
- Charmantier A. Gienapp P. Climate change and timing of avian breeding and migration: evolutionary versus plastic changes. Evolutionary Applications. 2013;7:15–28. doi: 10.1111/eva.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Deng G, Li Z, Tian G, Li Y, Jiao P, Zhang L, et al. The evolution of H5N1 influenza viruses in ducks in southern China. Proceedings of the National Academy of Science, USA. 2004;101:10452–10457. doi: 10.1073/pnas.0403212101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Smith GJD, Li KS, Wang J, Fan XH, Rayner JM, Vijaykrishna D, et al. Establishment of multiple sublineages of H5N1 influenza virus in Asia: implications for pandemic control. Proceedings of the National Academy of Science, USA. 2006;103:2845–2850. doi: 10.1073/pnas.0511120103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AL, Sæther BE. Røskaft E. Sex biases in avian dispersal, a reappraisal. Oikos. 1997;79:429–438. [Google Scholar]
- Clements JF, Schulenberg TS, Iliff MJ, Sullivan BL, Wood CL. Roberson D. 2013. , and The eBird/Clements checklist of birds of the world: Version 6.8 http://www.birds.cornell.edu/clementschecklist/ accessed 15 January 2015.
- Cornuet JM, Piry S, Luikart G, Estoup A. Solignac M. New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics. 1999;1531:989–2000. doi: 10.1093/genetics/153.4.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig DW, Pearson JV, Szelinger S, Sekar A, Redman M, Corneveaux JJ, Pawlowski TL, et al. Identification of genetic variants using bar-coded multiplexed sequencing. Nature Methods. 2008;5:887–893. doi: 10.1038/nmeth.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crochet PA. Can measures of gene flow help to evaluate bird dispersal? International Journal of Ecology. 1996;17:459–474. [Google Scholar]
- Cronn R, Liston A, Parks M, Gernandt DS, Shen R. Mockler T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Research. 2008;36:e122. doi: 10.1093/nar/gkn502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R. Posada D. jModeltest 2: more models, new heuristics and parallel computing. Nature Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Hoyo J, Elliott A. Sargatal J. Handbook of the Birds of the World, Vol. 3. Hoatzin to Auks. Barcelona: Lynx Edicions; 1996. [Google Scholar]
- Draheim HM, Miller MP, Baird P. Haig SM. Subspecific status and population genetic structure of Least Terns (Sternula antillarum) inferred by mitochondrial DNA control-region sequences and microsatellite DNA. The Auk. 2010;127:807–819. [Google Scholar]
- Dusek RJ, Hallgrimsson GT, Ip HS, Jónsson JE, Sreevatsan S, Nashold SW, TeSlaa JL, et al. North Atlantic migratory bird flyways provide routes for intercontinental movement of avian influenza viruses. PLoS ONE. 2014;9:e92075. doi: 10.1371/journal.pone.0092075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G, Regnaut S. Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE. Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes, application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Laval G. Schneider S. Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Fergus R, Fry M, Karesh WB, Marra PP, Newman S. Paul E. Migratory birds and avian flu. Science. 2006;312:845–846. doi: 10.1126/science.312.5775.845c. [DOI] [PubMed] [Google Scholar]
- Ferguson NM, Cummings DAT, Cauchemez S, Frazer C, Riley S, Meeyai A, Iamsirithaworn S, et al. Strategies for containing an emerging influenza pandemic in Southeast Asia. Nature. 2005;437:209–214. doi: 10.1038/nature04017. [DOI] [PubMed] [Google Scholar]
- Fernández G, Buchanan JB, Gill RE, Lanctot RB. Warnock N. Conservation Plan for Dunlin with Breeding Populations in North America (Calidris alpina arcticola, C. a. pacifica and C. a. hudsonia), Version 1.0. Manomet: Manomet Center for Conservation Sciences; 2008. [Google Scholar]
- Gabrielson IN. Lincoln FC. The Birds of Alaska. Washington D.C: The Stackpole Company, Harrisburg, and the Wildlife Management Institute; 1959. p. 922. [Google Scholar]
- Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. The New England Journal of Medicine. 2013;368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- Gates HR, Yezerinac S, Powell AN, Tomkovich PS, Valchuk OP. Lanctot RB. Differentiation of subspecies and sexes of Beringian Dunlins using morphometric measures. Journal of Field Ornithology. 2013;84:389–402. [Google Scholar]
- Gibbs HL, Dawson RJG. Hobson KA. Limited differentiation in microsatellite DNA variation among northern populations of the yellow warbler: evidence for male-biased gene flow? Molecular Ecology. 2000;9:2137–2147. doi: 10.1046/j.1365-294x.2000.01136.x. [DOI] [PubMed] [Google Scholar]
- Gilbert M, Xiao X, Domenech J, Lubroth J, Martin V. Slingenberg J. Anatidae migration in the western Palearctic and spread of highly pathogenic avian influenza H5N1 virus. Emerging Infectious Diseases. 2006;12:1650–1656. doi: 10.3201/eid1211.060223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill RE, Handel CM. Ruthrauff DR. Intercontinental migratory connectivity and population structuring in Dunlins (Calidris alpina) from western Alaska. Condor. 2013;115:525–534. [Google Scholar]
- Glaubitz JC. CONVERT: a user friendly program to reformat diploid genotypic data for commonly used population genetic software packages. Molecular Ecology Notes. 2004;4:309–310. [Google Scholar]
- Greenwood JG. Geographical variation and taxonomy of the Dunlin Calidris alpine (L.) Bulletin of the British Ornithologists' Club. 1986;106:43–56. [Google Scholar]
- Greenwood PJ. Harvey PH. Mating systems, philopatry and dispersal in birds and mammals. Animal Behaviour. 1976;28:1140–1162. [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W. Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology. 2010;59:301–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Haig SM, Gratto-Trevor CL, Mullins TD. Colwell MA. Population identification of western hemisphere shorebirds throughout the annual cycle. Molecular Ecology. 1997;6:413–427. [Google Scholar]
- Haig SM, Forsman ED. Mullins TD. Subspecies relationships and genetic structure in the Spotted Owl. Conservation Genetics. 2004;5:683–705. [Google Scholar]
- Haig SM, Beever EA, Chambers SM, Draheim HM, Dugger BD, Dunham S, Elliott-Smith E, et al. Taxonomic considerations in listing subspecies under the U.S. Endangered Species Act. Conservation Biology. 2006;20:1584–1594. doi: 10.1111/j.1523-1739.2006.00530.x. [DOI] [PubMed] [Google Scholar]
- Haig SM, Bronaugh W, Crowhurst R, D'Elia J, Eagles-Smith C, Epps C, Knaus B, et al. Perspectives in ornithology: applications of genetics in avian conservation. Auk. 2011;128:205–229. [Google Scholar]
- Hall JS, Franson JC, Gill RE, Meteyer CU, TeSlaa JL, Nashold S, Dusek RJ, et al. Experimental challenge and pathology of highly pathogenic avian influenza virus H5N1 in Dunlin (Calidris alpina), an intercontinental migrant shorebird species. Influenza and Other Respiratory Viruses. 2011;5:365–372. doi: 10.1111/j.1750-2659.2011.00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill BL. 2012. Fairbanks, AK University of Alaska Fairbanks Factors affecting survival of arctic-breeding Dunlin (Calidris alpina arcticola) adults and chicks. MS Thesis.
- Huelsenbeck JP. Ronquist F. MrBayes: Bayesian inference of phylogeny. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Ip HP, Flint PL, Franson JC, Dusek RJ, Derksen DV, Gill RE, Ely CR, et al. Prevalence of influenza A viruses in wild migratory birds in Alaska: patterns of variation in detection at the crossroads of intercontinental flyways. Virology Journal. 2008;5:71. doi: 10.1186/1743-422X-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro F, Takada N. Masuzawa R. Molecular evidence of the dispersal of Lyme disease Borrelia from the Asian continent to Japan via migratory birds. Japanese Journal of Infectious Diseases. 2005;58:184–186. [PubMed] [Google Scholar]
- Iverson SA, Takekawa JY, Schwarzbach S, Cardona CJ, Warnock N, Bishop MA, Schiratao GA, et al. Low prevalence of avian influenza virus in shorebirds on the Pacific Coast of North America. Waterbirds. 2008;31:602–610. [Google Scholar]
- Jackson DB. Breeding dispersal and site-fidelity in three monogamous wader species in the Western Isles, UK. Ibis. 1994;136:463–473. [Google Scholar]
- Jakobsson M. Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- Jefferies RL, Rockwell RF. Abraham KF. The embarrassment of riches: agricultural food subsidies, high goose numbers, and loss of Arctic wetlands – a continuing saga. Environmental Reviews. 2003;11:193–232. [Google Scholar]
- Jennings TN, Knaus BJ, Mullins TD. Haig SM. Multiplexed microsatellite recovery using massively parallel sequencing. Molecular Ecology Resources. 2011;11:1060–1067. doi: 10.1111/j.1755-0998.2011.03033.x. [DOI] [PubMed] [Google Scholar]
- Johnson SR. Herter DR. Birds of the Beaufort Sea. Anchorage: BP Exploration; 1989. p. 372. [Google Scholar]
- Johnson JA, DeCicco LH, Ruthrauff DR, Krauss S. Hall JS. Avian influenza virus antibodies in Pacific coast Red Knots (Calidris canutus roselaari. Journal of Wildlife Diseases. 2014;50:671–675. doi: 10.7589/2013-04-016. [DOI] [PubMed] [Google Scholar]
- Kalinowski ST. HP-Rare. A computer program for performing rarefaction on measures of allelic diversity. Molecular Ecology Notes. 2005;5:187–189. [Google Scholar]
- Kilpatrick AM, Chmura AA, Gibbons DW, Fleischer RC, Marra PP. Daszak P. Predicting the global spread of H5N1 avian influenza. Proceedings of the National Academy of Science, USA. 2006;103:19368–19373. doi: 10.1073/pnas.0609227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Sorte FA. Thompson FR. Poleward shifts in winter ranges of North American birds. Ecology. 2007;88:1803–1812. doi: 10.1890/06-1072.1. [DOI] [PubMed] [Google Scholar]
- Lanctot RB, Barter M, Chiang CY, Gill R, Johnson M, Haig S, Ma Z, et al. 2009. pp. 149–164. Nantou County, Republic of China (Taiwan) Endemic Species Research Institute Use of band resightings, molecular markers and stable isotopes to understand the migratory connectivity of Dunlin breeding in Beringia and wintering in the East Asian-Australasian Flyway. In Proceedings from the 2009 International Symposium on Coastal Wetlands and Water Birds Conservation.
- Lappo EG, Tomkovich PS. Syroechkovskiy EE., Jr . Atlas of Breeding Waders in the Russian Arctic. Moscow: UFOfsetnaya Pechat; 2012. [Google Scholar]
- Lewis P. Zaykin D. 2002. , and GDA: Genetic Data Analysis Computer software distributed by authors from http://www.eeb.uconn.edu/people/plewis/software.php (accessed on 15 January 2015)
- Li KS, Guan Y, Wang J, Smith GJD, Xu KM, Duan L, Rahardjo AP, et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature. 2004;430:209–213. doi: 10.1038/nature02746. [DOI] [PubMed] [Google Scholar]
- Longmire JL, Maltbie M. Baker RJ. Use of “lysis buffer” in DNA isolation and its implications for museum collections. Occasional Papers: Museum of Texas Tech University. 1997;163:1–3. [Google Scholar]
- Marthinsen G, Wennerberg L. Lifjeld JT. Phylogeography and subspecies taxonomy of Dunlin (Calidris alpina) in Western Palearctic analysed by DNA microsatellites and amplified fragment length polymorphism markers. Biological Journal of the Linnean Society. 2007;92:713–726. [Google Scholar]
- Miller MP, Mullins TD, Parrish JW, Jr, Walters JR. Haig SM. Variation in migratory behavior influences regional genetic diversity and structure among American kestrel populations (Falco sparverius) in North America. Journal of Heredity. 2012;103:503–514. doi: 10.1093/jhered/ess024. [DOI] [PubMed] [Google Scholar]
- Morshed MG, Scott JD, Fernando K, Beati L, Mazerolle DF, Geddes G. Durden LA. Migratory songbirds disperse ticks across Canada, and first isolation of the Lyme disease spirochete, Borrelia burgdorferi, from the avian tick, Ixodes auritulus. Journal of Parasitology. 2005;91:780–790. doi: 10.1645/GE-3437.1. [DOI] [PubMed] [Google Scholar]
- Nechaev VA. Tomkovich PS. A new subspecies of the Dunlin, Calidris aluina litoralis (Charadriidae, Aves), from the Sakhalin Island. Zoologichesky Zhurnal. 1987;66:1110–1113. [Google Scholar]
- Olsen B, Munster VJ, Wallenstein A, Waldenström J, Osterhaus ADME. Fouchier RAM. Global patterns of influenza A virus in wild birds. Science. 2006;312:384–388. doi: 10.1126/science.1122438. [DOI] [PubMed] [Google Scholar]
- Patten MA. Unitt P. Diagnosability versus mean differences of Sage Sparrow subspecies. Auk. 2002;119:26–35. [Google Scholar]
- Pearce JM, Ruthrauff DR. Hall JS. Paired serologic and polymerase chain reaction analyses of avian influenza prevalence in Alaskan shorebirds. Journal of Wildlife Diseases. 2012;48:812–814. doi: 10.7589/0090-3558-48.3.812. [DOI] [PubMed] [Google Scholar]
- Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L. Estoup A. GeneClass2: a software for genetic assignment and first-generation migrant detection. Journal of Heredity. 2004;95:536–539. doi: 10.1093/jhered/esh074. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M. Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannala B. Mountain JL. Detecting immigration by using multilocus genotypes. Proceedings of the National Academy of Sciences, USA. 1997;94:9197–9201. doi: 10.1073/pnas.94.17.9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappole JJ, Derrickson SR. Hubálek Z. Migratory birds and spread of West Nile virus in the Western Hemisphere. Emerging Infectious Diseases. 2000;6:319–328. doi: 10.3201/eid0604.000401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorey RI, Scribner KT, Kanefsky J, Samuel MD. Libants SV. Intercontinental gene flow among western arctic populations of lesser snow geese. Condor. 2011;113:735–746. [Google Scholar]
- Slatkin M. A measure of population subdivision based on microsatellite allele frequencies. Genetics. 1995;139:457–462. doi: 10.1093/genetics/139.1.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soikkeli M. Breeding cycle and population dynamics in the Dunlin Calidris alpina. Annales Zoologici Fennici. 1967;4:158–198. [Google Scholar]
- Soikkeli M. Mortality and reproductive rates in a Finnish population of Dunlin (Calidris alpina. Ornis Fennica. 1970;47:149–158. [Google Scholar]
- Sokal RR. Rohlf FJ. Biometry: The Principles and Practice of Statistics in Biological Research. 3rd edn. New York: W.H. Freeman and Co; 1995. p. 887. [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M. Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony method. Molecular Biology and Evolution. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AR, Lanctot RB, Powel AN, Kendall SJ. Nigro DA. Residence time and movement patterns of postbreeding shorebirds on Alaska's northern coast. Condor. 2011;113:779–794. [Google Scholar]
- Tomkovich PS. Geographical variability of the Dunlin in the Far East. Bulletin of the Moscow Society of Naturalists. 1986;91:3–15. [Google Scholar]
- Tomkovich PS. Site fidelity and spatial structure of populations of Rock Sandpiper Calidris ptilocnemis and Dunlin Calidris alpina on Chukotskiy Peninsula. Russian Journal of Ornithology. 1994;3:13–30. [Google Scholar]
- van Treuren R, Bijlsma R, Tinbergen JM, Heg D. Zande L. Genetic analysis of the population structure of socially organized oystercatchers (Haematopus ostralegus) using microsatellites. Molecular Ecology. 1999;8:181–187. doi: 10.1046/j.1365-294x.1999.00548.x. [DOI] [PubMed] [Google Scholar]
- United States Fish and Wildlife Service and United States Geological Survey (USFWS and USGS. 2007. Sampling results for highly pathogenic Asian H5N1 avian influenza in migratory birds in Alaska http://alaska.usgs.gov/science/biology/avian_influenza/pubs.php (accessed on 15 January 2015)
- Uyeki TM. Cox NJ. Global concerns regarding novel influenza A (H7N9) virus infections. The New England Journal of Medicine. 2013;368:1862–1864. doi: 10.1056/NEJMp1304661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villesen P. FaBox: an online toolbox for fasta sequences. Molecular Ecology Notes. 2007;7:965–968. [Google Scholar]
- Visser ME, Perdeck AC, van Balen JH. Both C. Climate change leads to decreasing bird migration distances. Global Change Biology. 2009;15:1859–1865. [Google Scholar]
- Warnock ND. Gill RE. Dunlin (Calidris alpina. In: Gill F, editor; Poole A, editor. The Birds of North America Online. 1996. Retrieved from the Birds of North America Online: http://bna.birds.cornell.edu/bna/species/203. doi: 10.2173/bna.203 (accessed on 2 April 2014) [Google Scholar]
- Webster RG, Bean WJ, Gorman OT, Chambers TM. Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiological Reviews. 1992;56:152–179. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MS, Marra PP, Haig SM, Bensch S. Holmes RT. Links between worlds: unraveling migratory connectivity. Trends in Ecology and Evolution. 2002;17:76–83. [Google Scholar]
- Wenink PW. Baker AJ. Mitochondrial DNA lineages in composite flocks of migratory and wintering Dunlin (Calidris alpina. Auk. 1996;113:744–756. [Google Scholar]
- Wenink PW, Baker AJ. Tilanus MGJ. Hypervariable-control-region sequences reveal global population structuring in a long-distance migrant shorebird: the Dunlin (Calidris alpina. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:94–98. doi: 10.1073/pnas.90.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenink PW, Baker AJ. Tilanus MGJ. Mitochondrial control-region sequences in two shorebird species, the turnstone and the Dunlin, and their utility in population genetic studies. Molecular Biology and Evolution. 1994;11:22–31. doi: 10.1093/oxfordjournals.molbev.a040089. [DOI] [PubMed] [Google Scholar]
- Wenink PW, Baker AJ, Rösner H-U. Tilanus MGJ. Global mitochondrial DNA phylogeography of Holarctic breeding Dunlins (Calidris alpina. Evolution. 1996;50:318–330. doi: 10.1111/j.1558-5646.1996.tb04495.x. [DOI] [PubMed] [Google Scholar]
- Wennerberg L. 2001a. Sweden Lund University Genetic variation and migration of waders. PhD thesis.
- Wennerberg L. Breeding origin and migration pattern of Dunlin (Calidris alpina) revealed by mitochondrial DNA analysis. Molecular Ecology. 2001b;10:1111–1120. doi: 10.1046/j.1365-294x.2001.01256.x. [DOI] [PubMed] [Google Scholar]
- Wennerberg L, Holmgren NMA, Jönsson PE. von Schantz TV. Genetic and morphological variation in Dunlin Calidris alpina breeding in the Palearctic tundra. Ibis. 1999;141:391–398. [Google Scholar]
- Wennerberg L, Marthinsen G. Lifjeld JT. Conservation genetics and phylogeography of southern Dunlins Calidris alpina schinzii. Journal of Avian Biology. 2008;39:423–437. [Google Scholar]
- Whitlock MC. Combining probability from independent tests: the weighted Z method is superior to Fisher's approach. Journal of Evolutionary Biology. 2005;18:1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- Wilson WH., Jr Bird feeding and irruptions of Northern finches: are migrations short-stopped? North American Bird Bander. 1999;24:113–121. [Google Scholar]
- Winker K. Gibson DD. The Asia-to-America influx of avian influenza wild bird hosts is large. Avian Diseases. 2010;54:477–482. doi: 10.1637/8741-032509-Reg.1. [DOI] [PubMed] [Google Scholar]
- Zink RM, Blackwell RC. Rojassoto O. Species limits in the Le Conte's Thrasher. Condor. 1997;99:132–138. [Google Scholar]