

Abstract
Biotin-dependent carboxylases are widely distributed in nature and have important functions in the metabolism of fatty acids, amino acids, carbohydrates, cholesterol and other compounds 1–6. Defective mutations in several of these enzymes have been linked to serious metabolic diseases in humans, and acetyl-CoA carboxylase (ACC) is a target for drug discovery against diabetes, cancer and other diseases 7–9. We report here the identification and biochemical, structural and functional characterizations of a novel single-chain (120 kD), multi-domain biotin-dependent carboxylase in bacteria. It has preference for long-chain acyl-CoA substrates, although it is also active toward short- and medium-chain acyl-CoAs, and we have named it long-chain acyl-CoA carboxylase (LCC). The holoenzyme is a homo-hexamer with molecular weight of 720 kD. The 3.0 Å crystal structure of Mycobacterium avium subspecies paratuberculosis LCC (MapLCC) holoenzyme revealed an architecture that is strikingly different compared to those of related biotin-dependent carboxylases 10,11. In addition, the domains of each monomer have no direct contacts with each other. They are instead extensively swapped in the holoenzyme, such that one cycle of catalysis involves the participation of four monomers. Functional studies in Pseudomonas aeruginosa suggest that the enzyme is involved in the utilization of selected carbon and nitrogen sources.
The reactions catalyzed by biotin-dependent carboxylases proceed in two steps and involve at least three different protein components (Extended Data Fig. 1). In the first step, a biotin carboxylase (BC) component catalyzes the carboxylation of the biotin cofactor, which is covalently linked to the biotin carboxyl carrier protein (BCCP) component. In the second step, the carboxylated biotin translocates to the carboxyltransferase (CT) active site and transfers the carboxyl group to the substrate. In bacteria, ACC has been well characterized as a multi-subunit enzyme, with a BC subunit, a BCCP subunit, and two subunits (α and β) for the CT activity (Extended Data Fig. 1). In contrast, ACC is a large (~250 kD), single-chain, multi-domain enzyme in most eukaryotes (Extended Data Fig. 1), with domains that are homologous to the bacterial subunits. Other members of this family include propionyl-CoA carboxylase (PCC) 10, 3-methylcrotonyl-CoA carboxylase (MCC) 11, pyruvate carboxylase (PC) 12,13, and urea carboxylase (UC) 14 (Extended Data Fig. 1).
By examining the sequence database, we identified a novel single-chain (~120 kD), multi-domain biotin-dependent carboxylase in bacteria 1. The enzyme contains a BC domain at the N terminus, a BCCP domain near the middle, and a CT domain that is homologous to that of ACC and PCC (Fig. 1a, Extended Data Fig. 1). Homologs of this enzyme are found in a large number of Gram-negative and Gram-positive bacteria, such as Rhodopseudomonas palustris, Mycobacterium avium subspecies paratuberculosis 15, and the human pathogen Pseudomonas aeruginosa (Extended Data Fig. 2), with highly conserved sequences (Extended Data Fig. 3). These homologs are in fact mis-annotated as PC 16 or carbamoyl-phosphate synthase (CPS) in the database, as was noted in an earlier report 17, probably because they have approximately the same size as PC and CPS. The CT domain of PC has a completely different sequence and structure 12,13 (Extended Data Fig. 1), whereas CPS is not a biotin-dependent enzyme and does not have a BCCP domain. These single-chain enzymes are somewhat related to a family of acyl-CoA carboxylases that have been characterized in Mycobacterium tuberculosis and other actinomycetes 3, where BC and BCCP are present in one subunit while CT is in a separate subunit (Extended Data Fig. 1, see below).
Figure 1.

Crystal structure of M. avium subspecies paratuberculosis long-chain acyl-CoA carboxylase (MapLCC). (a). Domain organization of MapLCC. The domains are labeled and given different colors. (b). Overlay of the structures of the two MapLCC monomers in the asymmetric unit (one in color, the other in gray). Residues (aa) that are missing in the linkers from BCCP are indicated with dashed lines. (c). Overall structure of the 720 kD hexameric holoenzyme of MapLCC. The six monomers are labeled. The domains in the three monomers in the top layer (numbered 1, 2, and 3) are colored as in panel a. The BC, BCCP, N and C CT domains in the three monomers in the bottom layer (numbered 4, 5, 6) are colored pink, pale blue, pale cyan and pale yellow, respectively. The disordered region of the BCCP-CT linker is indicated with the dashed line (black). The BC active sites are indicated with the asterisks (black). The CT active sites are on the side of the CT domain core, indicated with the black arrows. (d). Structure of the MapLCC holoenzyme viewed down the BC domain dimer (red arrow in panel c). (e). Structure of the MapLCC holoenzyme viewed down the blue arrow in panel c. The CT active sites are indicated with the asterisks (black). (f). Structure of the 750 kD α6β6 PCC holoenzyme 10. The view is equivalent to that of panel d. (g). Structure of the 750 kD α6β6 MCC holoenzyme 11. The structure figures were produced with PyMOL (www.pymol.org).
We over-expressed several of these single-chain enzymes in E. coli and purified them to homogeneity. The proteins migrated at the same position on a gel filtration column as the 750 kD α6β6 holoenzymes of PCC 10 and MCC 11, suggesting that these enzymes are hexamers, with a molecular weight of ~720 kD for the holoenzyme.
We characterized the catalytic activities of the enzyme from R. palustris, which is annotated as both PC and CPS in the database. Consistent with the sequence analysis, we found that this enzyme is an acyl-CoA carboxylase, and we did not observe any PC activity for it. The enzyme is active toward all the acyl-CoAs that we tested, with chain lengths from C2 to C16, but it prefers long-chain substrates (Extended Data Table 1). The kcat values for all the substrates are comparable, while the Km for palmitoyl-CoA is ~350-fold lower than that for acetyl-CoA. The high sequence conservation among these proteins (Extended Data Fig. 3) suggests that they have similar activity profiles. Therefore, we have named them long-chain acyl-CoA carboxylases (LCCs). Broad-spectrum activity has been observed for a few of the acyl-CoA carboxylases in actinomycetes 3,18.
To define the holoenzyme architecture, we determined the crystal structure at 3.0 Å resolution of M. avium subspecies paratuberculosis LCC (MapLCC, Figs. 1b–1e), which shares 52% amino acid sequence identity with R. palustris LCC (Extended Data Fig. 3). The holoenzyme hexamer is situated on a crystallographic three-fold axis, and there is a dimer in the asymmetric unit. The atomic model has good agreement with the crystallographic data and the expected geometric parameters (Extended Data Table 2). Several segments of the protein have poor or no electron density and are not included in the atomic model. These include parts of the linkers from the BCCP to the BC and CT domains (Fig. 1b), although there is no ambiguity in assigning the BCCP domain to a specific monomer. A different assignment will result in gaps that are too large to be bridged by the missing residues.
The overall structures of the two MapLCC monomers in the asymmetric unit are similar. With their CT domains superposed, a difference of 3° is seen in the orientation of their BC domains (Fig. 1b). The BCCP domains have a larger difference, corresponding to a rotation of 15°, indicating some asymmetry in the holoenzyme hexamer. Remarkably, the BC, BCCP, and CT domains of each monomer do not have any direct contacts with each other.
The structure of the holoenzyme hexamer of MapLCC has the shape of an equilateral triangle, obeying 32 symmetry and with a length of ~180 Å for each side (Fig. 1c) and a thickness of ~65 Å (Figs. 1d, 1e). A hexamer of the CT domain forms the central core of the structure, with three CT domains in each layer and CT dimers being formed by one domain from each layer. Dimeric BC domains are located at the vertices of the triangle, contacting CT domain dimers. The BCCP domains are situated between the BC and CT domains but not in the active site of either domain. The lysine residue that would be biotinylated is located on the surface, more than 15 Å away from the nearest BC or CT domain, and its side chain is disordered. The BCCP domain is not biotinylated in this holoenzyme, even though it was expressed under identical conditions as those for R. palustris LCC, which was completely biotinylated.
The overall architecture of the MapLCC holoenzyme is strikingly different from those of PCC (Fig. 1f) 10 and MCC (Fig. 1g) 11, even though all three enzymes are ~750 kD oligomers made up of homologous BC, CT and BCCP domains. The central CT domain core of MapLCC is similar to that of the β6 hexamer core of PCC, with an rms distance of 1.7 Å for 2,073 equivalent Cα atoms between them (Extended Data Fig. 4). In contrast, while the BC domains are located above and below the central core in PCC and MCC, they are positioned at the side of the central core in MapLCC. Moreover, the BC domain is a monomer in PCC and a weakly associated trimer in MCC, but it is a dimer in MapLCC. In fact, this dimer is similar to that for the BC subunit of E. coli ACC 19,20 (Extended Data Fig. 4) and the BC domain of PC 12,13.
A BT domain was identified in the structures of PCC (Fig. 1f) and MCC (Fig. 1g), located between the BC and BCCP domains in the primary sequence (Extended Data Fig. 1) and having an important role in mediating interactions between their α (BC) and β (CT) subunits 10,11. This domain does not exist in LCC, as there are not sufficient residues in the linker between BC and BCCP to form such a domain (Extended Data Fig. 1). On the other hand, the BC-BCCP linker and the BCCP-CT linker do participate in mediating interactions in the MapLCC holoenzyme, and there are also direct contacts between the BC and CT domains (see below).
The domains of the MapLCC monomers are swapped extensively in the holoenzyme hexamer. The CT domains of two monomers related by a BC domain dimer are located far from each other (Fig. 2a). Similarly, the BC domains of two monomers related by a CT domain dimer are also located far from each other (Fig. 2b). Therefore, interactions among these domains occur only in the context of the holoenzyme, and the structure of the monomer alone is unlikely to be stable (Fig. 1b).
Figure 2.

Extensive domain swapping in the structure of the MapLCC holoenzyme. (a). The CT domains of two monomers (in color) related by a BC dimer are located far away from each other (note that they are in opposite layers), and participate in dimerization with different monomers (in gray). The six monomers are labeled. (b). The BC domains of two monomers related by a CT dimer are located far away from each other.
A total of ~8,200 Å2 of the surface area of each MapLCC monomer is buried in the holoenzyme. The majority of this surface, 5,700 Å2, is buried by the interface among the CT domains in the central core, and 1,000 Å2 surface is buried in the BC dimer interface (Figs. 3a,3b). The remaining interfaces in the holoenzyme make smaller contributions to the surface area burial, 600 Å2 in the BC-CT interface, 500 Å2 for the linkers from BCCP, and 400 Å2 from the BCCP domain. Nonetheless, all of these smaller interfaces together may be important for stabilizing the holoenzyme.
Figure 3.

Interactions between the central core and the rest of the holoenzyme. (a) Two views of the molecular surface of a MapLCC monomer, colored by the domains. (b) Residues that mediate interactions in the MapLCC holoenzyme are indicated with the colors of the domains that they contact. For example, a pale cyan patch on the surface of the C domain of CT indicates residues that interact with the N domain of another monomer in the holoenzyme. The pink patch on BC indicates residues in the BC dimer interface, and the pale cyan patch indicates residues in the interface with CT. The major areas of contacts are among the CT domains and in the BC domain dimer. The views are identical to panel a. (c). Residues in the interface between the BC and CT domains. The domains and the monomers they belong to are labeled. (d). Residues in the interface between the BC-BCCP linker just prior to the BCCP domain and the rest of the holoenzyme. (e). Class average representing the top view (left) most similar to the crystal structure and the corresponding projection from the crystal structure filtered to 30 Å resolution (right). (f). Class average representing the side view (left) most similar to the crystal structure and the corresponding projection from the crystal structure filtered to 30 Å resolution (right). (g). Three panels showing that each of the peripheral BC domains can appear as a single or bilobal density, likely representing different orientations of the BC dimer. (h). Two panels illustrating that the BC domains can adopt different positions around the central CT core. The side length of the individual panels in c–f is 340 Å.
The primary BC-CT interface involves three monomers of MapLCC. A β-hairpin structure in the BC domain of monomer 4 contacts the CT domains of monomer 2 (N domain) and monomer 6 (C domain, Fig. 3c). Similarly, the BC-BCCP linker just prior to the BCCP domain in monomer 1 contacts the CT domain of monomer 2 and the BC domain of monomer 4 (Fig. 3d). These interactions also demonstrate the extensive connections among the monomers in the holoenzyme. The interfaces involve van der Waals contacts, hydrogen bonding and ionic interactions.
We also carried out electron microscopy (EM) studies on the MapLCC holoenzyme. Negative-stain EM images showed mono-dispersed particles of similar size but variable shapes (Extended Data Fig. 5). Classification in SPARX 21 of ~25,000 particles yielded 308 classes that represented 65% of the data set (Extended Data Fig. 5). Cross-correlation analysis identified class averages that are very similar to the top view (cross-correlation coefficient of 0.856, Fig. 3e) and side view (cross-correlation coefficient of 0.752, Fig. 3f) of the crystal structure, confirming the holoenzyme architecture seen in the crystal. At the same time, substantial variations in the three peripheral densities (corresponding to the BC domains) relative to the central core (the CT domains) are also observed (Extended Data Fig. 5). Each peripheral density can appear as a single or bilobed feature (Fig. 3g), likely representing different views of the BC dimer. The densities can also be located in different positions relative to the central core, breaking the alignment of the two-fold axes of the BC and CT dimers (Fig. 3h). Overall, the EM data indicate that the peripheral BC domains move as dimers and are flexibly tethered to the CT core (Supplementary video S1), consistent with the crystal structure that the BC-CT contact is relatively weak in MapLCC.
Each BC active site is located ~40 Å from a CT active site in the MapLCC holoenzyme (Fig. 4a). The BCCP domain, while not located in either active site, can readily access both of them, through conformational changes in its linkers. Remarkably, the crystal structure shows that the BCCP domain of monomer 1 visits the BC active site of monomer 4 and the CT active site at the interface of monomers 2 and 6 (Fig. 4a), suggesting that each cycle of catalysis requires the participation of 4 monomers. This again indicates the extensive communications among the monomers in this holoenzyme. The residues in both active sites are generally conserved with those in other biotin-dependent carboxylases, suggesting a similar catalytic mechanism 22 for LCC. The CT active site has a pocket that can accommodate short- and medium-chain acyl groups (Extended Data Fig. 6), and a conformational change is needed to bind long-chain substrates. This conformational flexibility may be important for the enzyme to adapt to and be active toward the broad collection of substrates.
Figure 4.
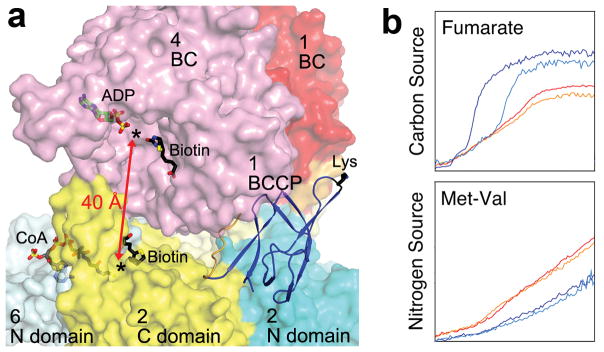
Catalysis and function of MapLCC. (a). The BC and CT active sites (black asterisks) of MapLCC are separated by ~40 Å (red arrow). Molecular surface of MapLCC is shown, colored as in Fig. 1c. The domains are labeled by the monomer they belong to. The Lys side chain of BCCP to which biotin would be connected to is shown in black. The binding modes of ADP (green) and biotin (black) to E. coli BC subunit are shown as stick models 20. The binding mode of CoA (gray) to the CT domain of yeast ACC 29 and biotin (black) in the β (CT) subunit of PCC 10 are also shown. (b). Phenotypic differences between wild-type and LCC knockout (ΔPA14_46320) P. aeruginosa strains, revealed by a colorimetric assay that monitors reduction of a tetrazolium dye. Assays were carried out twice in each medium for the wild-type (red and orange) and mutant (blue and cyan) strains. For each panel, the horizontal axis is time (24 hrs), and the vertical axis is OmniLog signal 27.
In most of the other biotin-dependent carboxylases, BCCP is located at the end of a polypeptide chain, and therefore there is only one linker to the rest of the protein. The LCCs, and the eukaryotic ACCs (Extended Data Fig. 1), are distinct in that BCCP is located in the middle of these proteins. Therefore, there are two linkers from BCCP to the rest of the protein. The amino acid sequences of these two linkers in the LCC enzymes are not conserved (Extended Data Fig. 3). Therefore, they are likely to be flexible and allow the movement of BCCP during catalysis, consistent with the fact that a portion of both linkers is disordered in the structure (Fig. 1b). Residues in both linkers that are included in the current atomic model also have high B values (Extended Data Fig. 4). In addition, the B domain of BC and a loop near the C terminus of the protein have high B values and are partially disordered.
The structure of MapLCC also has implications for the holoenzymes of other biotin-dependent carboxylases, especially the family of acyl-CoA carboxylases in M. tuberculosis, S. coelicolor and other actinomycetes 3. These enzymes contain two subunits, with BC-BCCP in the α subunit and CT in the β subunit, and there are not enough residues in one of the α subunits for a BT domain (Extended Data Fig. 1). The holoenzyme is an α6β6 dodecamer, and the structure of the β6 hexamer 23 is similar to that of the CT domain hexamer in MapLCC. Therefore, it is likely that the holoenzymes of such two-subunit carboxylases share a similar architecture as MapLCC (Fig. 1c) rather than PCC (Fig. 1f). Interestingly, M. tuberculosis does not have an LCC homolog, indicating some differences among these mycobacterial species.
Eukaryotic ACCs are also single-chain, multi-domain enzymes (Extended Data Fig. 1), although there are substantial differences compared to LCC. The eukaryotic ACCs contain ~1000 additional residues, including a unique central region of ~700 residues, and they are likely to carry a BT domain as well. Therefore, the overall architecture of the eukaryotic ACCs is likely to be different from that of MapLCC observed here.
We have begun to characterize the physiological functions of LCC, using Pseudomonas aeruginosa (strain PA14) as the model organism. The P. aeruginosa enzyme (locus name PA14_46320, homolog of PA1400 in P. aeruginosa PAO1) shares 59% amino acid sequence identity with R. palustris LCC. This organism also carries the multi-subunit ACC, MCC and geranyl-CoA carboxylase (GCC) 24,25, although it lacks a homolog for PCC. The multi-subunit ACC is likely essential, similar to the E. coli enzyme, as no transposon insertions in its subunits are found in a PA14 transposon mutant library 26. On the other hand, transposon insertions are found in LCC, MCC and GCC, suggesting that they are not essential for growth under the conditions used to produce this library 26.
We produced a markerless deletion of LCC/PA14_46320, confirming that it is not essential for cell survival. The activity profiles of the mutant under ~2000 conditions were characterized using a phenotype microarray, which monitored its ability to reduce a tetrazolium dye 27. The incubation conditions included different carbon or nitrogen sources, nutrient supplements, osmolytes, pH values, and antibiotics. While most of the conditions showed comparable profiles between the wild type and the LCC deletion mutant, phenotypic differences were observed for several conditions (Extended Data Fig. 7), and two of these were considered significant based on the phenotype microarray analysis – using fumarate as the sole carbon source and the Met-Val dipeptide as the sole nitrogen source (Fig. 4b). It is not clear how this enzyme is linked to selected carbon and nitrogen source utilization in P. aeruginosa. The metabolism of both Met and Val is likely to produce propionyl-CoA, and the PCC activity of LCC may be important for its further degradation, as is the case in many other organisms. This is also supported by the fact that P. aeruginosa lacks a proper PCC enzyme.
A long-chain acyl-CoA carboxylase activity is needed for the biosynthesis of mycolic acid in Mycobacterium and other actinomycetes 3,18,28. However, the LCC enzyme studied here is unlikely to be essential for this function, as its homolog is absent in M. tuberculosis. Moreover, mycolic acid is not known to be present in P. aeruginosa, R. palustris and other bacteria, suggesting that this LCC is likely to have different physiological functions.
The striking differences in the architectures of LCC, PCC and MCC holoenzymes are also linked to functional differences among them. Especially, the N and C domains of the β subunits of PCC and MCC are swapped relative to each other, and this is coupled to the different substrate specificity of the two enzymes. PCC carboxylates the α carbon of an acid (as a CoA ester), while MCC carboxylates the γ carbon of an α-β unsaturated acid (Extended Data Fig. 1). Therefore, the structural differences suggest that there are two lineages of the biotin-dependent carboxylases 11, one containing PCC, ACC and LCC, the other containing MCC and possibly GCC. At the same time, PCC and LCC share propionyl-CoA carboxylase activity, indicating that similar biochemical activities can be supported by holoenzymes with dramatically different architectures as well.
Overall, our studies have identified a new member of the biotin-dependent carboxylase family, and revealed a novel architecture for its holoenzyme. These observations are also relevant for other members of this family. Moreover, the differences in the architectures of LCC, PCC and MCC holoenzymes, despite their sharing domains with homologous structures, indicate that these domains can be arranged in remarkably different ways to form the various holoenzymes. This will have significant implications for other multi-domain proteins, especially eukaryotic ACCs, and protein structure and sequence conservation in general.
Online Methods
Protein expression and purification
Full-length LCCs from several different bacterial organisms, including R. palustris, M. avium subspecies paratuberculosis and P. aeruginosa, were amplified from genomic DNA by PCR and cloned into pET28a, pET26b and/or pET24d vectors (Novagen). The plasmids were transformed into BL21Star (DE3) cells (Invitrogen). Protein expression was induced with the addition of 1 mM IPTG, and the cells were grown at 16°C for 16–20 h.
To facilitate biotinylation, the recombinant enzyme was co-expressed with the E. coli biotin ligase BirA, and 15 mg/liter biotin was added to the medium. An avidin shift assay of the purified enzymes showed that R. palustris LCC was completely biotinylated. However, purified MapLCC did not show any biotinylation, possibly indicating some degree of selectivity of the BirA enzyme. Expression of P. aeruginosa LCC did not produce any soluble protein and was not pursued further.
Cells were lysed by sonication in a buffer containing 20 mM Tris (pH 8.0), 250 mM NaCl, 5% (v/v) glycerol, 10 mM β-mercaptoethanol, and 1 mM PMSF. Soluble enzyme was purified by Ni-NTA (Qiagen), anion exchange, and gel filtration (Sephacryl S-300, GE Healthcare) chromatography. The S-300 running buffer for MapLCC contained 25 mM Hepes (pH 7.4), 250 mM NaCl, and 2.5 mM dithiothreitol. The purified protein was concentrated to 6 mg/ml, and the solution was supplemented with 5% (v/v) glycerol before being flash-frozen in liquid nitrogen and stored at −80°C.
The selenomethionyl MapLCC protein was produced in B834 (DE3) cells (Novagen) that were grown in defined LeMaster medium supplemented with selenomethionine 30. The protein was purified with the same protocol as that for the native enzyme.
Protein crystallization
MapLCC was crystallized at 4°C using the microbatch method under paraffin oil. The protein solution was mixed with a precipitant solution containing 0.1 M Bis-tris propane (pH 7.5–8.5), and 1.5–2.0 M ammonium sulfate. Crystals took 4–6 weeks to grow to full size, and larger crystals were obtained by microseeding. They were cryo-protected with reservoir solution supplemented with 12–15% (v/v) glycerol and flash-frozen in liquid nitrogen for data collection at 100 K. The C-terminal His-tag on the protein was not removed before crystallization.
Data collection and structure determination
X-ray diffraction data for the native (wavelength 1.075 Å) and selenomethionyl (0.979Å) crystals were collected using a Q315 CCD (ADSC) at the X29A beamline of the National Synchrotron Light Source (NSLS). The diffraction images were processed with the HKL package 31. The crystals belong to space group P213, with cell dimensions of a=b=c=220.9 Å. There are two MapLCC monomers in the crystallographic asymmetric unit.
The structure of MapLCC was solved by a combination of molecular replacement and selenomethionyl SAD phasing. The orientation and position of the BC, CT and BCCP domains were located with the program Phaser 32. The Se sites were located with the program SHELX 33, and SOLVE/RESOLVE was used for phasing the reflections and automated model building 34. The atomic model was built with the program Coot 35. The structure refinement was carried out with the programs CNS 36. The crystallographic information is summarized in Extended Data Table 2.
We also obtained a second crystal form of MapLCC, with an entire hexamer in the asymmetric unit, and were able to collect an X-ray diffraction data set to 4.3 Å resolution (space group P212121, a=102 Å, b=292 Å, and c=314 Å). The structure of this crystal form was readily solved by the molecular replacement method, and it revealed essentially the same holoenzyme architecture (data not shown).
Electron microscopy and image processing
Purified MapLCC was prepared by conventional negative staining with 0.75% (w/v) uranyl formate 37. Images were collected with a Tecnai T12 electron microscope (FEI, Hillsboro, OR) equipped with an LaB6 filament and operated at an acceleration voltage of 120 kV. Images were recorded using low-dose procedures on an UltraScan 895 4K × 4K CCD camera (Gatan, Pleasanton, CA) using a defocus of −1.5 μm and a nominal magnification of 52,000x. The calibrated magnification was 70,527x, yielding a pixel size of 2.13 Å on the specimen level.
BOXER, the display program associated with the EMAN software package 38, was used to interactively select 24,535 particles from 270 CCD images, and the SPIDER software package 39 was used to window the particles into 160 × 160-pixel images. To perform iterative stable alignment and clustering (ISAC) 40 in SPARX 21, the size of the particle images was reduced to 64 × 64 pixels, and the particles were pre-aligned and centered. ISAC was run on the Orchestra High Performance Compute Cluster at Harvard Medical School (http://rc.hms.harvard.edu), specifying 200 images per group and a pixel error of 0.7. After 19 generations, 308 classes were obtained, accounting for 15,932 particles (65% of the entire data set) (Extended Data Fig. 4). Averages of these classes were calculated using the original 160 × 160-pixel images. To confirm that the ISAC averages are representative of the entire data set, the particles were also subjected to 10 cycles of multi-reference alignment in SPIDER. Each round of multi-reference alignment was followed by K-means classification, specifying 300 output classes (Extended Data Fig. 5).
To compare the class averages with the crystal structure, the crystal structure was Fourier transformed, filtered to 30 Å with a Butterworth low-pass filter, and transformed back. Evenly spaced projections were calculated at 4° intervals and subjected to 10 cycles of alignment with masked EM class averages. The class averages most similar to the top and side view of the crystal structure and their cross-correlation coefficients are presented in (Figs. 3e, 3f).
To visualize the structural heterogeneity of MapLCC in solution, 179 averages obtained by ISAC that showed three peripheral densities were selected (indicated by asterisks in Extended Data Fig. 5), ordered according to their correlation coefficients, and used to prepare Supplementary Video 1. This structural variability is likely the reason why it was not possible to calculate a 3D map from cryo-EM images of vitrified MapLCC samples.
Enzymatic assays
The kinetic assays monitored the hydrolysis of ATP by R. palustris LCC in the presence of various acyl-CoA substrates, using coupling enzymes to convert the ADP product to NADH oxidation 41. The reaction mixture contained 100 mM Hepes (pH 7.5), 40 mM KHCO3, 1.5 mM ATP, 0.4 mM NADH, 200 mM KCl, 10 mM MgCl2, 0.5 mM phosphoenolpyruvate, 3.5/3.7 units lactate dehydrogenase/pyruvate kinase (Sigma), 0.25 μM enzyme (except for MCC, which was at 1.2 μM), and varying concentrations of acyl-CoA. The absorbance at 340 nm was monitored for 1.5 min. The initial velocities were fitted to the Michaelis-Menten equation using the program Origin (OriginLab).
Construction of an LCC deletion mutant in P. aeruginosa
A markerless deletion was generated for the gene PA14_46320 in P. aeruginosa PA14 using previously described methods 42. Briefly, ~1 kb flanking regions for PA14_46320 were amplified using primers listed in Extended Data Table 3 and recombined into the allelic-replacement vector pMQ30 through gap repair cloning in the yeast strain InvSc1 43. This plasmid was transformed into Escherichia coli BW29427 and moved into PA14 using biparental conjugation. LB agar containing 100 μg/ml gentamicin was used to select for P. aeruginosa single recombinants. Markerless deletions in PA14_46320 (double recombinants) were then selected using LB agar plates devoid of NaCl and containing 10% (w/v) sucrose as a counter-selection and their genotypes were confirmed by PCR.
Phenotype microarrays
The phenotype microarray screening was carried out by Biolog, Inc. as described 27.
Extended Data
Extended Data Fig. 1.

Domain organization of biotin-dependent carboxylases. (a). Reactions catalyzed by biotin-dependent carboxylases. Biotin is linked to the side chain of a Lys residue in BCCP, and this flexible arm has a maximal length of ~16 Å. The BCCP domain must also translocate to reach both active sites, separated by distances of 40–85 Å based on known holoenzyme structures (swinging domain model). (b). Domain organizations of several representative biotin-dependent carboxylases. Homologous domains are given the same colors. The CT domain of PC has a completely different sequence and structure compared to that of ACC and PCC. The proteins are drawn to scale, and a scale bar is shown at the bottom. BT: BC-CT interaction domain; PT: PC tetramerization domain, also known as allosteric domain. (c). Chemical structures of the substrates of ACC, PCC, LCC, MCC and GCC. The site of carboxylation is indicated with the red arrow.
Extended Data Fig. 2.

Phylogenetic trees for selected biotin-dependent carboxylases. (a). Phylogenetic tree for LCC homologs in a collection of organisms. The three homologs studied in this paper are shown in red. (b). Phylogenetic tree for PCC homologs in a collection of organisms, based on a sequence alignment of the β subunit. Modified from an output from the Phylogeny.fr server44.
Extended Data Fig. 3.

Sequence alignment of long-chain acyl-CoA carboxylases (LCCs) from M. avium subspecies paratuberculosis (MapLCC), R. palustris (RpLCC), and P. aeruginosa (PaLCC). The various domains in the proteins are labeled. The BCCP domain has two linkers to the rest of the protein. Modified from an output from ESPript45.
Extended Data Fig. 4.

Structural comparisons of domains in LCC with related enzymes. (a). Stereo drawing of the overlay of the structure of the BC domain dimer of MapLCC (in color) with that of BC subunit dimer of E. coli ACC (in gray)46. The bound positions of biotin (black) and ADP (green) in the E. coli BC structure are also shown. The two-fold axis of the dimer is indicated with the black oval. With the two monomers at the bottom overlaid, a difference of 21° in the orientations of the two monomers at the top is observed. Most of the B domain of BC is ordered in one of the two monomers of MapLCC. In the other monomer, only weak electron density is observed for a few segments, and the B domain is not modeled. (b). Overlay of the structures of the CT domain hexamer of MapLCC (in color) and the β subunit of PCC (in gray)47. Each enzyme is highly conserved across species, and therefore the overlay should be meaningful. (c). Stereo drawing of the overlay of the CT domain dimer of MapLCC (in color) and the β subunit of PCC (in gray). The view is down the red arrow of panel b. The bound position of biotin in the holoenzyme is shown (black). The position of CoA is modeled based on that of CoA bound in the active site of the CT domain of yeast ACC48. (d). Plot of the temperature factor value of each Cα atom in the two monomers (in red and blue). Several linker regions with high temperature factor values are indicated.
Extended Data Fig. 5.

Electron microscopy studies of LCC. (a). Representative raw image of negatively stained MapLCC. “S” marks a side view of the holoenzyme, and “C” indicates a contaminant. Scale bar: 500 Å. (b). The 308 class averages of negatively stained MapLCC obtained from 19 generations of the iterative stable alignment and clustering (ISAC) procedure49 implemented in SPARX50. These class averages represent 65% (15,932 particles) of the entire data set (24,535 particles). Averages representing side views are marked with a “S”, averages that were used to create Supplementary Video S1 are marked with a “*”, and averages that represent a contaminant are marked with “C”. The side length of the individual panels is 340 Å. (c). The averages obtained by classifying all 24,535 particles of negatively stained MapLCC into 300 classes using K-means classification in SPIDER51. Averages are shown in rows with the most populous class at the top left and the least populous class at the bottom right. The side length of the individual panels is 340 Å.
Extended Data Fig. 6.

The CT active site of LCC. (a). Stereo drawing of the overlay of the CT active site (cyan and yellow) of MapLCC with that of PCC (gray)47. The model of CoA was obtained from the structure of the complex with yeast ACC CT domain48. The α6 helix in the N domain of monomer 6 shows a more closed conformation (indicated with the red arrow), and clashes with the CoA model. There is also a clash with the adenine base of CoA. There may be a conformational change in this region of MapLCC for CoA binding. (b). Molecular surface of the CT active site region of MapLCC. The α6 helix in the N domain of monomer 6 was removed for a clearer view of the active site.
Extended Data Fig. 7.

Phenotypic differences between wild-type and LCC knockout (ΔPA14_46320) P. aeruginosa strains, revealed by a colorimetric assay that monitors reduction of a tetrazolium dye. The conditions were identified from a screen that sampled 1920 different media (Biolog Inc, Hayward, CA). Assays were carried out twice in each medium for the wild-type (red and orange) and mutant (blue and cyan) strains. Shown are activity profiles for strains incubated with Gly-Pro as the sole carbon source (top panel) and Asp-Phe, Glu-Val, and Met-Asp as the sole nitrogen source (bottom panels). For each panel, the horizontal axis is time (24 hrs), and the vertical axis is OmniLog signal52.
Extended Data Table 1.
Kinetic parameters of R. palustris LCC toward various substrates
| Substrate | Km (mM) | kcat (s−1) |
|---|---|---|
| Acetyl-CoA (C2) | 2.0±0.3 | 0.77±0.05 |
| Propionyl-CoA (C3) | 2.2±0.5 | 0.54±0.07 |
| Butyryl-CoA (C4) | 0.82±0.16 | 0.35±0.03 |
| Hexanoyl-CoA (C6) | 0.20±0.04 | 0.30±0.01 |
| Octanoyl-CoA (C8) | 0.37±0.06 | 0.33±0.01 |
| Decanoyl-CoA (C10) | 0.033±0.005 | 0.28±0.01 |
| Lauroyl-CoA (C12) | 0.019±0.003 | 0.24±0.005 |
| Myristoyl-CoA (C14) | 0.026±0.002 | 0.45±0.01 |
| Palmitoyl-CoA (C16) | 0.0058±0.0005 | 0.16±0.003 |
| 3-methylcrotonyl-CoA | 1.1±0.1 | 0.074±0.004 |
The errors are standard deviations from fitting one titration curve to the Michaelis-Menten equation.
Extended Data Table 2.
Data collection and refinement statistics
| Primer | Sequence (5′ to 3′) |
|---|---|
| ΔPA14_46320 flank 1F | CCAGGCAAATTCTGTTTTATCAGACCGCTTCTGCGTTCTGATGCTGCCTGCTCTACATGCT |
| ΔPA14_46320 flank 1R | CCTTCAACGCCTTGCTGATCCAGCTACCTGGAGATCGAC |
| ΔPA14_46320 flank 2F | GTCGATCTCCAGGTAGCTGGATCAGCAAGGCGTTGAAGG |
| ΔPA14_46320 flank 2R | GGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGCTGGCGCGACCAGTAGAGATT |
Two crystals were used for data collection.
Highest resolution shell is shown in parenthesis.
Extended Data Table 3.
Primers used for making the LCC deletion mutant
| MapLCC | |
|---|---|
| Data collection | |
| Space group | P213 |
| Cell dimensions | |
| a, b, c (Å) | 220.9, 220.9, 220.9 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 50-3.0 (3.1-3.0)* |
| Rmerge | 9.6 (44.6) |
| I/σI | 10.3 (1.9) |
| Completeness (%) | 91 (72) |
| Redundancy | 3.3 (2.1) |
| Refinement | |
| Resolution (Å) | 50-3.0 |
| No. reflections | 64,953 |
| Rwork/Rfree | 20.9/26.2 |
| No. atoms | |
| Protein | 14,632 |
| Ligand/ion | 0 |
| Water | 0 |
| B-factors | |
| Protein | 66.4 |
| Ligand/ion | – |
| Water | – |
| R.m.s deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.4 |
Supplementary Material
{kind=link}
Acknowledgments
We thank Christine Huang for carrying out some initial studies in this project; Alexa Price-Whelan for discussions on P. aeruginosa physiology; Rick Jackimowicz, Neil Whalen, Annie Heroux and Howard Robinson for access to the X29A beamline. The in-house instrument for X-ray diffraction screening was purchased with an NIH grant to LT (S10OD012018). This research is supported by grants from the NIH (R01DK067238 to LT and R01AI103369 to LEPD) and from the Protein Structure Initiative of the NIH (U54GM094597 to LT). The Orchestra High Performance Compute Cluster at Harvard Medical School is a shared facility partially supported by NIH grant NCRR 1S10RR028832-01. We thank Zongli Li for EM support and Pawel Penczek for help with SPARX. TW is an investigator with the Howard Hughes Medical Institute.
Footnotes
Author Contributions. Protein expression, purification and crystallization: THT and CYC. Crystallography: THT and LT. Electron microscopy: YSH and TW. Kinetic assays: THT and CYC. P. aeruginosa experiments: JJ and LEPD. THT, LEPD, TW and LT wrote the paper.
Coordinates and structure factors have been deposited in Protein Data Bank under accession number 4RCN.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Tong L. Structure and function of biotin-dependent carboxylases. Cell Mol Life Sci. 2013;70:863–891. doi: 10.1007/s00018-012-1096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waldrop GL, Holden HM, St Maurice M. The enzymes of biotin dependent CO2 metabolism: what structures reveal about their reaction mechanisms. Prot Sci. 2012;21:1597–1619. doi: 10.1002/pro.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gago G, Diacovich L, Arabolaza A, Tsai SC, Gramajo H. Fatty acid biosynthesis in actinomycetes. FEMS Microbiol Rev. 2011;35:475–497. doi: 10.1111/j.1574-6976.2010.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jitrapakdee S, et al. Structure, mechanism and regulation of pyruvate carboxylase. Biochem J. 2008;413:369–387. doi: 10.1042/BJ20080709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tong L. Acetyl-coenzyme A carboxylase: crucial metabolic enzyme and attractive target for drug discovery. Cell Mol Life Sci. 2005;62:1784–1803. doi: 10.1007/s00018-005-5121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cronan JE, Jr, Waldrop GL. Multi-subunit acetyl-CoA carboxylases. Prog Lipid Res. 2002;41:407–435. doi: 10.1016/s0163-7827(02)00007-3. [DOI] [PubMed] [Google Scholar]
- 7.Polyak SW, Abell AD, Wilce MCJ, Zhang L, Booker GW. Structure, function and selective inhibition of bacterial acetyl-CoA carboxylase. Appl Microbiol Biotechnol. 2012;93:983–992. doi: 10.1007/s00253-011-3796-z. [DOI] [PubMed] [Google Scholar]
- 8.Abramson HN. The lipogenesis pathway as a cancer target. J Med Chem. 2011;54:5615–5638. doi: 10.1021/jm2005805. [DOI] [PubMed] [Google Scholar]
- 9.Wakil SJ, Abu-Elheiga LA. Fatty acid metabolism: target for metabolic syndrome. J Lipid Res. 2009;50:S138–S143. doi: 10.1194/jlr.R800079-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang CS, et al. Crystal structure of the a6b6 holoenzyme of propionyl-coenzyme A carboxylase. Nature. 2010;466:1001–1005. doi: 10.1038/nature09302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang CS, Ge P, Zhou ZH, Tong L. An unanticipated architecture of the 750-kDa a6b6 holoezyme of 3-methylcrotonyl-CoA carboxylase. Nature. 2012;481:219–223. doi: 10.1038/nature10691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.St Maurice M, et al. Domain architecture of pyruvate carboxylase, a biotin-dependent multifunctional enzyme. Science. 2007;317:1076–1079. doi: 10.1126/science.1144504. [DOI] [PubMed] [Google Scholar]
- 13.Xiang S, Tong L. Crystal structures of human and Staphylococcus aureus pyruvate carboxylase and molecular insights into the carboxyltransfer reaction. Nat Struct Mol Biol. 2008;15:295–302. doi: 10.1038/nsmb.1393. [DOI] [PubMed] [Google Scholar]
- 14.Fan C, Chou CY, Tong L, Xiang S. Crystal structure of urea carboxylase provides insights into the carboxyltransfer reaction. J Biol Chem. 2012;287:9389–9398. doi: 10.1074/jbc.M111.319475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, et al. The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc Natl Acad Sci USA. 2005;102:12344–12349. doi: 10.1073/pnas.0505662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stover CK, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 17.Lai H, Kraszewski JL, Purwantini E, Mukhopadhyay B. Identification of pyruvate carboxylase genes in Pseudomonas aeruginosa PAO1 and development of a P. aeruginosa-based overexpression system for a4- and a4b4-type pyruvate carboxylases. Appl Environmental Microbiol. 2006;72:7785–7792. doi: 10.1128/AEM.01564-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grande R, et al. The two carboxylases of Corynebacterium glutamicum essential for fatty acid and mycolic acid synthesis. J Bacteriol. 2007;189:5257–5264. doi: 10.1128/JB.00254-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waldrop GL, Rayment I, Holden HM. Three-dimensional structure of the biotin carboxylase subunit of acetyl-CoA carboxylase. Biochem. 1994;33:10249–10256. doi: 10.1021/bi00200a004. [DOI] [PubMed] [Google Scholar]
- 20.Chou CY, Yu LPC, Tong L. Crystal structure of biotin carboxylase in complex with substrates and implications for its catalytic mechanism. J Biol Chem. 2009;284:11690–11697. doi: 10.1074/jbc.M805783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hohn M, et al. SPARX, a new environment for Cryo-EM image processing. J Struct Biol. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Knowles JR. The mechanism of biotin-dependent enzymes. Ann Rev Biochem. 1989;58:195–221. doi: 10.1146/annurev.bi.58.070189.001211. [DOI] [PubMed] [Google Scholar]
- 23.Lin TW, et al. Structure-based inhibitor design of AccD5, an essential acyl-CoA carboxylase carboxyltransferase domain of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:3072–3077. doi: 10.1073/pnas.0510580103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forster-Fromme K, Jendrossek D. Catabolism of citronellol and related acyclic terpenoids in pseudomonads. Appl Microbiol Biotechnol. 2010;87:859–869. doi: 10.1007/s00253-010-2644-x. [DOI] [PubMed] [Google Scholar]
- 25.Aguilar JA, et al. Substrate specificity of the 3-methylcrotonyl coenzyme A (CoA) and geranyl-CoA carboxylases from Pseudomonas aeruginosa. J Bacteriol. 2008;190:4888–4893. doi: 10.1128/JB.00454-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberati NT, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci USA. 2006;103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shea A, Wolcott M, Daefler S, Rozak DA. Biolog phenotype microarrays. Methods Mol Biol. 2012;881:331–373. doi: 10.1007/978-1-61779-827-6_12. [DOI] [PubMed] [Google Scholar]
- 28.Takayama K, Wang C, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H, Yang Z, Shen Y, Tong L. Crystal structure of the carboxyltransferase domain of acetyl-coenzyme A carboxylase. Science. 2003;299:2064–2067. doi: 10.1126/science.1081366. [DOI] [PubMed] [Google Scholar]
- 30.Hendrickson WA, Horton JR, LeMaster DM. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J. 1990;9:1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheldrick GM. A short history of SHELX. Acta Cryst. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 34.Terwilliger TC. SOLVE and RESOLVE: Automated structure solution and density modification. Meth Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan KD. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Brunger AT, et al. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 37.Ohi M, Li Y, Cheng Y, Walz T. Negative staining and image classification - powerful tools in modern electron microscopy. Biol Proced Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 39.Frank J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 40.Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20:237–247. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blanchard CZ, Lee YM, Frantom PA, Waldrop GL. Mutations at four active site residues of biotin carboxylase abolish substrate-induced synergism by biotin. Biochem. 1999;38:3393–3400. doi: 10.1021/bi982660a. [DOI] [PubMed] [Google Scholar]
- 42.Recinos DA, et al. Redundant phenazine operons in Pseudomonas aeruginosa exhibit environment-dependent expression and differential roles in pathogenicity. Proc Natl Acad Sci USA. 2012;109:19420–19425. doi: 10.1073/pnas.1213901109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shanks RM, Caiazza NC, Hinsa SM, Toutain CM, O’Toole GA. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from gram-negative bacteria. Appl Environmental Microbiol. 2006;72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dereeper A, et al. Phylogeny. fr: robust phylogenetic analysis for the non-specialist. Nucl Acid Res. 2008;36:W465–469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 46.Chou CY, Yu LPC, Tong L. Crystal structure of biotin carboxylase in complex with substrates and implications for its catalytic mechanism. J Biol Chem. 2009;284:11690–11697. doi: 10.1074/jbc.M805783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang CS, et al. Crystal structure of the a6b6 holoenzyme of propionyl-coenzyme A carboxylase. Nature. 2010;466:1001–1005. doi: 10.1038/nature09302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H, Yang Z, Shen Y, Tong L. Crystal structure of the carboxyltransferase domain of acetyl-coenzyme A carboxylase. Science. 2003;299:2064–2067. doi: 10.1126/science.1081366. [DOI] [PubMed] [Google Scholar]
- 49.Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20:237–247. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hohn M, et al. SPARX, a new environment for Cryo-EM image processing. J Struct Biol. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Frank J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 52.Shea A, Wolcott M, Daefler S, Rozak DA. Biolog phenotype microarrays. Methods Mol Biol. 2012;881:331–373. doi: 10.1007/978-1-61779-827-6_12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.