

Abstract
We evaluated the effect of AAV2- and 17-AAG (17-N -allylamino-17-demethoxygeldanamycin)-mediated upregulation of Hsp70 expression on the survival of retinal ganglion cells (RGCs) injured by optic nerve crush (ONC). AAV2-Hsp70 expression in the retina was primarily observed in the ganglion cell layer. Approximately 75% of all transfected cells were RGCs. RGC survival in AAV2-Hsp70 injected animals was increased by an average of 110% 2 weeks after the axonal injury compared to the control. The increase in cell numbers was not even across the retinas with a maximum effect of approximately 306% observed in the inferior quadrant. 17-AAG-mediated expression of Hsp70 has been associated with cell protection in various models of neurodegenerative diseases. We show here that a single intravitreal injection of 17-AAG (0.2 ug/ul) results in an increased survival of ONC injured RGCs by approximately 49% compared to the vehicle-treated animals. Expression of Hsp70 in retinas of 17-AAG-treated animals was upregulated approximately by 2-fold compared to control animals. Our data support the idea that the upregulation of Hsp70 has a beneficial effect on the survival of injured RGCs, and the induction of this protein could be viewed as a potential neuroprotective strategy for optic neuropathies.
Keywords: retina, ganglion cells, optic nerve, Hsp70, 17-AAG, neuroprotection, neurodegeneration
INTRODUCTION
Heat shock proteins (Hsps) belong to a superfamily of stress proteins, which also include glucose regulating protein and lectin chaperons - calnexin and calreticulin. Stress proteins, in particular Hsps, are critical constituents of a complex defense mechanism that enhances cell survival under adverse environmental conditions. Hsps comprise a heterogeneous group of proteins and are classified into six families based on their molecular weight: small Hsps (12–43 kDa), Hsp40, Hsp60, Hsp70, Hsp90, and large Hsps (100–110 kDa). Under normal conditions, these proteins act as molecular chaperones and play a critical role in protein homeostasis by assisting protein folding, the assembly and disassembly of protein complexes, protein repair or degradation, reduction of protein aggregation, subcellular localization of newly synthesized proteins, protein transport across membranes, and cytoskeletal organization.1–6 Stressful stimuli, such as hyperthermia,7,8 hypothermia,9 ischemia,10 hypoxia,11 depletion of ATP,12 free radicals,13 desiccation,14 viral infection,15 steroid hormones,16 and ethanol,17 induce expression of Hsps, which in turn assist in the refolding and repair of denatured proteins and facilitate the synthesis of new proteins to repair damage. Hsps have also been shown to suppress apoptotic pathways by interacting with proteins associated with signal transduction in active cell death. Hsps, Hsp70 in particular, have been shown to have cell protective effects in response to various insults. The Hsp70 family includes Hsc70 (the constitutive form), Hsp70 (the inducible form, also known as Hsp72), Grp75 (a constitutively expressed glucose-regulated protein found in the endoplasmic reticulum) and mtHsp70 (mitochondrial Hsp70). Hsp70 is synthesized at high levels in response to insults and has been recognized as a potential cell-protective protein.18,19 Hsp70 overexpression has been shown to protect cells from both apoptotic and necrotic death induced by various insults.19–21 However, survival of cells with induced Hsp70 expression also depends on the severity of injury, indicating that Hsp70 expression may not always be sufficient to ensure cell survival.
The cell protective role of Hsp70 has been demonstrated in various models of neurodegenerative diseases associated with toxicity from misfolded/aggregated proteins, such as Alzheimer’s disease, Parkinson disease, amyotrophic lateral sclerosis, Creutzfeld-Jacob Disease, Gerstmann-Straussler-Scheinker syndrome, Huntington’s disease, and Spinocerebellar ataxias. Hsp70 induction in an animal model of glaucomatous neurodegeneration by systemic administration of the divalent cation zinc or geranylgeranylacetate (GGA) was associated with increased survival of RGCs. 22,23 Degeneration of RGCs and their axons is the cause of visual impairment in optic neuropathies, including glaucoma that affects millions of people worldwide. Various neuroprotective strategies to preserve RGCs and their axons from glaucomatous damage have been proposed and studied over the last decade, and promising results have been reported in laboratory settings. These studies were performed mainly on animal models that were characterized by RGC degeneration induced by damage to the optic nerve, excitotoxicity, or ocular hypertension. It is anticipated that some of these neuroprotective strategies will demonstrate beneficial clinical effects and might be used in combination with controlling intraocular pressure (IOP) to treat glaucoma. We believe that the cytoprotective/anti-apoptotic characteristics of Hsp70 and the possibility of its pharmacological induction in cells experiencing stress make this protein an attractive therapeutic target for RGC protection. The ability of Hsp70 to protect cells from a variety of stress stimuli is an important factor in designing a strategy to preserve RGCs in glaucoma, since the cellular damage in glaucoma may be caused by different molecular mechanisms leading to optic nerve damage. In the current study, we evaluated the effect of viral (AAV2) and pharmacological (17-AAG, 17- N -allylamino-17-demethoxygeldanamycin) mediated upregulation of Hsp70 on the survival of RGCs after optic nerve crush (ONC) injury.
RESULTS
The cell protective effect of AAV2-Hsp70 and 17-AAG was evaluated in mouse and rat ONC models respectively, characterized by rapid and specific degeneration of RGCs due to physical damage to the optic nerve. RGC numbers, in both control and experimental animals, were determined by counting Rbpms-positive cells on whole mounted retinas two weeks after ONC.
AAV2-induced Hsp70 expression in the retina
AAV-mediated Hsp70 expression and cell protective effect of AAV2-Hsp70 based therapy was evaluated in a mouse ONC model four weeks after intravitreal vector injection (two weeks after ONC). This time period has been reported to be sufficient for an efficient transgene expression in the rodent retina.24,25 Expression of Hsp70 was primarily observed in the GCL of the retina. Since the GCL of rodent retinas contain both RGCs and displaced amacrine cells in approximately 1:1 ratio, double staining with antibodies against Hsp70 and markers for RGCs (Rbpms) and amacrine cell (calbindin) was performed to determine relative number of RGCs, a primary targets of the therapy, and amacrine cells transfected with AAV2-Hsp70 (Fig. 1A). By colacalization of Rbpms- and Hsp70-positive cells we found, that 76.4% of all transfected cells in the GCL were RGCs, indicating preferential transfection of RGCs with AAV serotype 2 vectors by intravitreous delivery. Western blot analysis of the retinal extract showed an approximately 1.2 fold increase in Hsp70 level in AAV2-Hsp70-treated animals compared to vehicle (PBS)-injected controls (n=6; P<0.05; Figs. 1B and C). The Hsp70 expression level in PBS- and AAV2-GFP-injected animals was similar.
Figure 1.

AAV2-Hsp70-mediated expression of HSP70 in the mouse retina. A. Colocalization of Hsp70 positive cells with RGCs and amacrine cells labeled with Rbpms and calbindin, respectively (long arrows). Short arrows point at Hsp70-positive cells that are not labeled with calbindin. Approximately 75% of all AAV2-Hsp70 transfected cells in the retina 4 weeks following intravitreal injection were RGCs. B and C. Western blot analysis showed approximately 1.2 fold increase in the level of Hsp70 protein in retinas 4 weeks after transfection with AAV2-Hsp70 compared to vehicle (PBS)-injected control group (*P<0.05).
AAV2-mediated Hsp70 expression increases survival of RGCs
To determine the cell protective effect of Hsp70 induction, RGCs were counted in uninjured controls injected with PBS or AAV2-GFP, uninjured controls injected with AAV2-Hsp70, ONC animals injected with PBS and ONC animals injected with AAV2-Hsp70. No significant difference in RGC numbers was observed between uninjured animals (n=8 per group) injected with PBS (2824±488 cells/mm2), AAV2-GFP (2841±486 cells/mm2) and AAV2-Hsp70- (2877±445 cells/mm2). In ONC animals, RGC survival in AAV2-Hsp70 treated group (636±168 cells/mm2) was increased by an average of 110% (n=7; P=0.029) 2 weeks after axonal injury compared to the vehicle-injected control (303±129 cells/mm2; Figs. 2A and B). The largest difference in RGC numbers in AAV2-Hsp70-injected versus control animals, a 306% increase, was observed in the inferior retinal quadrant (n=7; P=0.002). This could be explained by more efficient cell transfection near the injection site compared to other regions of the retina that we observed in our study. Fig. 3 shows a regional distribution of AAV2-GFP transfected cells 4 weeks after injection.
Figure 2.

AAV2-Hsp70 protects RGCs from axonal injury. A. Representative images of Rbpms-labeled RGCs in uninjured and ONC retinas. RGC densities in mice with no ONC damage were similar in vehicle (PBS)-injected and AAV2-Hsp70-injected animals. The number of RGCs two weeks after ONC was reduced by approximately 80% compared to uninjured controls. B. Quantitative analysis of RGCs in whole mount retinas showed approximately 110% increase in cell survival in animals injected with AAV2-Hsp70 two weeks after ONC compared to vehicle-injected ONC control group (*P=0.029).
Figure 3.

AAV2-mediated GFP expression in the retina. A. GFP fluorescence was observed in RGC axons four weeks after intravitreal administration of AAV2-GFP. Regional GFP expression was present in the inferior (a), temporal (b), inferior-temporal (c) and inferior-nasal (d) retina. Images were taken approximately at the margin of the optic nerve head or 1 mm from the center of the optic nerve head. B. GFP positive cells were detected primarily in superior (a) and inferior (c) retina. A few GFP positive axons were noted in the temporal (b) retina. GFP positive expression was rarely seen in the nasal retina. Images were taken approximately at 3 to 4 mm from the center of optic nerve head.
17-AAG protects RGCs from axonal injury
Cell protective effect of 17-AAG-induced Hsp70 expression on RGC survival was analyzed similar to AAV2-Hsp70 study. RGC density in Vehicle/ONC group was decreased to approximately 355±63 cells/mm2, which is about 19% of total number of cells in vehicle-treated animals with no damage to RGC axons (1860±175 cells/mm2; Figs. 4A and B). In 17-AAG injected rats with no ONC injury, RGC density was 2050±157 cells/mm2, slightly higher (4.8%) than in vehicle-treated uninjured animals although not statistically significant. The number of remaining RGCs in retinas of 17-AAG/ONC animals was approximately 527±30 cells/mm2. Compared to the Vehicle/ONC group, the administration of 17-AAG preserved approximately 49% more cells (n=8; P=0.007). RGC numbers in 17-AAG-treated animals were higher in all three retinal quadrants (inferior, superior and temporal) used in this study for RGC counting, with the most notable increase in cell density observed in the temporal region (~120%; P=0.01). With respect to the retinal location, RGC densities at 1, 2, 3 and 4 mm from the center of optic nerve head were higher by 61% (P=0.01), 57% (P=0.01), 26% (P=0.13) and 52% (P=0.01), respectively in the 17-AAG/ONC group compared to the Vehicle/ONC group.
Figure 4.

RGC density in vehicle- and 17-AAG-treated animals. A. Representative images of flat mounted rat retinas used for quantification of RGCs. RGCs immunolabeled with Rbpms antibodies were counted in three sampling fields (0.32 X 0.24 mm each) within the superior, temporal, inferior and nasal retinal quadrants at 1, 2, 3 and 4 mm from the optic disc. B. Two weeks after ONC, RGC density in vehicle-treated rats was decreased approximately 80% compared to the uninjured vehicle-treated animals. The number of surviving RGCs in retinas of 17-AAG/ONC animals was approximately 49% higher than in Vehicle/ONC retinas (*P=0.007).
17-AAG-mediated modulation of heat shock protein expression
Interaction of 17-AAG with Hsp90 leads to the dissociation and activation of Hsf1, which in turn can induce transcription of various Hsp genes. Since cell protective effect of 17-AAG is generally associated with its induction of Hsp70 expression, we first analyzed the modulation of Hsp70 in the retina two weeks after a single intravitreal administration of 17-AAG. An approximately 2-fold (n=6; P=0.01) induction of Hsp70 was observed by Western blot analysis in retinas of uninjured and ONC animals, compared to that of control animals that received a vehicle (Figs. 5A and E). The immunoblot data were further corroborated with the IHC analysis of Hsp70 expression in vehicle- and 17-AAG treated retinas (Fig. 6A). In vehicle-injected animals, Hsp70 immunoreactivity was primarily observed in the photoreceptor inner segments (IS) and outer nuclear layer (ONL). A significant upregulation of Hsp70 expression was seen in retinal sections obtained from 17-AAG treated animals. Hsp70 staining in these retinas was increased across all layers, and particularly, in the IS, ONL and GCL. The Hsp70 expression pattern after ONC was similar to that of uninjured animals. 17-AAG mediated upregulation of Hsp70 in retinas of these animals was primarily observed in the IS and ONL. Hsp70 staining was also more prominent in the GCL of 17-AAG-treated animals compared to the vehicle-injected ONC animals. The intensity of Hsp70 staining in the GCL of 17-AAG-treated animals was lower in the ONC group than in the uninjured group most likely due to dramatic reduction in RGC numbers. We also analyzed the expression level of Hsp90, Hsf1 and Hsf2 in vehicle- and 17-AAG injected animals. However, no significant change in the levels of these proteins was observed in 17-AAG-treated retinas (both uninjured and ONC) compared to the controls (Figs. 5B, C, D and E and Fig. 6B; IHC data for Hsp90 and Hsf2 are not shown).
Figure 5.

Quantitative analysis of Hsp70 expression in retinas of animals treated with 17-AAG. A. Western blot analysis of Hsp70 in retinal extracts from ONC and uninjured rats treated with vehicle or 17-AAG showed a significant change in Hsp70 expression level associated with the administration of 17-AAG. B. A chart demonstrates densitometric values of corresponding band intensities normalized against beta-actin. Expression of Hsp70 in 17-AAG-treated animals with and without optic nerve injury was upregulated ~2.2 fold (*P=0.01) compared to vehicle-injected uninjured and ONC animals. No significant changes in Hsp90, Hsf1 or Hsf2 expression levels were observed in retinas of 17-AAG-injected animals. Protein levels in this study were quantified 2 weeks after ONC (a single intravitreal injection of 17-AAG or vehicle was given on the day of ONC).
Figure 6.
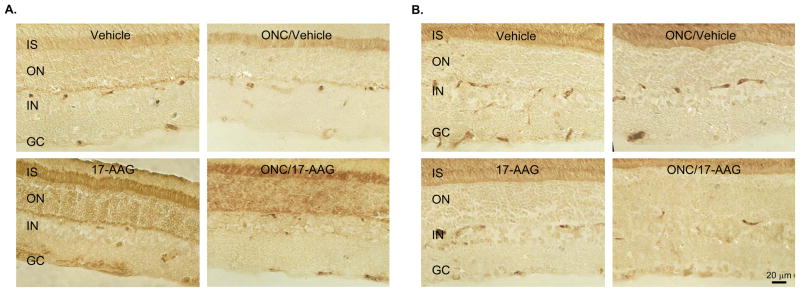
IHC analysis of Hsp70 (A) and Hsf1 (B) in control and experimental rat retinas. Retinal sections used for IHC were obtained 2 weeks after ONC. Increase in Hsp70 expression in retinas from animals injected with 17-AAG is observed primarily in photoreceptor inner segment (IS), outer nuclear (ON) and ganglion cell (GC) layers.
DISCUSSION
The present study evaluates the neuroprotective effects of AAV2-Hsp70- and 17-AAG-mediated stimulation of Hsp70 expression as a new strategy to promote RGC survival following an axonal injury. Earlier published results from our group implicate heat stress, Zn2+ and GGA induced Hsp70 upregulation in protection of RGCs in experimental model of glaucoma generated by elevation of intraocular pressure.22,23 From the standpoint of designing new neuroprotective strategies to control the progression of glaucomatous damage, it is important to target both IOP and non-IOP components of glaucoma pathophysiology, since the exact mechanisms underlying the pathogenesis of glaucoma are unknown and multiple factors and pathways were proposed to be involved in glaucomatous RGC degeneration. This suggests that enhancing the overall cell defense mechanism (for instance, by inducing expression of Hsps), rather than targeting specific mechanisms, including protein aggregation, excytotoxcicity, inflammation, oxidative stress and mitochondrial dysfunction, could be viewed as a potentially viable strategy to protect RGCs from the multifactorial nature of this disease.26–31 Here, we showed that AAV2-Hsp70 therapy increases the survival of RGCs injured by ONC by more than 100% compared to the control retinas. The cell protective effect of Hsp70 upregulation in the retinal inferior regions was even more impressive than that calculated for the entire retinas: more than 300% compared to controls. These regional differences in RGC survival could be due to the fact that the viral transduction in our experiments was mostly localized to a region near the injection site. Although AAV2 does not target RGCs specifically,32–34 the transfected cells were distributed predominantly within the GCL, where approximately 75% of these cells, as expected, were colocalized with Rbpms-positive RGCs. Preferential transduction of RGCs by AAV2 has been reported earlier and for that reason we used this serotype in our study, in which RGCs are the main target for the therapy.35 Quantitative analysis of Hsp70 levels showed approximately 1.2 fold increase in AAV2-Hsp70-treated retinas compared to the controls. This number does not adequately reflect the increase in Hsp70 level in individual transduced RGCs, since the quantification of Hsp70 was performed using proteins isolated from the entire retina where RGCs constitute a very small percent of total retinal cell population. In rat retinas, for instance, there is approximately 1 RGC per 260 photoreceptors (3×107 photoreceptors in total).36 Furthermore, we think that actual increase in Hsp70 expression level in the transduced RGCs is higher than 1.2 fold since the cell transfection in the retina was regional.
The observed neuroprotective effect of AAV2-Hsp70 can serve as a proof-of-principal that upregulation of Hsp70 expression can be applied to protect RGCs from axonal injury. Although AAV-based gene therapy is an attractive strategy for the treatment of different ocular diseases and has been evaluated in clinical trials, we believe that pharmacological induction of Hsp70 could be an alternative to viral-mediated approach with some important advantages: a) the drug administration could be less invasive than intravitreal injection (oral or i.v); b) the treatment regimen can be easily modified or interrupted if necessary; and c) the systemic induction of Hsp expression may exert its beneficial effect by targeting both ocular and non-ocular tissues involved in glaucomatous neurodegeneration, including neurons and glial cells in the retina, lateral geniculate nucleus (LGN) and primary visual cortex (V1), and trabecular meshwork (TM) cells. This was the rationale of using 17-AAG, the cell protective effect of which is generally associated with an induction of Hsp expression. 17-AAG, a potent Hsp90 inhibitor, can specifically bind Hsp90 ATP-binding site resulting in the dissociation and activation of Hsf1, which in resting cells forms a complex with Hsp90.37–39 Upon its activation, Hsf1 induces transcription of various Hsps.40,41 This ability of 17-AAG to imitate the cellular heat shock response and induce the expression of Hsps (Hsp70 in particular) has been associate with its cell protective effects in various models of degenerative diseases. In addition to its cell protective effect, 17-AAG inhibition of Hsp90 has been extensively studied in cancer research as a new strategy for the proteosomal degradation of oncoproteins.39 Results of the current study indicate that a single intravitreal injection of 17-AAG can preserve approximately 49% more RGCs injured by ONC compared to vehicle-injected animals. We found that an increase in RGC survival is correlated with a 2-fold increase in the level of Hsp70. To our knowledge, there is only one published report that assessed the effect of intravitreal injection of 17-AAG on cell survival: 17-AAG was used to rescue photoreceptors in a murine model of autosomal dominant retinitis pigmentosa caused by a mutation in the IMPDH1 gene.42 Expression of Hsp70 in experimental retinas was shown to be upregulated by ~1.8 fold 24 hours after 17-AAG injection, whereas the increase in Hsp40 and Hsp90 levels was marginal and not statistically significant.33 No upregulation of these proteins was observed 48 and 72 h after injection compared to control retinas. In agreement with these data, we also observed a 17-AAG-induced increase in the Hsp70 level (~2-fold in uninjured and ONC animals) in rat retinas, whereas no significant change in Hsp90 expression was detected. Interestingly, the observed Hsp70 upregulation was registered 2 weeks after injection. Alternative to the commonly accepted mechanism underlying 17-AAG-mediated Hsp induction through Hsf activation, the expression of Hsps could be upregulated by stimulation of Hsf1 expression. To test this possibility, we analyzed the levels of Hsf1 in animals treated with 17-AAG. However, both western blot analysis and IHC showed no detectable difference in Hsf1 and Hsf2 expression in retinas of drug-injected versus controls.
Although both AAV2-Hsp70 and 17-AAG-mediated protection of RGCs from ONC injury is associated with elevated Hsp70 levels, it appears that an approximately 2-fold increase in Hsp70 expression in the retinas of 17-AAG-treated animals is less efficient compared to a 1.2-fold Hsp70 upregulation in the AAV2-Hsp70 treated group: a 49% increase in RGC survival in 17-AAG group vs 110% in AAV-Hsp70 group. This apparent discrepancy can be explained by imunohistochemical data presented in Figs. 1 and 6 indicating that AAV2-Hsp70-mediated Hsp70 upregulation is taking place in GCL cells, and primarily in RGCs, whereas 17-AAG induces expression of this protein in the GCL as well as in other retinal cells, including photoreceptors. Therefore, the observed 2-fold increase in Hsp70 expression in retinas from 17-AAG-treated animals is a contribution not only from RGCs but also from other retinal cells.
The effect of Hsp70 induction on the survival of RGCs injured by ONC presented here is well correlated with the earlier published studies on the roles of Hsps in ocular hypertension, optic nerve axotomy and ischemia/reperfusion (I/R) models of RGC degeneration. In animals with experimental glaucoma, the induction of Hsp70 by heat stress, administration of zinc or GGA was shown to be associated with approximately 48%, 80% and 57% increased RGC survival, respectively, compared to the control group.22,23 In comparison with these data, heat stress treatment and administration of zinc in rats with acute glaucoma led to a modest 10% and 13%, respectively increase in RGC survival.43,44 Upregulation of Hsps was also associated with hydrogen sulfide (Hsp90) or carbon monoxide (Hsp70) preconditioning that have attenuated RGC death after I/R injury by approximately 42% and 52%, respectively.45,46 Furthermore, small Hsps, such as Hsp27, and alphaA and alphaB crystallins, were also reported to have a cell protective effect in the optic nerve transection (ONT) model. Administration of simvastatin (3-Hydroxy-3-methylglutaryl-CoA reductase inhibitor) has been shown to enhance RGC survival 7 and 14 days after axotomy by 90% and 19%, respectively, and this effect of simvastatin was associated with the induction of Hsp27 expression.47 We had shown earlier that overexpression of alphaA and alphaB increases the number of survived RGCs by 40% 1 week after axotomy and by 95% and 75%, respectively 2 weeks after ONT.48
In summary, the present study demonstrates that both viral and pharmacological upregulation of Hsp70 expression stimulate the survival of RGCs injured by ONC. These data, together with earlier observations on RGC protective effect of Hsp70 in experimental glaucoma model suggest that Hsp70-based therapy could be viewed as a promising strategy to stimulate cell defense mechanisms against insults of various nature.
METHODS
ONC injury animal model and intravitreal injections
The ONC model was generated in adult C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) and Brown Norway rats (Charles River Laboratories, Wilmington, MA). The use of animals and all experimental procedures with animals were approved by the Animal Research Committee of the University of California at Los Angeles and were in compliance with the National Institutes of Health Guide for the Care and Use of Animals and the ARVO (The Association for Research in Vision and Ophthalmology) Statement for the Use of Animals in Ophthalmic and Vision Research. Rats were housed with standard food and water provided ad libitum. ONC procedure was performed according to the previously described protocol with care to avoid damage to the blood circulation.49 Briefly, the left optic nerve of the anesthetized animals was exposed through a lateral conjunctival incision and crushed with a pair of fine self-closing forceps approximately 1 to 2 mm behind the posterior pole of the eye for 5 sec. Animals with noticeable changes in the lens and retina due to interrupted blood supply after ONC were excluded from the study. ONC was performed on the left eye of each animal. Since ONC of one eye affects the morphology of the contralateral retina, the right eyes of these animals were not served as untreated controls.50 The eyes of uninjured animals were used as untreated control.
AAV2-Hsp70 or 17-AAG were injected into the vitreous chamber using a 10-μl Hamilton syringe as described earlier.51 To evaluate the effect of AAV2-induced Hsp70 expression on RGC survival, mice received a single intravitreal injection (2 ul) of AAV2-Hsp70, AAV2-GFP (green fluorescent protein) or vehicle (phosphate-buffered saline; PBS) 2 weeks prior to ONC procedure. AAV2-GFP- and PBS-injected animals served as controls. To evaluate the RGC protective effect of 17-AAG, a single intravitreal injection (4 ul) of the drug (0.2 ug/ul in 2% DMSO) or vehicle (2% DMSO) was administered to rats on the day of ONC procedure. The animals were euthanized two weeks after ONC, and the retinas were then dissected and used for cell counting, protein extraction, and immunohistochemistry (IHC).
Preparation of AAV2 vectors
The human Hsp70 cDNA was inserted into pTR-UF-SB plasmid downstream of the hybrid CMV immediate early enhancer/chicken beta actin promoter in the UF-SB plasmid, containing the AAV2 inverted terminal repeat sequences and a SV-40 polyadenylation sequence. Recombinant AAV2 vector were prepared as previously described.52 Briefly, plasmid DNA was amplified and purified by cesium chloride gradient centrifugation and then packaged into AAV2 capsids by transfection into human HEK 293 cells. Real-time PCR was used to determine genome titers of the recombinant AAV. This protocol also was used to prepare a control AAV2-GFP. Titers for AAV2-Hsp70 and AAV2-GFP were 7.88E12 and 2.15E12 genomic copies (gc) per ml, respectively.
Quantification of RGCs
RGCs were identified by immunolabeling of flat mounted retinas with Rbpms (RNA binding protein with multiple splicing) antibodies according to a published protocol.53 Briefly, animals were deeply anesthetized with intramuscular injections of 80 mg/kg sodium pentobarbital and then transcardially perfused with 4% paraformaldehyde in 0.1 M phosphate buffer. The eyes were enucleated, immersed in fixative for 0.5 hours, and the lenses were removed. The eyecups were postfixed for another 0.5 hours. The retinas were dissected, incubated with 20% fetal bovine serum for 1 hour to block nonspecific staining, and then with the primary antibody against Rbpms (1:500) in PBS containing 1% triton, 0.5% BSA, and 0.9% sodium chloride (PBS-T-BSA) overnight at 4°C. Following incubation with secondary Alexa Fluor 488 goat anti-rabbit IgG antibody (1/1000) overnight at 4°C, retinas were flat mounted for topographical analysis of RGC density under a fluorescence microscope (LSM410; Carl Zeiss, Oberkochen, Germany). Immunolabeled cells were counted in three sampling fields within the superior, temporal, inferior and nasal retinal quadrants. Sampling locations at 1, 2, 3 and 4 mm from the optic disc were selected for rat retinas, and at 0.5, 1, 1.5 and 2 mm for mouse retinas. Three adjacent microscopic fields were imaged at each location along the axis of each retinal quadrant. In total, 48 fields were recorded per retina. Rbpms-positive cells were counted in each field (0.32 X 0.24 mm each) and the density of RGCs was calculated. RGC quantification was performed in a masked manner.
Immunoblot analysis
Immunoblot analysis was performed according to a standard protocol. Briefly, after determining protein concentrations (BCA Protein Assay Kit, Pierce, Rockford, IL), 1ug of detergent-soluble retinal proteins was separated on precast 12% SDS-polyacrylamide gels (Mini-Protein TGX, BIO-RAD, Hercules, CA) and transferred onto PVDF membranes (Immobilon-P, Millipore, Billerica, MA). Membranes were blocked with 5% non-fat milk and incubated with primary antibodies for Hsp70 (1:5000; Enzo, Farmingdale, NY), Hsp90 (1:5000; Enzo), Hsf1 (1:1000; Enzo) or Hsf2 (1:1000; Enzo) at 4°C overnight. Membranes were subsequently incubated with species-specific biotinylated antibodies (1:500; Enzo) and Horseradish Peroxidase (1:400; Enzo) for 1 hour at 4°C. Immunoreactive bands were detected with ECL Western Blotting Detection Reagents (GE Healthcare, Piscataway, NJ), and their intensities were quantified with NIH image software. Reprobing blots with a monoclonal antibody to a housekeeping beta-actin (1:10000; Sigma, St. Louis, MO) was used as an internal reference to normalize protein expression levels.
IHC analysis
IHC was performed according to a standard procedure published elsewhere.54 Briefly, enucleated eyeballs were immersed in fixative for 1 hour, bisected, and postfixed for 3 hours. The eye cups were incubated with 30% sucrose at 4°C overnight and embedded in an optimal cutting temperature compound (O.C.T.; Sakura Finetec, Torrance, CA). Ten-um thick sections were obtained along the vertical meridian through the optic nerve head. The sections were washed three times in 0.01M PBS and twice in 0.01M PBS with Triton-X (T-PBS), followed by blocking with 20% bovine serum albumin and 5% goat serum in T-PBS for 1-hour at 4°C. After blocking, they were incubated with primary antibodies at 4°C overnight. The primary antibodies recognizing the following proteins were used in this study: Hsp70 (1:100; Enzo) or Hsf1 (1:500; Enzo), Rbpms (1:500), and calbindin (1:500; Chemicon). For fluorescent staining, following an incubation with the primary antibodies, sections were incubated with rhodamine-conjugated anti rabbit IgG antibody or FITC-conjugated anti mouse IgG antibody (Cappel Research Products, Durham, NC). After mounting, images were taken with a fluorescence microscope (LSM510, Carl Zeiss, Oberkochen, Germany). 4′6-diamino-2-phenylindole dihydrochloride (DAPI, Molecular Probes, Eugen, OR) was used for nuclear staining. The distribution of GFP-positive cells was analyzed with fluorescence microscopy. For colorimetric staining, Avidin-Biotin Complex (ABC) method was used. Following incubation with primary antibodies, sections were incubated with biotinylated secondary antibodies according to a protocol provided by Vectastain ABC kit (Vector Labs, Burlingame, CA). Sections were then labeled with DAB Peroxidase Substrate Kit (Vector Labs) and mounted. Negative-control sections were performed by replacing the primary antibody with serum.
Statistical analysis
Data are presented as the mean ± standard deviation (SD). Differences among groups were analyzed by a Student’s t-test. P < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) Grant EY018644 (NP) and Research to Prevent Blindness (JC). WWH acknowledges NIH grants P30EY021721 and R01EY17549 and grants from the Macular Vision Research Foundation, Foundation Fighting Blindness, Usher3 Initiative, Overstreet Fund and Research to Prevent Blindness, Inc. for partial support of this work.
Footnotes
CONFLICT OF INTEREST
WWH and the University of Florida have a financial interest in the use of AAV therapies, and own equity in a company (AGTC Inc.) that might, in the future, commercialize some aspects of this work. The remaining authors declare no conflict of interest.
References
- 1.Zimmerman SB, Minton AP. Macromolecular crowding: biochemical, biophysical and physiological consequences. Annu Rev Biophys Biomol Struct. 1993;22:27–65. doi: 10.1146/annurev.bb.22.060193.000331. [DOI] [PubMed] [Google Scholar]
- 2.Saibil H. Molecular chaperones: containers and surfaces for folding, stabilizing or unfolding proteins. Curr Opin Struct Biol. 2000;10:251–258. doi: 10.1016/s0959-440x(00)00074-9. [DOI] [PubMed] [Google Scholar]
- 3.Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 4.Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- 5.Ohtsuka K, Hata M. Molecular chaperone function of mammalian Hsp70 and Hsp40 – a review. Int J Hyperthermia. 2000;16:231–245. doi: 10.1080/026567300285259. [DOI] [PubMed] [Google Scholar]
- 6.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 7.Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- 8.Ostberg JR, Kaplan KC, Repasky EA. Induction of stress proteins in a panel of mouse tissues by fever-range whole body hyperthermia. Int J Hyperthermia. 2002;18:552–562. doi: 10.1080/02656730210166168. [DOI] [PubMed] [Google Scholar]
- 9.Norton PM, Latchman DS. Levels of 90kd heat shock protein and resistance to glucocorticoid mediated cell killing in a range of human and murine lymphocyte cell lines. Genes Dev. 1989;33:149–154. doi: 10.1016/0022-4731(89)90288-4. [DOI] [PubMed] [Google Scholar]
- 10.Richard V, Kaeffer N, Thuillez C. Delayed protection of the ischemic heart from pathophysiology to therapeutic applications. Fundam Clin Pharmacol. 1996;10:409–415. doi: 10.1111/j.1472-8206.1996.tb00595.x. [DOI] [PubMed] [Google Scholar]
- 11.Patel BA, Khaliq J, Evans J. Hypoxia induces hsp70 gene expression in human hepatoma (HEP G2) cells. Biochem Mol Biol Int. 1995;36:907–912. [PubMed] [Google Scholar]
- 12.Kabakov AE, Gabai VL. Heat-shock proteins maintain the viability of ATP-deprived cells: what is the mechanism? Trends Cell Biol. 1994;4:193–196. doi: 10.1016/0962-8924(94)90135-x. [DOI] [PubMed] [Google Scholar]
- 13.Kukreja RC, Kontos MC, Loesser KE, Batra SK, Qian YZ, Gbur CJ, Jr, et al. Oxidant stress increases heat shock protein 70 mRNA in isolated perfused rat heart. Am J Physiol. 1994;267:2213–2219. doi: 10.1152/ajpheart.1994.267.6.H2213. [DOI] [PubMed] [Google Scholar]
- 14.Yang XM, Baxter GF, Heads RJ, Yellon DM, Downey JM, Cohen MV. Infarct limitation of the second window of protection in a conscious rabbit model. Cardiovasc Res. 1996;31:777–783. doi: 10.1016/0008-6363(96)00026-0. [DOI] [PubMed] [Google Scholar]
- 15.Ravindran RK, Tablin F, Crowe FJ, Oliver AE. Resistance to dehydration damage in HeLa cells correlates with the presence of endogenous heat shock proteins. Cell Preserv Tech. 2005;3:155–164. [Google Scholar]
- 16.Collins P, Hightower LE. Newcastle disease virus stimulates the cellular accumulation of stress (heat shock) mRNAs and proteins. J Virol. 1982;44:703–707. doi: 10.1128/jvi.44.2.703-707.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plesset J, Palm C, McLaughlin CS. Induction of heat shock proteins and thermotolerance by ethanol in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1982;108:1340–1345. doi: 10.1016/0006-291x(82)92147-7. [DOI] [PubMed] [Google Scholar]
- 18.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 19.Jaattela M. Heat shock proteins as cellular lifeguards. Ann Med. 1999;31:261–271. doi: 10.3109/07853899908995889. [DOI] [PubMed] [Google Scholar]
- 20.Vayssier M, Polla BS. Heat shock proteins chaperoning life and death. Cell Stress Chaperones. 1998;3:221–227. doi: 10.1379/1466-1268(1998)003<0221:hspcla>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buzzard KA, Giaccia AJ, Killender M, Anderson RL. Heat shock protein 72 modulates pathways of stress-induced apoptosis. J Biol Chem. 1998;273:17147–17153. doi: 10.1074/jbc.273.27.17147. [DOI] [PubMed] [Google Scholar]
- 22.Park KH, Cozier F, Ong OC, Caprioli J. Induction of heat shock protein 72 protects retinal ganglion cells in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2001;42:1522–1530. [PubMed] [Google Scholar]
- 23.Ishii Y, Kwong JM, Caprioli J. Retinal ganglion cell protection with geranylgeranylacetone, a heat shock protein inducer, in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2003;44:1982–1992. [PubMed] [Google Scholar]
- 24.Bennet J, Duan D, Engelhardt JF, Maguire AM. Cross-species comparison of in vivo reporter gene expression after recombinant adeno-associated virus-mediated retinal transduction. Methods Enzymol. 2000;316:777–789. doi: 10.1016/s0076-6879(00)16762-x. [DOI] [PubMed] [Google Scholar]
- 25.Cheng L, Sapieha P, Kittlerova P, Hauswirth WW, Di Polo A. TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J Neurosci. 2002;22:3977–3986. doi: 10.1523/JNEUROSCI.22-10-03977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen SD, Wang L, Zhang XL. Neuroprotection in glaucoma: present and future. Chin Med J (Engl) 2013;126:1567–1577. [PubMed] [Google Scholar]
- 27.Guo L, Salt TE, Luong V, Wood N, Cheung W, Maass A, et al. Targeting amyloid-beta in glaucoma treatment. Proc Natl Acad Sci U S A. 2007;104:13444–13449. doi: 10.1073/pnas.0703707104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai Y, Du L, Dunsmore KE, Jenkins LW, Wong HR, Clark RS. Selectively increasing inducible heat shock protein 70 via TAT-protein transduction protects neurons from nitrosative stress and excitotoxicity. J Neurochem. 2005;94:360–366. doi: 10.1111/j.1471-4159.2005.03212.x. [DOI] [PubMed] [Google Scholar]
- 29.Borges TJ, Wieten L, van Herwijnen MJ, Broere F, van der Zee R, Bonorino C, et al. The anti-inflammatory mechanisms of Hsp70. Front Immunol. 2012;3:95. doi: 10.3389/fimmu.2012.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalmar B, Greensmith L. Induction of heat shock proteins for protection against oxidative stress. Adv Drug Deliv Rev. 2009 Apr 28;61(4):310–8. doi: 10.1016/j.addr.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Pan P, Urban MJ, Zhao H, Blagg BS, Dobrowsky RT. Heat shock protein 70 is necessary to improve mitochondrial bioenergetics and reverse diabetic sensory neuropathy following KU-32 therapy. J Pharmacol Exp Ther. 2014;348:281–292. doi: 10.1124/jpet.113.210435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Auricchio A, Kobinger G, Anand V, Hildinger M, O’Connor E, Maguire AM, et al. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics. Hum Mol Genet. 2001;10:3075–3081. doi: 10.1093/hmg/10.26.3075. [DOI] [PubMed] [Google Scholar]
- 33.Harvey AR1, Kamphuis W, Eggers R, Symons NA, Blits B, Niclou S, et al. Intravitreal injection of adeno-associated viral vectors results in the transduction of different types of retinal neurons in neonatal and adult rats: a comparison with lentiviral vectors. J Mol Cell Neurosci. 2002;21:141–157. doi: 10.1006/mcne.2002.1168. [DOI] [PubMed] [Google Scholar]
- 34.Yin L, Greenberg K, Hunter JJ, Dalkara D, Kolstad KD, Masella BD, et al. Intravitreal injection of AAV2 transduces macaque inner retina. Invest Ophthalmol Vis Sci. 2011;52:2775–2783. doi: 10.1167/iovs.10-6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harvey AR, Hellström M, Rodger J. Gene therapy and transplantation in the retinofugal pathway. Prog Brain Res. 2009;175:151–161. doi: 10.1016/S0079-6123(09)17510-6. [DOI] [PubMed] [Google Scholar]
- 36.Mayhew TM, Astle D. Photoreceptor number and outer segment disk membrane surface area in the retina of the rat: stereological data for whole organ and average photoreceptor cell. J Neurocytol. 1997;26:53–61. doi: 10.1023/a:1018563409196. [DOI] [PubMed] [Google Scholar]
- 37.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90–pp60v–src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 39.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 40.Guettouche T, Boellmann F, Lane WS, Voellmy R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005;6:4. doi: 10.1186/1471-2091-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shamovsky I, Ivannikov M, Kandel ES, Gershon D, Nudler E. RNA-mediated response to heat shock in mammalian cells. Nature. 2006;440:556–560. doi: 10.1038/nature04518. [DOI] [PubMed] [Google Scholar]
- 42.Tam LC, Kiang AS, Campbell M, Keaney J, Farrar GJ, Humphries MM, et al. Prevention of autosomal dominant retinitis pigmentosa by systemic drug therapy targeting heat shock protein 90 (Hsp90) Hum Mol Genet. 2010;19:4421–4436. doi: 10.1093/hmg/ddq369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biermann J, Lagrèze WA, Dimitriu C, Stoykow C, Goebel U. Preconditioning with inhalative carbon monoxide protects rat retinal ganglion cells from ischemia/reperfusion injury. Invest Ophthalmol Vis Sci. 2010;51:3784–3791. doi: 10.1167/iovs.09-4894. [DOI] [PubMed] [Google Scholar]
- 44.Biermann J, Lagrèze WA, Schallner N, Schwer CI, Goebel U. Inhalative preconditioning with hydrogen sulfide attenuated apoptosis after retinal ischemia/reperfusion injury. Mol Vis. 2011;17:1275–1286. [PMC free article] [PubMed] [Google Scholar]
- 45.Qing G, Duan X, Jiang Y. Induction of heat shock protein 72 in RGCs of rat acute glaucoma model after heat stress or zinc administration. Yan Ke Xue Bao. 2004;20:30–33. [PubMed] [Google Scholar]
- 46.Qing G, Duan X, Jiang Y. Heat shock protein 72 protects retinal ganglion cells in rat model of acute glaucoma. Yan Ke Xue Bao. 2005;21:163–168. [PubMed] [Google Scholar]
- 47.Kretz A, Schmeer C, Tausch S, Isenmann S. Simvastatin promotes heat shock protein 27 expression and Akt activation in the rat retina and protects axotomized retinal ganglion cells in vivo. Neurobiol Dis. 2006;21:421–430. doi: 10.1016/j.nbd.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 48.Munemasa Y, Kwong JM, Caprioli J, Piri N. The role of alphaA- and alphaB-crystallins in the survival of retinal ganglion cells after optic nerve axotomy. Invest Ophthalmol Vis Sci. 2009;50:3869–3875. doi: 10.1167/iovs.08-3138. [DOI] [PubMed] [Google Scholar]
- 49.Nadal-Nicolas FM, Jimenez-Lopez M, Sobrado-Calvo P, Nieto-Lopez L, Canovas-Martinez I, Salinas-Navarro M, et al. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve–injured retinas. Invest Ophthalmol Vis Sci. 2009;50:3860–3868. doi: 10.1167/iovs.08-3267. [DOI] [PubMed] [Google Scholar]
- 50.Panagis L, Thanos S, Fischer D, Dermon CR. Unilateral optic nerve crush induces bilateral retinal glial cell proliferation. Eur J Neurosci. 2005;21:2305–2309. doi: 10.1111/j.1460-9568.2005.04046.x. [DOI] [PubMed] [Google Scholar]
- 51.Mansour-Robaey S, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci U S A. 1994;91:1632–1636. doi: 10.1073/pnas.91.5.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hauswirth WW, Lewin AS, Zolotukhin S, Muzyczka N. Production and purification of recombinant adeno-associated virus. Methods Enzymol. 2000;316:743–761. doi: 10.1016/s0076-6879(00)16760-6. [DOI] [PubMed] [Google Scholar]
- 53.Kwong JM, Caprioli J, Piri N. RNA binding protein with multiple splicing: a new marker for retinal ganglion cells. Invest Ophthalmol Vis Sci. 2010;51:1052–1058. doi: 10.1167/iovs.09-4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munemasa Y, Ahn JH, Kwong JM, Caprioli J, Piri N. Redox proteins thioredoxin 1 and thioredoxin 2 support retinal ganglion cell survival in experimental glaucoma. Gene Ther. 2009;16:17–25. doi: 10.1038/gt.2008.126. [DOI] [PubMed] [Google Scholar]