

Abstract
Glioblastoma multiforme (GBM), the most common type of malignant brain tumor is highly fatal. Limited understanding of its rapid progression necessitates additional approaches that integrate what is known about the genomics of this cancer. Using a discovery set (n=348) and a validation set (n=174) of GBM patients, we performed genome-wide analyses that integrated mRNA and microRNA expression data from GBM as well as associated survival information, assessing coordinate variability in each as this reflects their known mechanistic functions. Cox proportional hazards models were used for the survival analyses, and nonparametric permutation tests were performed for the microRNAs to investigate the association between the number of associated genes and its prognostication. We also utilized mediation analyses for microRNA-gene pairs to identify their mediation effects. Genome-wide analyses revealed a novel pattern: microRNAs related to more gene expressions are more likely to be associated with GBM survival (P=4.8×10−5). Genome-wide mediation analyses for the 32,660 microRNA-gene pairs with strong association (FDR<0.01%) identified 51 validated pairs with significant mediation effect. Of the 51 pairs, miR-223 had 16 mediation genes. These 16 mediation genes of miR-223 were also highly associated with various other microRNAs and mediated their prognostic effects as well. We further constructed a gene signature using those 16 genes, which was highly associated with GBM survival in both the discovery and validation sets (P=9.8×10−6). This comprehensive study discovered mediation effects of microRNA to gene expression and GBM survival and provided a new analytic framework for integrative genomics.
Keywords: integrative genomics, glioblastoma multiforme, cancer survival, mediation analysis
INTRODUCTION
Glioblastoma multiforme (GBM) is the most common type of malignant brain tumor [Louis, et al. 2007]. GBM is rapidly fatal with median survival time between 12 and 15 months depending, in some cases, upon the type of treatment [Stupp, et al. 2005]. The lack of well-established environmental risk factors for GBM emphasizes the importance of discovering the genetic and epigenetic drivers of this disease in order to further our understanding of both its etiology and progression [Bondy, et al. 2008]. Due to the current incomplete understanding of GBM as well as its poor prognosis, there is a pressing need to find new ways to use the rapidly growing library of GBM genomic alterations to summarize relevant patterns in an effort to potentially target treatment efforts and help improve patient survival.
MicroRNAs are endogenous noncoding RNA molecules, usually 22 nucleotides in length. They take part in the epigenetic regulation of gene expression by inhibiting translation or inducing cleavage of mRNA in the targeted genes. Studies suggest that alterations in microRNA regulation contribute to cancer pathogenesis via inhibition of tumor suppressor genes [Bartels and Tsongalis 2009; Xiao and Rajewsky 2009]. MicroRNAs have also been reported to be associated with survival of GBM patients [Srinivasan, et al. 2011]. By understanding the biology of the relevant microRNAs, one can design small interfering RNAs (siRNA) that function in similar fashion to microRNAs, but can target and repress the expression of specific genes of interest [Castanotto and Rossi 2009]. The translational application of microRNA as potential biomarkers of prognosis, and as part of personalized therapy has made it a primary focus for future research in cancer biology [Jacobsen, et al. 2013].
There have been emerging interests in integrating multiplatform genomic data in studying clinical outcomes [Ben-Hamo and Efroni 2013; Jacobsen, et al. 2013; Suzuki, et al. 2013; Wang, et al. 2013]. Ben-Hamo and Efroni (2013) have published an algorithm that integrates microRNA and gene expression to classify GBM patients’ prognosis while Wang et al. (2013) have published another algorithm that can perform network analyses using microRNA and gene expression. Suzuki et al. (2013) proposed to use microRNA target genes for better selection of prognostic microRNAs. Despite the wide availability of these prediction algorithms, the mining of relevant features in the GBM genome for potential clinical insights and applications is still very sparse.
We utilized a novel approach applying mediation modeling [MacKinnon 2008; Pearl 2001; Robins and Greenland 1992] to investigate the joint effect of microRNA and gene expression on GBM survival. The mediation model has been applied to analyze genome-wide association studies (GWAS) data to disentangle the effects of genetic variants of a nicotinic acetylcholine receptor gene, using findings derived from multiple GWAS looking at the effects of cigarette smoking [VanderWeele, et al. 2012]. We also developed a method to integrate genetic and gene expression data in GWAS under the framework of mediation model [Huang, et al. 2014]. These applications illustrate the utility of a mediation analysis that employs mechanistic insights from genomic profiles in understanding and predicting clinical outcomes. In addition to its use for continuous or categorical outcomes, mediation analysis has recently been proposed for survival data where the outcome is the time-to-event [Lange and Hansen 2011; VanderWeele 2011].
In this study, we conducted thorough genome-wide analyses of microRNAs and gene expression along with GBM survival in a dataset of more than 500 patients. We sought specifically to examine the correlation of each microRNA-gene pair, guided in this by the known mechanisms tying these effects together, and we were able to summarize the number of genes that are associated with each microRNA. Using this large and complete profile enabled us to further investigate their implications of coordinated variability at these loci, positing that they define phenotypes that affect GBM survival. We specifically hypothesized that microRNA regulates gene expression which consequently affects GBM survival and set up mediation models to evaluate our hypothesis. Our findings may shed light on the drivers of now poorly understood GBM progression.
MATERIAL AND METHODS
Genomic data and clinical information of glioblastoma multiforme
There are 522 patients of glioblastoma multiforme with complete genomic data on gene expression (UNC AgilentG4502A-07) and microRNA expression (UNC H-miRNA 8×15K) archived in The Cancer Genome Atlas (TCGA), a research project that maps the genome of many types of cancer (http://cancergenome.nih.gov/)[International Cancer Genome, et al. 2010]. Patients who carried the IDH1 or MGMT mutations were not included in our analyses in order to restrict the application of our models to tumors thought to be more homogenous in terms of their genomic origin. In addition to genomic data, these patients have available clinical and survival information, including vital status, time to death, time to last follow-up, age, gender and race. Throughout the paper, we analyzed the level 3 data that has been preprocessed and normalized. We randomly divided the 522 subjects into a discovery set (n=348) and a validation set (n=174). To obtain robust findings and avoid false positives from multiple comparisons, genome-wide mediation analyses and survival analyses of the gene signature followed a two-stage discovery-validation process; the findings first observed in the discovery set need to then be confirmed in the validation set. We summarized our analysis procedure using a schematic in Figure 1.
Figure 1.

The schematic of data analyses.
Genome-wide microRNA-gene associations and survival analyses
For each microRNA, we conducted a genome-wide association analysis with 17,814 genes. With use of ordinary least square estimators, we regressed the expression value on natural log-transformed microRNA expression adjusting for age, gender and race as covariates. To avoid undue influence of outliers, we restricted our analyses to the middle 95% of the mRNA expression values. The genome-wide survival analyses with 17,814 genes were performed using Cox proportional hazards model [Cox 1972], adjusting for the same covariates. Similarly, the Cox model was also fit for the 534 log-transformed microRNAs. False Discovery Rate (FDR) [Benjamini and Hochberg 1995; Storey 2002] was calculated to address multiple comparisons.
Comparing microRNA-gene and microRNA-survival associations
We compared the number of associated genes between microRNAs associated with survival and those unassociated using the following steps. We first divided the 534 microRNAs into 27 survival-related microRNAs (P<0.05) and 507 survival-unrelated ones (P≥0.05). With a pre-specified P-value cut-off (e.g., 10−8), we compared the number of genes that were associated with each microRNA in the two groups. To formally evaluate the difference in the associated genes between the two groups, we then performed permutation analyses. For each permutation, we performed a genome-wide survival analysis by shuffling the survival outcome and obtained n1 survival-related microRNA (P<0.05) and n0 survival-unrelated ones (P≥0.05). We denoted zj as the number of associated genes (less than the pre-specified P-value cut-off) for microRNA j. To compare the number of associated genes between the two groups {z1, …, zn0} (the survival-unrelated microRNAs) vs. {zn0+1, …, zn0+n1} (the survival-related microRNAs), we calculated the Wilcoxon rank sum test statistic wb (b is the index of permutation). The reason to choose the rank-based statistic instead of other parametric statistics such as the t-statistic is to avoid signals driven by outliers. With 1,000 permutations, we approximated the distribution of the test statistics {wb, b = 1, …, 1000} using a Gaussian mixture model with three mixtures [Cai, et al. 2012] and compared the statistic from the original dataset to this distribution to obtain the permutation P-value. The sampling scheme was with respect to subjects and provided more meaningful interpretation of the findings [Goeman and Buhlmann 2007]. Lastly, the analyses were repeated for different significance cutoffs of microRNA-gene associations, from 0.1 to 10−40.
In order to compare the tail from the distribution of the associated genes between two groups (i.e., survival-related and unrelated microRNAs), we further dichotomized the number of associated genes at the third quartile (Q3) for the number of associated genes in all 534 microRNAs. We performed the testing procedure on whether the proportions of greater than Q3 between survival-related and survival-unrelated microRNAs differed. We first calculated the difference in two proportions, Δ. We then obtained 1,000 Δ’s from permutations where we again shuffled the survival outcome and repeated the genome-wide survival analyses and the above steps. We calculated the permutation P-value by comparing the observed Δ from the original dataset and its null distribution, approximated from the 1,000 permutation Δ’s with the Gaussian mixture model described above. Finally, the analyses were performed for different significance cut-offs of microRNA-gene associations.
Mediation analyses
We identified genes that mediate the effect of prognostic microRNAs on GBM survival by mediation analyses [VanderWeele 2011]. For each microRNA-gene pair, we fit the following two regression models, one a linear model and the other an accelerated failure time model [Wei 1992]:
where G, M and X are mRNA expression value of a gene, microRNA expression value and covariates, respectively; εG, the error term for gene expression follow normal distribution with mean 0 and variance ; ν and εT, the scale parameter and the error term for survival time; α’s and β’s, the regression parameters. We proposed an AFT model instead of a Cox model because of the rare outcome assumption for mediation analyses using Cox model [VanderWeele 2011], which does not hold in GBM mortality. The mediation (indirect) effect (ME) and the alternative (direct) effect (AE) can be calculated as:
and their variances were calculated using the delta method. The marginal effect of microRNA can be decomposed into mediation and alternative effects. Although direct and indirect effects have been defined and used in causal inference literature, we renamed them here as alternative and mediation effects to better reflect their biological interpretation. The indirect effect or mediation effect in our setting is the effect of a microRNA on the GBM survival mediated through gene expression (the red path in Figure 2) whereas the direct effect or alternative effect is the effect on the survival independent of expression of the gene, but perhaps through other genes or other mechanisms not related to the gene (the blue path in Figure 2). The genome-wide mediation analyses were performed in the discovery set, and the top 1,000 microRNA-gene pairs were cross-checked using the validation set. Only the microRNA-gene pairs that were validated at P<0.05 and had the mediation effect in the same direction for both discovery and validation sets were presented.
Figure 2. Mediation model.
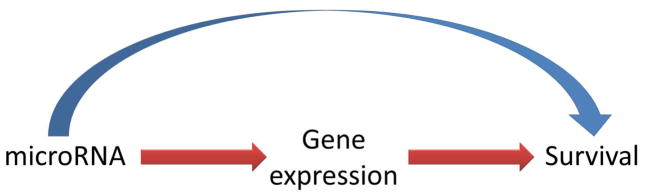
Directed acyclic diagraph (DAG) illustrating mediation process from microRNA to gene expression and then to cancer survival with the red path for mediation effect (ME, or indirect effect) and blue path for alternative effect (AE, or direct effect), the effect of microRNA on survival through other mechanisms independent of gene expression.
We constructed a gene signature from the 16 mediation genes of miR-223 using hierarchical clustering. Kaplan-Meier survival probabilities and 95% confidence intervals (CI) were estimated for the subjects, and log-rank tests were performed to evaluate the difference in the survival probabilities.
Network analysis of the mediation genes of miR-223
We further investigated the 16 mediation genes of miR-223 and microRNAs by network analyses. We plotted in gray the microRNA-gene association at a cut-off of FDR<0.01% (or equivalently, P<1.02×0−6), and varied the size of nodes for genes and microRNAs in proportion to −log10 P to represent their marginal association with GBM survival. We superimposed with the red edges the microRNA-gene pairs with significant mediation effect on GBM survival in the genome-wide mediation analyses.
RESULTS
The analysis procedure was illustrated in Figure 1. We first investigated the genome-wide association of the mRNA expression of 17,814 genes with 534 microRNAs in tumor tissues of glioblastoma multiforme. The distribution of z-statistics obtained from the 9,512,676 (17,814×534) microRNA-mRNA associations has heavy tails (gray histogram in Figure 3a), which indicates enriched associations between mRNAs and microRNAs in GBM. The enrichment was even more prominent in the top 107 (the top 20 percentile) microRNAs that were associated with the most genes (red histogram in Figure 3a). The distribution for the z-statistics of the bottom 160 (bottom 30 percentile) microRNAs (the blue histogram) is very close to the standard normal (the black line). The microRNA associated with the most gene expression was miR-222, and there were 1,425 genes associated with its value at P<10−10. miR-142-5p and miR-223, the second and third most gene-associated microRNAs were associated with 1,332 and 1,262 genes, respectively, under the same cut-off. For genes associated with miR-222, 928 genes had positive association and 497 genes had negative association (Figure 3b); for miR-142-5p, 1,105 genes had positive association and 227 genes had negative association (Figure 3c); for miR-223, 1,068 genes had positive association and 194 genes had negative association (Figure 3d). The Manhattan plots of the genome-wide association of gene expression with miR-222, miR-142-5p and miR-223 are provided in the Supplement (Supplementary Figures 1–3).
Figure 3. Genome-wide association of microRNA with gene expression in glioblastoma multiforme.

A, z-statistics of microRNA-gene expression association. B, Heatmap of the gene expression associated with miR-222. C, Heatmap of the gene expression associated with miR-142-5p. D, Heatmap of the gene expression associated with miR-223. The row is sorted by the value of microRNAs from low (left) to high (right)
MicroRNAs associated with more gene expression are more likely to be associated with GBM survival
We next studied whether microRNA expression, mRNA expression of genes and survival are coordinately variable when the analysis uses microRNAs as the anchor comparison. We found that the two associations (microRNA-mRNA and microRNA-survival) were related to each other in different aspects. First, dividing the 534 microRNAs into two groups at the median number of genes associated with the microRNA (>5 vs. ≤5 with association cut-off P<10−8), microRNAs associated with more genes had enriched signals in their association with GBM survival as well (P=0.068, Figure 4a). Second, dividing the 534 microRNAs into those that were survival-related (P<0.05) and survival-unrelated (P≥0.05), the distribution of the number of genes associated with the microRNA was significantly right shifted among survival-related microRNAs. Such differences were robust across different cut-offs of mRNA-microRNA associations with permutation test (smallest P=4.8×10−5, Figure 4b). Finally, if we dichotomized the distribution for the number of associated genes into greater or less than the third quartile, the difference between survival-related and survival-unrelated microRNAs was also significant (smallest P=5.6×10−4, Figure 4c). Therefore, we concluded from the above findings a robust pattern that microRNAs associated with more genes were more likely to be prognostic for GBM survival.
Figure 4. Gene expression-associated microRNAs are more likely to be survival-related.

A, histogram of P-values for the microRNA-survival association by the number of associated genes. B, The 3rd quartile of numbers of genes associated among survival-related (P<0.05) and survival-unrelated microRNAs (blue lines) and the P-values testing for the difference between the two (black line). C, The proportion of associated genes > 3rd quartile among survival-related (P<0.05) and survival-unrelated microRNAs (blue lines); and the P-values testing for the difference between the two (black line).
Genome-wide mediation analyses reveal genes that mediate the effects of miR-223, miR-33 and miR-142-5p on GBM survival
Although results in the previous section seemed to suggest a coordinated microRNA-gene expression-survival association, it was not until we performed mediation analyses that we were able to directly investigate such a mediation effect. We selected 32,660 microRNA-gene pairs with the significance for association less than 0.01% false discovery rate. For the 32,660 pairs, we performed a genome-wide mediation analyses in the discovery set and selected the top 1,000 pairs with the most significant mediation effect to confirm using the validation set. Ninety-seven pairs from the top 1,000 were confirmed with P<0.05 in the validation set. For the 97 microRNA-gene pairs, we excluded those with opposite directions in the discovery and validation sets or those with consistent association but with a P-value of association in the total subjects not smaller than that in either single set. These criteria gave us a final result of 51 pairs (Supplementary Table 1). Of these 51 pairs, there are 16 mediation genes for miR-223 on the effect of GBM survival (Table 1); 7 genes for miR-33; 5 genes for miR-142-5p; 4 genes for miR-130b; 2 genes each for miR-124a, miR-129, miR-142-3p, miR-338 and miR-93; and 1 gene each for miR-128a, miR-128b, miR-139, miR-181c, miR-181d, miR-29a, miR-34a, miR-9 and miR-9*. The results of the mediation analyses for miR-223, miR-33, miR-142-5p, miR-130b as well as the two known prognostic microRNAs, miR-221 and miR-222 [Medina, et al. 2008; Quintavalle, et al. 2012; Zhang, et al. 2010] were presented in Supplementary Tables 2–7.
Table 1.
16 mediation genes of miR-223.
| Gene | P-value of ME | AE of miR-223* | 95% CI* | P-value of AE* | ME* | 95% CI* | P-value of ME* | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Discovery Set | Validation Set | ||||||||||
| 1 | NFKBIZ | 0.0041 | 0.0010 | 3.44 | 1.32 | 8.99 | 0.012 | 0.29 | 0.17 | 0.50 | 7.8E-06 |
| 2 | PSCD4 | 0.0029 | 0.0025 | 2.90 | 1.14 | 7.38 | 0.025 | 0.30 | 0.18 | 0.51 | 8.9E-06 |
| 3 | BCL3 | 0.0043 | 0.0017 | 2.90 | 1.14 | 7.36 | 0.025 | 0.31 | 0.19 | 0.53 | 1.1E-05 |
| 4 | SLC16A3 | 0.00047 | 0.016 | 2.42 | 0.99 | 5.90 | 0.053 | 0.36 | 0.23 | 0.58 | 1.6E-05 |
| 5 | PLEKHQ1 | 0.0021 | 0.0089 | 2.41 | 0.98 | 5.93 | 0.056 | 0.37 | 0.23 | 0.59 | 2.9E-05 |
| 6 | LHFPL2 | 0.0029 | 0.010 | 2.56 | 1.00 | 6.57 | 0.051 | 0.33 | 0.20 | 0.56 | 4.1E-05 |
| 7 | LSP1 | 0.00055 | 0.030 | 2.00 | 0.83 | 4.82 | 0.12 | 0.46 | 0.32 | 0.67 | 5.0E-05 |
| 8 | URP2 | 0.0052 | 0.0088 | 2.21 | 0.88 | 5.56 | 0.090 | 0.35 | 0.21 | 0.58 | 5.7E-05 |
| 9 | CTSL1 | 0.0024 | 0.0097 | 2.30 | 0.92 | 5.74 | 0.075 | 0.36 | 0.22 | 0.60 | 6.6E-05 |
| 10 | ISG20 | 0.0068 | 0.017 | 1.73 | 0.74 | 4.04 | 0.21 | 0.52 | 0.37 | 0.74 | 0.00020 |
| 11 | HMOX1 | 0.0038 | 0.034 | 1.88 | 0.77 | 4.56 | 0.16 | 0.45 | 0.29 | 0.68 | 0.00021 |
| 12 | IL17RA | 0.0079 | 0.039 | 1.58 | 0.68 | 3.70 | 0.29 | 0.53 | 0.37 | 0.74 | 0.00026 |
| 13 | RBMX | 0.0077 | 0.039 | 1.78 | 0.74 | 4.25 | 0.20 | 0.52 | 0.37 | 0.74 | 0.00027 |
| 14 | FZD7 | 0.0031 | 0.048 | 1.55 | 0.66 | 3.66 | 0.32 | 0.52 | 0.36 | 0.74 | 0.00030 |
| 15 | CCNB1IP1 | 0.0070 | 0.036 | 1.76 | 0.74 | 4.16 | 0.20 | 0.54 | 0.38 | 0.75 | 0.00030 |
| 16 | LITAF | 0.0048 | 0.050 | 1.67 | 0.71 | 3.95 | 0.24 | 0.54 | 0.39 | 0.76 | 0.00030 |
For total subjects.
The mediation effects from the same microRNA through different genes showed the same direction of the effect. For example, the 16 mediation effects of miR-223 were all hazardous, i.e., the survival time decreased with the increase of miR-223 (Table 1). The effect of miR-223 increasing from 9.3 (the first quartile) to 10.5 (the third quartile) through the most significant mediation gene, NFKBIZ showed a decrease in the survival time by more than 70% (7.8×10−6). In contrast, the 7 mediation effects of miR-33 were all protective, i.e., the elevated expression of miR-33 increased the survival time. Another interesting finding was that most of the mediation genes of miR-33 also mediated the effect of miR-223, and their opposite mediation effects resulted from the opposite directions of microRNA-gene associations for miR-223 and miR-33.
The microRNAs that showed up in the mediation analyses are not necessarily marginally prognostic. For example, the marginal association with GBM survival were not significant in miR-223 (P=0.88), miR-33 (P=0.51) and miR-142-5p (P=0.60). From mediation analyses, we discovered that there existed alternative effects independent of these gene expressions and opposite to the mediation effects through their mediation genes (Tables 1 and Supplementary Tables S2–S4). The opposite directions of the mediation effects and the alternative effects lead to attenuation or even negation and thus non-significant marginal effects, which demonstrates the advantage of using mediation analyses that disentangle the effect from different mechanisms.
GBM survival can be predicted by a gene signature constructed from the 16 mediation genes of miR-223
Hierarchical clustering of the 16 mediation genes of miR-223 revealed a gene signature (Figure 5a). We then used the 16-gene signature to predict overall survival of GBM patients, again following the two-step discovery-validation process. The 16-gene signature provided a significant discrimination of overall survival in the discovery set (P=0.0019, Figure 5b), which was confirmed in the validation set (P=7.7×10−4, Figure 5c) and led to a very significant association for the total subjects (P=9.8×10−6, Figure 5d). The subjects carrying the gene signature had a one-year survival rate of 70.7% (95% CI: 63.1–79.2%); those without the gene signature had only 52.3% (47.2–58.0%).
Figure 5. Signature of 16 mediation genes of miR-223 and its association with GBM survival.

A, Hierarchical clustering of the 16 mediation genes of miR-223. Kaplan-Meier survival probabilities by the 16-gene signature in the discovery set (n=348) (B), the validation set (n=174) (C) and the total subjects (n=522) (D).
Network analysis of the 16 mediation genes and microRNAs
In order to visualize and better summarize the results from the microRNA-gene association and the microRNA-gene-survival mediation analyses, we performed a network analysis using only the 16 mediation genes of miR-223, as shown in Figure 6. The 16 genes were highly connected to the top microRNAs with significant mediation effects as well as the two known prognostic microRNAs, miR-221 and miR-222. There were 107 microRNA-gene associations with FDR<0.01%, of which 31 pairs had significant mediation effect on GBM survival. Most of the microRNAs with significant mediation effects did not have marginal association with the rates of survival, and their mediation effects came from the strong microRNA-gene association in conjunction with the significant prognostic effects of the genes. Although the 16 genes were identified as the mediation genes of miR-223, many of them were also associated with or were even mediation genes of other microRNAs with mediation effects.
Figure 6. Network analysis of 16 mediation genes of miR-223 and 11 microRNA with prognostic effects.

The gray edges indicate strong microRNA-gene association (FDR<0.01%); nodes with blue and gray colors represent microRNAs and genes, respectively; the size of nodes represents the significance of the association with survival and is proportional to −log10 P, the significance of marginal association with survival; the red edges indicate the microRNA-gene pairs with significant mediation effect on GBM survival.
Another noteworthy finding was that the microRNA-gene association (gray edge) is necessary but not sufficient to insure the presence of a mediation effect (red edge). In other words, a strong microRNA-gene association cannot be readily assumed to be a mediation effect, even if the microRNAs or genes are marginally prognostic. For example, miR-221 and miR-222, both known to be prognostic for GBM, are associated with many of the 16 genes tested; however, none of these strong associations turned out to have a mediation effect even though both of the microRNAs and all of the 16 genes are marginally associated with the survival.
DISCUSSION
We showed that microRNAs related to more gene expressions such as miR-221, miR-222, miR-223, miR-33 and miR-142-5p were more likely to be associated with GBM survival (P= 4.8×10−5). In other words, coordinated variability in gene and microRNA expression defines loci associated with GBM survival. Although the finding supported our mediation hypothesis (Figure 2), the evidence was too oblique to draw a definite conclusion. Therefore, we further conducted genome-wide mediation analyses to explicitly study the mediation effect from microRNAs to gene expression as it related to GBM survival. The mediation analyses suggested two types of prognostic microRNAs, both associated with significant variation in gene expression. One type of prognostic microRNAs such as miR-222 and miR-221 is associated with survival as well as many gene expressions but its prognostic effect is not mediated through the gene expressions associated with it. The other type of prognostic microRNAs, such as miR-223, miR-142-5p and miR-33, is not necessarily marginally associated with survival, but the prognostic effect is mediated through genes they are associated with. We then constructed a gene signature using the 16 mediation genes of miR-223, which was highly associated with patients’ survival. As the set of mediation genes was identified from a biology-driven hypothesis rather than an agnostic gene set from pure statistical association, we expected to see a stronger biological relevance and a promising clinical utility of the gene set. However, the mechanistic action represented by the gene set in relation to microRNAs and tumor progression remains elusive and will require further work.
Wang et al. (2013)[Wang, et al. 2013] proposed another graphical approach using Gaussian graphical model to characterize undirected co-expression of microRNA and gene, which does not necessarily have the same interpretation as the directed mediation effects. Due to the difference rooted in the nature of undirected co-expression and directed mediation effect, the mediation genes found here (Table 1) were not reported in their paper. Wang et al. (2013) assumed a steady-state network whereas we focus here on causal mediation model that requires unmeasured confounding assumptions [VanderWeele 2011]. Additionally, while our mediation approach performs survival analyses using accelerated failure time model, Wang’s approach is not able to directly handle time-to-event survival outcome and requires ad hoc imputation of censored survival time, which would not be easily applicable to a dataset with more censored subjects. Compared to Ben-Hamo and Efroni’s approach[Ben-Hamo and Efroni 2013], our study also has several fundamental differences. First, we focused on a common profile of microRNA-gene-survival relationship where they are more interested in different features of microRNA-gene pairs between two prognosis groups. Secondly, we aimed to classify genes and microRNAs according to their association with survival and biological functions where Ben-Hamo and Efroni (2013) classified patients for their prognosis. Lastly, our goal was to discover a new genomic feature related to survival while theirs was to develop a prediction model. Results based on different hypotheses and approaches may provide a complementary view to GBM genomics.
Although the mediation model with uni-directional arrows in Figure 2 is supported by biological knowledge, what we observed from the data is merely statistical association. Causal interpretation can be established provided a series of no unmeasured confounding assumptions [VanderWeele 2011]. If confounding is a concern, one may interpret the mediation effect as the statistical association of the coordinated variability of microRNA and mRNA expressions with GBM survival, rather than overstating the causality. While enumerating all possible confounders with proper adjustment is an ideal practice to perform causal mediation modeling, a more realistic approach may be to conduct independent biological experiments in cell lines or animal models to establish biological causality.
There have been a growing number of discoveries of microRNAs and their capability of regulating downstream gene expression in GBM [Karsy, et al. 2012]. Here we discovered genes that are not only associated with microRNAs but also mediate their effect on tumor progression or survival. The mediation genes of miR-223, for example, all increase with poor prognosis of GBM, but very few have been heavily researched in that context. However, most of these genes have been observed in some important immune or cytoregulatory role that contributes to other, more prominent types of cancer. The most significant mediation gene of miR-223, NFKBIZ encoding a protein that controls IL-6 production and, as such, has a central role in regulating cytokine production that leads to the inflammation response [Lee, et al. 2007]. Though it commonly is involved in transcriptional misregulations in cancer, there exists no explanation for the mechanism, so further research pertaining to this protein is needed. PCSD4 (or CYTH4) encodes a cytohesin that mediates protein sorting and movement of solutes across the cell membrane [Ogasawara, et al. 2000]. Mutations of this protein have been linked to some types of breast cancer, but with little information linking to glioblastomas or other types of brain cancers. BCL3 is a B-cell lymphoma-associated gene and a proto-oncogene candidate that inhibits cell growth and increases DNA damage, while decreasing apoptosis in specific types of cells with damaged DNA [Wakefield, et al. 2013]. Though mostly associated with lymphomas, the gene is highly associated with solid non-hematopoietic tumors and metastasis as well. SLC16A3 is a promoter for a monocarboxylate transporter, responsible for moving lactate and other monocarboxylates across cell membranes [Fisel, et al. 2013]. It has been linked to cancer via its regulation of lactate transport, which results in antiapoptotic effects in the cell. PLEKHQ1 (or PLEKHO2) codes for a plekstrin homology domain, and controls intracellular signaling. Little information on it regarding cancer has surfaced, so further research is required.
Supplementary Material
Acknowledgments
The research is supported by NIH/NCI 5R03CA182937-02, NIH/NIA 1R01AG048825-01 and Salomon Research Fund at Brown University.
References
- Bartels CL, Tsongalis GJ. MicroRNAs: novel biomarkers for human cancer. Clin Chem. 2009;55(4):623–31. doi: 10.1373/clinchem.2008.112805. [DOI] [PubMed] [Google Scholar]
- Ben-Hamo R, Efroni S. MicroRNA-Gene Association As a Prognostic Biomarker in Cancer Exposes Disease Mechanisms. PLoS Comput Biol. 2013;9(11):e1003351. doi: 10.1371/journal.pcbi.1003351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, Series B. 1995;57:289–300. [Google Scholar]
- Bondy ML, Scheurer ME, Malmer B, Barnholtz-Sloan JS, Davis FG, Il’yasova D, Kruchko C, McCarthy BJ, Rajaraman P, Schwartzbaum JA, et al. Brain tumor epidemiology: consensus from the Brain Tumor Epidemiology Consortium. Cancer. 2008;113(7 Suppl):1953–68. doi: 10.1002/cncr.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T, Lin X, Carroll RJ. Identifying genetic marker sets associated with phenotypes via an efficient adaptive score test. Biostatistics. 2012;13(4):776–90. doi: 10.1093/biostatistics/kxs015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457(7228):426–33. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D. Regression models and life-tables. Journal of the Royal Statistical Society, Series B. 1972;34:187–220. [Google Scholar]
- Fisel P, Kruck S, Winter S, Bedke J, Hennenlotter J, Nies AT, Scharpf M, Fend F, Stenzl A, Schwab M, et al. DNA methylation of the SLC16A3 promoter regulates expression of the human lactate transporter MCT4 in renal cancer with consequences for clinical outcome. Clin Cancer Res. 2013;19(18):5170–81. doi: 10.1158/1078-0432.CCR-13-1180. [DOI] [PubMed] [Google Scholar]
- Goeman JJ, Buhlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics. 2007;23(8):980–7. doi: 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
- Huang YT, VanderWeele TJ, Lin X. Joint analysis of SNP and gene expression data in genetic association studies of complex diseases. Annals of Applied Statistics. 2014;8:352–376. doi: 10.1214/13-AOAS690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Cancer Genome C. Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, Bhan MK, Calvo F, Eerola I, et al. International network of cancer genome projects. Nature. 2010;464(7291):993–8. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen A, Silber J, Harinath G, Huse JT, Schultz N, Sander C. Analysis of microRNA-target interactions across diverse cancer types. Nat Struct Mol Biol. 2013;20(11):1325–32. doi: 10.1038/nsmb.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsy M, Arslan E, Moy F. Current Progress on Understanding MicroRNAs in Glioblastoma Multiforme. Genes Cancer. 2012;3(1):3–15. doi: 10.1177/1947601912448068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange T, Hansen JV. Direct and indirect effects in a survival context. Epidemiology. 2011;22(4):575–81. doi: 10.1097/EDE.0b013e31821c680c. [DOI] [PubMed] [Google Scholar]
- Lee S, Medina D, Tsimelzon A, Mohsin SK, Mao S, Wu Y, Allred DC. Alterations of gene expression in the development of early hyperplastic precursors of breast cancer. Am J Pathol. 2007;171(1):252–62. doi: 10.2353/ajpath.2007.061010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon D. Introduction to statistical mediation analysis. New York: Taylor and Francis; 2008. [Google Scholar]
- Medina R, Zaidi SK, Liu CG, Stein JL, van Wijnen AJ, Croce CM, Stein GS. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 2008;68(8):2773–80. doi: 10.1158/0008-5472.CAN-07-6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara M, Kim SC, Adamik R, Togawa A, Ferrans VJ, Takeda K, Kirby M, Moss J, Vaughan M. Similarities in function and gene structure of cytohesin-4 and cytohesin-1, guanine nucleotide-exchange proteins for ADP-ribosylation factors. J Biol Chem. 2000;275(5):3221–30. doi: 10.1074/jbc.275.5.3221. [DOI] [PubMed] [Google Scholar]
- Pearl J. Direct and indirect effects. San Francisco, CA: Morgan Kaufmann; 2001. pp. 411–420. [Google Scholar]
- Quintavalle C, Garofalo M, Zanca C, Romano G, Iaboni M, del Basso De Caro M, Martinez-Montero JC, Incoronato M, Nuovo G, Croce CM, et al. miR-221/222 overexpession in human glioblastoma increases invasiveness by targeting the protein phosphate PTPmu. Oncogene. 2012;31(7):858–68. doi: 10.1038/onc.2011.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3(2):143–55. doi: 10.1097/00001648-199203000-00013. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Patric IR, Somasundaram K. A ten-microRNA expression signature predicts survival in glioblastoma. PLoS One. 2011;6(3):e17438. doi: 10.1371/journal.pone.0017438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey J. A direct approach to false discovery rate. Journal of the Royal Statistical Society, Series B. 2002;64:479–498. [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Suzuki HI, Mihira H, Watabe T, Sugimoto K, Miyazono K. Widespread inference of weighted microRNA-mediated gene regulation in cancer transcriptome analysis. Nucleic Acids Res. 2013;41(5):e62. doi: 10.1093/nar/gks1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderWeele TJ. Causal mediation analysis with survival data. Epidemiology. 2011;22(4):582–5. doi: 10.1097/EDE.0b013e31821db37e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderWeele TJ, Asomaning K, Tchetgen Tchetgen EJ, Han Y, Spitz MR, Shete S, Wu X, Gaborieau V, Wang Y, McLaughlin J, et al. Genetic variants on 15q25.1, smoking, and lung cancer: an assessment of mediation and interaction. Am J Epidemiol. 2012;175(10):1013–20. doi: 10.1093/aje/kwr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield A, Soukupova J, Montagne A, Ranger J, French R, Muller WJ, Clarkson RW. Bcl3 selectively promotes metastasis of ERBB2-driven mammary tumors. Cancer Res. 2013;73(2):745–55. doi: 10.1158/0008-5472.CAN-12-1321. [DOI] [PubMed] [Google Scholar]
- Wang W, Baladandayuthapani V, Holmes CC, Do KA. Integrative network-based Bayesian analysis of diverse genomics data. BMC Bioinformatics. 2013;14(Suppl 13):S8. doi: 10.1186/1471-2105-14-S13-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei LJ. The accelerated failure time model: a useful alternative to the Cox regression model in survival analysis. Stat Med. 1992;11(14–15):1871–9. doi: 10.1002/sim.4780111409. [DOI] [PubMed] [Google Scholar]
- Xiao C, Rajewsky K. MicroRNA control in the immune system: basic principles. Cell. 2009;136(1):26–36. doi: 10.1016/j.cell.2008.12.027. [DOI] [PubMed] [Google Scholar]
- Zhang CZ, Zhang JX, Zhang AL, Shi ZD, Han L, Jia ZF, Yang WD, Wang GX, Jiang T, You YP, et al. MiR-221 and miR-222 target PUMA to induce cell survival in glioblastoma. Mol Cancer. 2010;9:229. doi: 10.1186/1476-4598-9-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.