

Abstract
Singleton-Merten syndrome (SMS) is an autosomal-dominant multi-system disorder characterized by dental dysplasia, aortic calcification, skeletal abnormalities, glaucoma, psoriasis, and other conditions. Despite an apparent autosomal-dominant pattern of inheritance, the genetic background of SMS and information about its phenotypic heterogeneity remain unknown. Recently, we found a family affected by glaucoma, aortic calcification, and skeletal abnormalities. Unlike subjects with classic SMS, affected individuals showed normal dentition, suggesting atypical SMS. To identify genetic causes of the disease, we performed exome sequencing in this family and identified a variant (c.1118A>C [p.Glu373Ala]) of DDX58, whose protein product is also known as RIG-I. Further analysis of DDX58 in 100 individuals with congenital glaucoma identified another variant (c.803G>T [p.Cys268Phe]) in a family who harbored neither dental anomalies nor aortic calcification but who suffered from glaucoma and skeletal abnormalities. Cys268 and Glu373 residues of DDX58 belong to ATP-binding motifs I and II, respectively, and these residues are predicted to be located closer to the ADP and RNA molecules than other nonpathogenic missense variants by protein structure analysis. Functional assays revealed that DDX58 alterations confer constitutive activation and thus lead to increased interferon (IFN) activity and IFN-stimulated gene expression. In addition, when we transduced primary human trabecular meshwork cells with c.803G>T (p.Cys268Phe) and c.1118A>C (p.Glu373Ala) mutants, cytopathic effects and a significant decrease in cell number were observed. Taken together, our results demonstrate that DDX58 mutations cause atypical SMS manifesting with variable expression of glaucoma, aortic calcification, and skeletal abnormalities without dental anomalies.
Main Text
Singleton-Merten syndrome (SMS [MIM 182250]) was first described in two unrelated females who presented with abnormal dentition, distal-limb osteoporosis, bilateral glaucoma, and marked calcifications of the aortic arch and valve, the latter of which can eventually be fatal.1 Since then, a few SMS-affected families have been described, and joint laxity, muscle weakness, and psoriasis were often reported in individuals with SMS.1–4 One of the main characteristics of classic SMS is the presence of dental anomalies, including delayed exfoliation or eruption of the primary teeth and early loss of permanent teeth.1,2 Despite an apparent autosomal-dominant pattern of inheritance, the genetic background of SMS and information about its phenotypic heterogeneity remain unknown.2
DDX58, also known as retinoic-acid-inducible gene I (RIG-I), is a 925-residue cytoplasmic viral RNA receptor and a member of the RIG-I-like receptor (RLR) family, which also includes IFIH1 and DHX58.5 DDX58 is an essential intracellular sensor for several viruses and elicits antiviral interferon (IFN) responses by recognizing viral double-stranded RNAs (dsRNAs).6 DDX58 comprises a helicase domain, a C-terminal domain, and N-terminal caspase activation recruitment domains (CARDs) involved in activating mitochondrial antiviral signaling protein (MAVS).6 The cooperative binding of ATP and RNA substrates to the helicase domain induces conformational changes and downstream signaling to MAVS via CARDs.7
Recently, we identified a family (family A) harboring aortic calcification, glaucoma, and skeletal abnormalities (Figure 1 and Table 1). The proband (II:6) was found to have severe calcification of the aorta and coronary arteries and mild aortic valve stenosis (Figure 1C). She was diagnosed with bilateral glaucoma at approximately 6 years of age and was treated medically. At 17 years of age, she became blind. In early adulthood, she experienced arthritis of the hands with contracture between phalangeal and metacarpal joints. Skeletal X-ray demonstrated calcific tendinitis and mild calcified ligament of the inter-phalangeal and metacarpo-phalangeal joints. Radiographs of both hands showed mild erosive changes in the terminal tufts of the distal phalanges. She also had dry skin with scabbing. In a random skin biopsy taken from the thigh, papillomatosis and hyperkeratosis were observed. She had no dental anomalies (Figure 1D), and neurologic examination revealed no muscle weakness or hypotonia.
Figure 1.
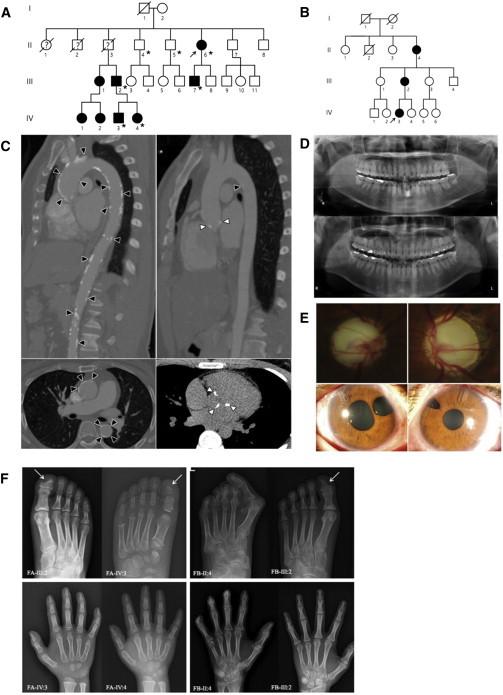
Families Affected by DDX58 Mutations
(A) Pedigree of family A, affected by the c.1118A>C (p.Glu373Ala) DDX58 mutation. An asterisk indicates that exome sequencing was performed.
(B) Pedigree of family B, affected by the c.803G>T (p.Cys268Phe) DDX58 mutation.
(C) Computed tomographic angiography shows aortic calcification (black arrowheads) and calcification of the aortic valves (white arrowheads) in II:6 (left) and III:7 (right) of family A.
(D) Orthopantomograms of II:6 (upper) and III:7 (lower) of family A. These subjects show no dental anomalies except mild dental caries.
(E) Photographs of both eyes of family A individual III:7, who was diagnosed with glaucoma at 3 years of age, show severe optic nerve head cupping (upper left and right). Iris defects due to surgical iridectomy, which was performed with filtering surgeries and additional subsequent bilateral glaucoma implant surgeries, are visible (lower left and right).
(F) Skeletal findings in the affected families. In family A (FA), foot radiographs of III:2 at age 33 years and his son (IV:3) at age 8 years show acro-osteolysis of the distal phalanges and the resulting triangular shape of the great toes (arrows). Hand radiographs of the son (IV:3) and daughter (IV:4, age 5 years) also reveal erosive changes of the terminal tufts and the resulting hypoplastic appearance of all distal phalanges. Carpal bone ossification is delayed in IV:3, who is approximately 3–4 years of age. Expanded medullary cavities of the metacarpals and metatarsals are not evident in any affected member of family A. In family B (FB), the foot radiograph of II:4 at age 61 years shows marked subluxation of the hallux, erosions, and acro-osteolysis of the distal phalanges and the resulting short toes (upper). Note the similar but lesser degree of erosive changes of the distal phalanges, which show a triangular shape of the great toe, in III:2 of family B (upper, arrow). In the hand radiographs of II:4 and III:2 of family B, note the severe acro-osteolysis of the distal phalanges and flexion contractures of II:4 and mild erosive changes of the distal phalanges in III:2. The medullary cavities of the metatarsals and metacarpals are also not widened in this family.
Table 1.
Clinical Findings of the Present Subjects with DDX58-Related Disease Phenotypes
|
Family A |
Family B |
Total n/Na(%) from This Study | Total n/Na(%) from Feigenbaum et al.2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| II:6 | III:7 | III:1 | III:2 | IV:1 | IV:2 | IV:3 | IV:4 | II:4 | III:2 | IV:3 | |||
| Gender | female | male | female | male | female | female | male | female | female | female | female | NA | NA |
| Current age (years) | 56 | 22 | 35 | 33 | 16 | 14 | 8 | 5 | 61 | 38 | 20 | NA | NA |
| Glaucomab | + | + | + | − | + | + | + | + | + | + | + | 10/11 (91) | 5/10 (50) |
| Age at glaucoma diagnosisc (years) | 6 | 3 | 18 | − | 6 | 4 | 4 | 2 | 10 | 3 | 5 | NA | NA |
| Intraocular pressured (mmHg, right/left) | ND | ND | ND | ND | 32/30 | 30/32 | 29/38 | 38/34 | ND | ND | ND | NA | NA |
| Short stature | − | − | − | − | − | − | + | + | − | − | − | 2/11 (18) | 6/9 (67) |
| Aortic and valvular calcification | + | + | + | + | ND | ND | ND | ND | +/−e | − | − | 5/7 (71) | 10/11 (91) |
| Cardiac arrhythmia | − | − | − | − | − | − | − | − | − | − | − | 0/11 (0) | 6/11 (55) |
| Osteopenia | − | − | ND | − | ND | ND | − | − | + | − | − | 1/8 (13) | 9/10 (90) |
| Acro-osteolysis or tuft erosion of distal phalanx | + | + | ND | + | ND | ND | + | + | + | + | + | 8/8 (100) | 6/9 (67) |
| Wide medullary cavities in the phalanges | − | − | − | − | − | − | − | − | − | − | − | 0/11 (0) | 9/10 (90) |
| Subungal calcifications | − | − | − | − | − | − | − | − | − | − | − | 0/11 (0) | 3/8 (38) |
| Tendon rupture | − | − | − | − | − | − | − | − | − | − | − | 0/11 (0) | 6/11 (55) |
| Joint subluxation | − | − | − | − | − | − | − | − | + | − | − | 1/11 (9) | 8/9 (89) |
| Thick neurocranium | − | − | − | − | − | − | − | − | − | − | − | 0/11 (0) | 8/9 (89) |
| Scoliosis | − | − | − | − | − | − | − | − | − | − | − | 0/11 (0) | 3/10 (30) |
| Dental problemsf | − | − | − | − | − | − | − | − | − | − | − | 0/11 (0) | 10/11 (91) |
| Unusual faceg | − | − | − | − | − | − | − | − | − | − | − | 0/11 (0) | 7/7 (100) |
| Weakness or hypotonia | − | − | − | − | − | − | − | − | − | − | − | 0/11 (0) | 8/10 (80) |
| Psoriasiform rash | − | +h | − | + | + | + | + | + | − | − | + | 7/11 (64) | 8/9 (89) |
Abbreviations are as follows: NA, not applicable; ND, no data.
n/N indicates the number of positive individuals out of the total number of examined individuals.
Glaucoma was diagnosed when intraocular pressure was higher than 21 mmHg in at least one eye before treatment in the presence of optic nerve head change.
Age at onset of glaucoma could not be estimated exactly.
The intraocular pressure was measured by either Goldmann tonometry or Perkins applanation tonometry before treatment. Except for four individuals (IV:1, IV:2, IV:3, and IV:4) in family A, other family members with glaucoma were already managed with surgical intervention at the time of visit in our hospital.
This person had small calcifications in the aortic arch and abdominal aorta, but the causes were nonspecific given her age.
Dental problems in individuals II:6 and III:7 from family A were assessed by an orthopantomogram performed by a dentist. Other subjects were confirmed with a physical examination.
Symptoms included a high hair line, a broad forehead, mild ptosis, a smooth philtrum, and a thin upper vermilion.
The skin lesion was confirmed as psoriasiform rash by skin biopsy. Other individuals were evaluated by physical examination.
The son of the proband (III:7) was also diagnosed with bilateral glaucoma at the age of 3 years and underwent three glaucoma surgeries (Figure 1E). He experienced atopic dermatitis in babyhood. Over the following years, the psoriatic skin lesion developed, especially in the elbow and axilla, and was confirmed as psoriasiform hyperplasia in a skin biopsy. Like his mother, he showed healthy permanent dentition (Figure 1D). At 21 years of age, he was found to have calcification of the aorta and coronary arteries as well as of the aortic valve (Figure 1C). After 1 year, the highly calcified valve lesion led to acute severe mitral regurgitation as a result of chordae rupture, and pulmonary edema subsequently ensued. He underwent mitral valvuloplasty with annuloplasty and aortic valve decalcification. A skeletal survey showed hypoplastic and cone-shaped distal phalanges due to terminal tuft erosion. Additional neurologic and skeletal exams revealed no abnormal findings.
The niece of the proband (III:1) presented with glaucoma at 18 years of age. There were small calcifications in the proximal ascending aorta and coronary artery. Her aortic valve showed mild calcification without stenosis. Her two daughters (IV:1 and IV:2) also had glaucoma, although they did not complain of any dental problems. The nephew of the proband (III:2) did not have glaucoma but did have calcification of the aorta, subclavian, and iliac arteries. Radiologic examination of his hands demonstrated erosion of the terminal tufts of all distal phalanges without widening of the medullary spaces. Both feet showed acro-osteolysis of the distal phalanges, resulting in a triangular shape (Figure 1F). His son (IV:3) and daughter (IV:4) developed bilateral glaucoma and underwent glaucoma surgery. In particular, the son (IV:3) showed delayed carpal bone age and short stature (less than the third percentile). The daughter (IV:4) also showed short stature (less than the third percentile), but her bone age was within the normal range. Similar to those of the father, radiographs of the hands and feet showed erosive changes of the terminal tufts (Figure 1F). No one in this family presented with dental problems or dysmorphic facial features.
To identify genetic causes of the disease, we performed exome sequencing with approval from the Samsung Medical Center institutional review board (no. 2013-09-063). After obtaining informed consent, we extracted genomic DNA from peripheral-blood leukocytes in five individuals (II:6, III:2, III:7, IV:3, and IV:4) and two healthy individuals (II:4 and II:5) of family A and subjected it to whole-exome capture and sequencing on the Agilent SureSelect Human All Exon V3 (Agilent Technologies) and the HiSeq2000 (Illumina) sequencing platform, respectively. The raw sequence reads were processed and aligned to the UCSC Genome Browser hg19 human reference sequence with the Burrows-Wheeler Aligner (version 0.6.2). Duplicate reads were removed with Picard, and local alignment optimization was performed with the Genome Analysis Toolkit. ANNOVAR was used for annotation against dbSNP. After filtering steps based on segregation status and variant databases (dbSNP 137 and the NHLBI Exome Sequencing Project Exome Variant Server [EVS]), ten variants co-segregating with the disease remained, and then intronic variants and synonymous exonic variants were excluded (Tables S1 and S2). We identified a missense variant (c.1118A>C [p.Glu373Ala]) in DDX58 (MIM 609631; RefSeq accession number NM_014314.3), which encodes a cytoplasmic helicase that mediates the induction of the IFN response to viral RNA (Figure S1A).6 We subsequently validated this variant by Sanger sequencing (Figure S1B). The c.1118A>C (p.Glu373Ala) variant was considered most likely pathogenic because the affected residue was strictly conserved across taxa from zebrafish to human (Figure 2A) and because the variant was predicted to be deleterious by in silico analysis with SIFT and PolyPhen-2. In addition, this variant was absent in 1,000 control chromosomes from individuals of Korean descent and was not found in 13,006 chromosomes in the NHLBI Exome Sequencing Project EVS (see Web Resources). We did not find any other strong candidate genes in the results of exome sequencing.
Figure 2.
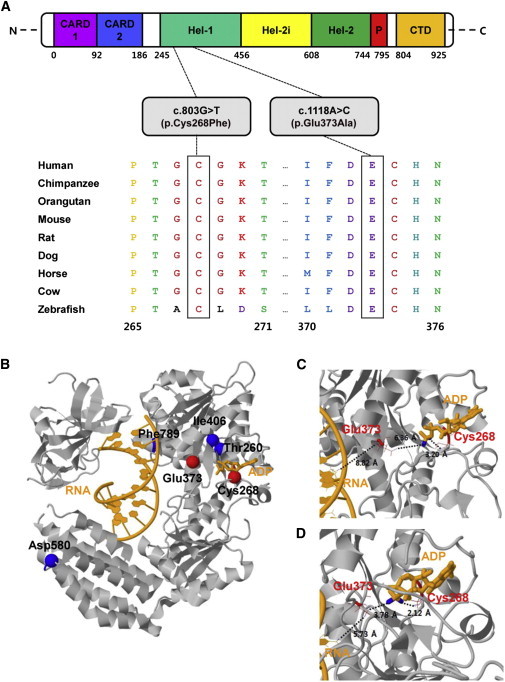
Ortholog Conservation of DDX58 Mutations and Molecular Helicase Structure of DDX58
(A) Schematic representation of DDX58 mutations relative to the affected protein domains. Also, sequence alignment of DDX58 is shown in vertebrate species. The alignment regions corresponding to the missense mutations are shown. Cys at codon 268 and Glu at codon 373 are highly conserved across all species. The Cys and Glu residues are indicated by the closed white boxes. The Ensembl IDs for DDX58 sequences aligned in this study are as follows: human, ENSP00000369197; chimpanzee, ENSPTRT00000038562; orangutan, ENSPPYT00000022324; mouse, ENSMUSP00000115052; rat, ENSRNOT00000008465; dog, ENSCAFT00000002841; horse, ENSECAT00000023725; cow, ENSBTAP00000053514; and zebrafish, ENSDART00000058276. Abbreviations are as follows: CARD, caspase activation recruitment domain; CTD, C-terminal domain; Hel, helicase domain (Hel-1 and Hel-2 are the two conserved core helicase domains, and Hel-2i is an insertion domain conserved in DDX58-like helicase family); and P, pincer of bridge region connecting Hel-2 to the CTD involved in binding dsRNA.
(B) The protein structure is drawn in gray. The RNA and ADP molecules are shown in orange. The locations of the amino acid changes caused by four known SNPs (rs35527044 [p.Thr260Pro], rs951618 [p.Ile406Thr], rs17217280 [p.Asp580Glu], and rs35253851 [p.Phe789Leu]) and two most likely pathogenic variants (p.Cys268Phe and p.Glu373Ala) are shown by blue and red spheres, respectively. The Thr260, Ile406, and Phe789 sites are hidden by the protein. PDB ID 3ZD7 was used for the wild-type (WT) structure of DDX58.8 This structure contains 73 experimentally missing residues. All protein structural analyses were executed with the program CHARMM.9
(C and D) Focused views of the WT X-ray structure (C) and 10-ns (ns) molecular dynamics (MD) final structure (D). The 10-ns MD simulation was performed with an implicit solvation model, i.e., the generalized Born model with a simple switch.10 Two residues (Cys268 and Glu373) affected by mutations are drawn as red wireframe models. Close distances are shown as black dotted lines with distance values. The used atom pairs for the distance measurements are shown in Table S3.
Because glaucoma was one of the core manifestations of the disease in family A, we decided to Sanger sequence all of the coding exons of DDX58 in 100 unrelated individuals with diagnoses of congenital glaucoma. We collected blood samples from each person after obtaining informed consent. Most of the individuals with congenital glaucoma have previously been screened for CYP1B1 (MIM 601771) and MYOC (MIM 601652) mutations.11 Primers were designed to amplify the coding exons of DDX58 (primers are available upon request). Purified PCR amplification products were sequenced with the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems) and an ABI 3730xl Genetic Analyzer (Applied Biosystems). Interestingly, we identified another variant (c.803G>T [p.Cys268Phe]) in a female individual, her mother, and her maternal grandmother (family B; Figures 1B and S1C). The Cys268 residue is highly conserved (Figure 2A), and the p.Cys268Phe variant is absent from 1,000 Korean chromosomes and the EVS.
The detailed clinical features of family B are summarized in Table 1 and Figure 1F. In brief, the maternal grandmother of the proband (II:4) developed glaucoma during childhood and was blind as an adult. She complained of severe arthralgia of the phalangeal joints of the hands and feet. Radiologic examination revealed advanced phalangeal osteoarthropathy and acro-osteolysis of the distal phalanges and the resulting flexion contracture of the hands, subluxation, and hallux valgus deformity of the feet (Figure 1F). She was found to have small calcifications in the aortic arch and abdominal aorta, but the causes were nonspecific given her age. Other examinations did not reveal any abnormal findings. The mother of the proband (III:2) was diagnosed with bilateral glaucoma and went blind at the age of 3 years. She had neither aortic calcifications nor dental problems. Radiologic examination of her hands and feet demonstrated significant osteoarthropathy, including acro-osteolysis (Figure 1F). The proband (IV:3) was diagnosed with bilateral glaucoma at the age of 5 years and underwent two glaucoma surgeries. She had dry skin with scabbing from childhood, although she did not have aortic calcification or dental abnormalities. Radiologic findings showed mild erosive changes of the distal phalanges of her hands. These three individuals in family B did not have muscle weakness or hypotonia and showed normal facial features.
Mapping of the altered residues onto 3D molecular structures is shown for the identified variants (p.Cys268Phe and p.Glu373Ala) and four other known SNPs: rs35527044 (c.778A>C [p.Thr260Pro]), rs951618 (c.1217T>C [p.Ile406Thr]), rs17217280 (c.1740T>A [p.Asp580Glu]), and rs35253851 (c.2367C>A [p.Phe789Leu]) (Figure 2B). We selected these DDX58 SNPs for structural analysis because they did not affect DDX58 activity according to functional analysis in cell culture.12 Because the DDX58 structure includes RNA and ADP as a reactant and a cofactor, respectively, we measured the closest contact distances of any variant residues from these two molecules. Three types of DDX58 structures—the wild-type (WT) and the p.Cys268Phe and p.Glu373Ala altered proteins—were prepared for structural analyses. The Cys268 and Glu373 residues of DDX58 belong to ATP-binding motifs I and II,7 respectively, and these residues are predicted to be located much closer to the ADP and RNA molecules than other nonpathogenic missense variants according to both X-ray and molecular dynamic final structures (Figures 2C and 2D and Table S3). The locations of the altered residues might affect the ATP binding of DDX58, leading to their functional effects on DDX58.
To interpret the effects of DDX58 alterations on RLR-mediated signaling pathways, we tested for activity at the basal level or activity induced by the dsRNA analog polyI:C of IFN regulatory factor (IRF) and NF-κB by using luciferase assays in HEK293FT cells transfected with WT or altered DDX58. Elevated amounts of the altered DDX58 structures were associated with significantly enhanced PRDIII-I and NF-κB reporter gene activity at the basal level, and this activity was further increased by polyI:C stimulation (Figure 3A). In particular, high amounts of the altered DDX58 structures were sufficient to induce IRF3 phosphorylation and IRF3 dimerization at the basal level (Figures 3B and 3C). As a result, DDX58 p.Glu373Ala and p.Cys268Phe led to increased expression of IFNB1 and ISG15 in both basal and stimulated cells (Figure 3D). Altered DDX58 also increased CCL5 expression, but expression of the proinflammatory cytokine TNF was not significantly affected by the alterations. All together, these results suggest that altered DDX58 confers constitutive activation leading to increased IFN activity and IFN-stimulated gene expression.
Figure 3.
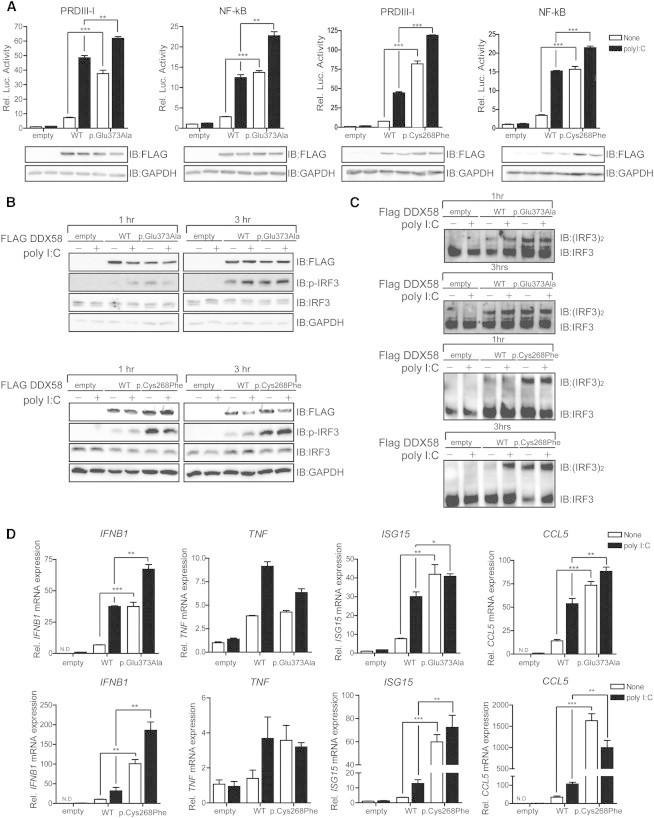
DDX58 Mutations Constitutively Activate RLR-IFN Production Pathways
(A) In HEK293FT cells, FLAG-DDX58 (WT), FLAG-DDX58-p.Glu373Ala, or FLAG-DDX58-p.Cys268Phe was transfected along with reporter construct pLuc-PRDIII-I or pLuc-NF-κB for 24 hr. A dual-luciferase assay was performed after 12 hr of polyI:C (1 μg/ml) stimulation.
(B and C) HEK293FT cells were transfected with the indicated plasmids for 30 hr and then stimulated with polyI:C (1 μg/ml) for the indicated time. After stimulation, the lysates were separated by SDS-PAGE (B) or on native gels (C). Immunoblot assays were performed with the indicated antibodies.
(D) HEK293FT cells were transfected with the indicated plasmids for 24 hr. After stimulation with polyI:C (1 μg/ml) for 12 hr, the expression of IFNB1, TNF, ISG15, and CCL5 was measured by quantitative real-time PCR and normalized to β-actin expression. The primers, which were used for quantitative real-time PCR, were described previously.13 Statistical analysis using t tests was conducted in GraphPad Prism 4 (∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.001).
We then investigated the effect of altered DDX58 on primary human trabecular meshwork (HTM) cells. Previous studies have shown that mutations in MYOC, encoding myocilin, cause glaucoma and that altered myocilins can lead to cytotoxicity in HTM cells.14–16 Because HTM cells are responsible for draining the aqueous humor from the eye, the death of HTM cells might represent an underlying mechanism for elevated intraocular pressure leading to glaucoma.17 To test potential cytotoxicity of altered DDX58 on HTM cells, we transduced HTM cells with empty vector (null) or expression vectors for the WT, two alterations (p.Cys268Phe and p.Glu373Ala), and two other known SNPs by using replication-deficient recombinant adenoviral vectors (Figure 4). The two SNPs, rs11795404 (c.548G>T [p.Ser183Ile]) and rs951618 (c.1217T>C [p.Ile406Thr]), were chosen because their functional activities have been documented.12 In particular, p.Ser183Ile is known to be a loss-of-function variant by CARD inactivation, whereas p.Ile406Thr has no functional effect.12 After 6 days, images of phase-contrast microscopy were obtained from the cells, and the images demonstrated the cytopathic effects of p.Cys268Phe and p.Glu373Ala (Figure 4A). Immunohistochemistry further revealed diffuse green fluorescence throughout the cytoplasm and prominent shrinkage of vimentin (red fluorescence) in HTM cells transduced with either p.Cys268Phe or p.Glu373Ala in comparison to HTM cells transduced with WT or the two known SNPs (Figure 4B). Vimentin is one of five major groups of intermediate filament proteins, and thus vimentin shrinkage highlights the contracted morphology of stressed cells.18 To assess HTM cell viability, we stained transduced HTM cells with trypan blue, and we counted only cells excluding the dye by using a disposable hemocytometer. We observed a significant decrease in the number of cells transduced with p.Cys268Phe and p.Glu373Ala (Figure 4C). These findings suggest that DDX58 mutations, similar to MYOC mutations, have cytotoxic effects on HTM cells.
Figure 4.
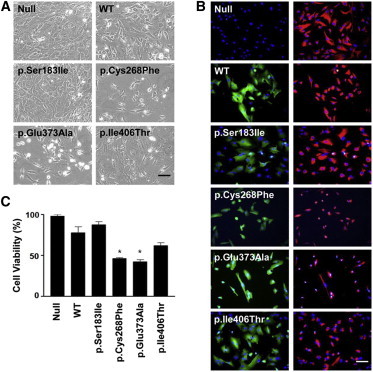
Potential Cytotoxicity of Altered DDX58 in HTM cells
(A) Phase-contrast photographs of HTM cells transduced with empty vector (null) or expression vectors for the WT, two DDX58 alterations (p.Cys268Phe and p.Glu373Ala), and two other known SNPs (rs11795404 [c.548G>T (p.Ser183Ile)] and rs951618 [c.1217T>C (p.Ile406Thr)]) are shown. The scale bar represents 100 μm.
(B) HTM cells were incubated with anti-FLAG antibody diluted 200× or Cy3-conjugated anti-vimentin antibody diluted 500×. The cells were washed and reacted for 2 hr with FITC-conjugated secondary antibody. After the nuclei were counterstained for 20 min with DAPI, the cells were viewed under a fluorescence microscope with the appropriate filter sets and a 200× objective. FITC fluorescence (green) of DDX58 staining merged with DAPI (blue) is shown in the left column, and Cy3 fluorescence (red) of vimentin staining merged with DAPI is shown in the right column. The scale bar represents 100 μm.
(C) Cell viability (%) was calculated as the percentage of cells surviving in comparison to the null. The data were statistically analyzed by one-way ANOVA followed by the Newman-Keuls multiple-comparison test, and p < 0.05 was considered significant. The data are presented as the mean percentage ± the SEM (∗p < 0.01 versus WT).
The type 1 IFN system is essential to human antiviral immunity. However, the excessive production or defective negative regulation of type 1 IFNs might be related to a predisposition to inflammatory disease or autoimmune disease.19,20 In individuals with Aicardi-Goutières syndrome (AGS [MIM 225750]), Gillian et al. recently identified IFIH1 (encoding MDA5) mutations, which lead to the inappropriate stimulation of type 1 IFN.20 AGS is an inflammatory disease affecting the brain and skin and demonstrates consistently higher expression of genes induced by type I IFN, such as IFI27 (MIM 600009), IFI44L (MIM 613975), IFIT1 (MIM 147690), ISG15 (MIM 147571), RSAD2 (MIM 607810), and SIGLEC1 (MIM 600751).20 Funabiki et al. reported that dysregulation of IFIH1 function causes autoimmune disorders without viral infection.19 In that study, mutant mice harboring a single Ifih1 missense mutation (c.2461G>A [p.Gly821Ser]) generated by N-ethyl-N-nitrosourea mutagenesis spontaneously developed lupus-like nephritis and systemic autoimmune symptoms without viral infection. These findings suggest that the IFIH1 mutation activates downstream signaling in the absence of its ligand but is paradoxically defective for ligand- and virus-induced signaling, indicating that the mutation induces a conformational change in IFIH1.
On the basis of the results of our DDX58 cellular assay in HEK293FT cells, we hypothesized that the clinical phenotypes of our individuals possessing DDX58 mutations were associated with a gain of function of DDX58, just as AGS due to IFIH1 mutations is associated with MDA5 gain of function.20 Although systemic-inflammation-related symptoms or signs were not evident in our individuals with DDX58 mutations, glaucoma was a core manifestation in all but one individual, and three (II:6 in family A and II:4 and III:2 in family B) were totally blind. Aortic and vascular calcification was observed only in family A, whereas skeletal abnormalities such as acro-osteolysis were more prominent in family B.
The pathogenesis of the aortic and valvular calcification observed in family A, who harbored DDX58 c.1118A>C (p.Glu373Ala), remains unknown. Once believed to be a passive degenerative disease, cardiovascular calcification is now recognized as an active process.21 The chronic inflammatory state represented by the increased production of proinflammatory cytokines induced by the gain of function of DDX58 most likely leads to mineralization of the aorta and valves. Indeed, inflammation is a key contributor to the pathogenesis of vascular calcification, and some degree of vascular inflammation is observed in most cases of human arterial calcification.22 The late stages of large vessel vasculitis, such as Takayasu’s arteritis or giant cell arteritis, are characterized by heavy calcification of the aorta. In addition, in a diabetic Ldlr−/− murine model of calcific vasculopathy, the local augmentation of inflammatory cytokines, such as IL-6 and TNF, in the aortic wall was shown to activate osteogenic transcriptional factors (e.g., SOX9, RUNX2, MSX2, and OSX).22,23
Of particular note, dental anomalies, which are core manifestations of classic SMS, were absent in all of our individuals with DDX58 mutations. The overall disease phenotype of our individuals was less aggressive and showed a better prognosis than classic SMS. Why DDX58 mutations do not cause systematic inflammation or autoimmune disease is not well understood, but a negative regulatory mechanism might play a role in controlling activated RLR-IFN signaling. Further studies are needed to reveal the exact mechanisms underlying these genotype-phenotype correlations.
In conclusion, we identified DDX58 mutations that are responsible for atypical SMS manifesting with variable phenotypes, including glaucoma, aortic calcification, and skeletal abnormalities but no dental manifestations.
Acknowledgments
This study was supported by the Samsung Medical Center Clinical Research fund (CRO112031) and by a National Research Foundation of Korea grant (no. NRF-2012R1A4A1028200) funded by the Korean Ministry of Science, ICT, and Future Planning. J.L. was supported by the Korea Research Institute of Bioscience and Biotechnology Research Initiative Program.
Contributor Information
Changwon Kee, Email: ckee@skku.edu.
Chang-Seok Ki, Email: changski@skku.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/index.html
GraphPad, http://www.graphpad.com/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
PDB, http://www.pdb.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Singleton E.B., Merten D.F. An unusual syndrome of widened medullary cavities of the metacarpals and phalanges, aortic calcification and abnormal dentition. Pediatr. Radiol. 1973;1:2–7. doi: 10.1007/BF00972817. [DOI] [PubMed] [Google Scholar]
- 2.Feigenbaum A., Müller C., Yale C., Kleinheinz J., Jezewski P., Kehl H.G., MacDougall M., Rutsch F., Hennekam R.C. Singleton-Merten syndrome: an autosomal dominant disorder with variable expression. Am. J. Med. Genet. A. 2013;161A:360–370. doi: 10.1002/ajmg.a.35732. [DOI] [PubMed] [Google Scholar]
- 3.Gay B.B., Jr., Kuhn J.P. A syndrome of widened medullary cavities of bone, aortic calcification, abnormal dentition, and muscular weakness (the Singleton-Merten syndrome) Radiology. 1976;118:389–395. doi: 10.1148/118.2.389. [DOI] [PubMed] [Google Scholar]
- 4.Valverde I., Rosenthal E., Tzifa A., Desai P., Bell A., Pushparajah K., Qureshi S., Beerbaum P., Simpson J. Singleton-merten syndrome and impaired cardiac function. J. Am. Coll. Cardiol. 2010;56:1760. doi: 10.1016/j.jacc.2010.02.078. [DOI] [PubMed] [Google Scholar]
- 5.Liu F., Gu J. Retinoic acid inducible gene-I, more than a virus sensor. Protein Cell. 2011;2:351–357. doi: 10.1007/s13238-011-1045-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 7.Kowalinski E., Lunardi T., McCarthy A.A., Louber J., Brunel J., Grigorov B., Gerlier D., Cusack S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 8.Kohlway A., Luo D., Rawling D.C., Ding S.C., Pyle A.M. Defining the functional determinants for RNA surveillance by RIG-I. EMBO Rep. 2013;14:772–779. doi: 10.1038/embor.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks B.R., Brooks C.L., 3rd, Mackerell A.D., Jr., Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Im W., Lee M.S., Brooks C.L., 3rd Generalized born model with a simple smoothing function. J. Comput. Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 11.Kim H.J., Suh W., Park S.C., Kim C.Y., Park K.H., Kook M.S., Kim Y.Y., Kim C.S., Park C.K., Ki C.S., Kee C. Mutation spectrum of CYP1B1 and MYOC genes in Korean patients with primary congenital glaucoma. Mol. Vis. 2011;17:2093–2101. [PMC free article] [PubMed] [Google Scholar]
- 12.Shigemoto T., Kageyama M., Hirai R., Zheng J., Yoneyama M., Fujita T. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: implications for resistance to type I diabetes. J. Biol. Chem. 2009;284:13348–13354. doi: 10.1074/jbc.M809449200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang S.Y., Sun H.Y., Lee K.H., Oh B.H., Cha Y.J., Kim B.H., Yoo J.Y. 5′-Triphosphate-RNA-independent activation of RIG-I via RNA aptamer with enhanced antiviral activity. Nucleic Acids Res. 2012;40:2724–2733. doi: 10.1093/nar/gkr1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stone E.M., Fingert J.H., Alward W.L., Nguyen T.D., Polansky J.R., Sunden S.L., Nishimura D., Clark A.F., Nystuen A., Nichols B.E. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275:668–670. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- 15.Alward W.L., Fingert J.H., Coote M.A., Johnson A.T., Lerner S.F., Junqua D., Durcan F.J., McCartney P.J., Mackey D.A., Sheffield V.C., Stone E.M. Clinical features associated with mutations in the chromosome 1 open-angle glaucoma gene (GLC1A) N. Engl. J. Med. 1998;338:1022–1027. doi: 10.1056/NEJM199804093381503. [DOI] [PubMed] [Google Scholar]
- 16.Joe M.K., Sohn S., Hur W., Moon Y., Choi Y.R., Kee C. Accumulation of mutant myocilins in ER leads to ER stress and potential cytotoxicity in human trabecular meshwork cells. Biochem. Biophys. Res. Commun. 2003;312:592–600. doi: 10.1016/j.bbrc.2003.10.162. [DOI] [PubMed] [Google Scholar]
- 17.Alvarado J., Murphy C., Juster R. Trabecular meshwork cellularity in primary open-angle glaucoma and nonglaucomatous normals. Ophthalmology. 1984;91:564–579. doi: 10.1016/s0161-6420(84)34248-8. [DOI] [PubMed] [Google Scholar]
- 18.Minin A.A., Moldaver M.V. Intermediate vimentin filaments and their role in intracellular organelle distribution. Biochemistry Mosc. 2008;73:1453–1466. doi: 10.1134/s0006297908130063. [DOI] [PubMed] [Google Scholar]
- 19.Funabiki M., Kato H., Miyachi Y., Toki H., Motegi H., Inoue M., Minowa O., Yoshida A., Deguchi K., Sato H. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity. 2014;40:199–212. doi: 10.1016/j.immuni.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 20.Rice G.I., del Toro Duany Y., Jenkinson E.M., Forte G.M., Anderson B.H., Ariaudo G., Bader-Meunier B., Baildam E.M., Battini R., Beresford M.W. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 2014;46:503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.New S.E., Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ. Res. 2011;108:1381–1391. doi: 10.1161/CIRCRESAHA.110.234146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shao J.S., Cheng S.L., Sadhu J., Towler D.A. Inflammation and the osteogenic regulation of vascular calcification: a review and perspective. Hypertension. 2010;55:579–592. doi: 10.1161/HYPERTENSIONAHA.109.134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Aly Z., Shao J.S., Lai C.F., Huang E., Cai J., Behrmann A., Cheng S.L., Towler D.A. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr-/- mice. Arterioscler. Thromb. Vasc. Biol. 2007;27:2589–2596. doi: 10.1161/ATVBAHA.107.153668. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.