

Abstract
Primary coenzyme Q10 (CoQ10) deficiencies are rare, clinically heterogeneous disorders caused by mutations in several genes encoding proteins involved in CoQ10 biosynthesis. CoQ10 is an essential component of the electron transport chain (ETC), where it shuttles electrons from complex I or II to complex III. By whole-exome sequencing, we identified five individuals carrying biallelic mutations in COQ4. The precise function of human COQ4 is not known, but it seems to play a structural role in stabilizing a multiheteromeric complex that contains most of the CoQ10 biosynthetic enzymes. The clinical phenotypes of the five subjects varied widely, but four had a prenatal or perinatal onset with early fatal outcome. Two unrelated individuals presented with severe hypotonia, bradycardia, respiratory insufficiency, and heart failure; two sisters showed antenatal cerebellar hypoplasia, neonatal respiratory-distress syndrome, and epileptic encephalopathy. The fifth subject had an early-onset but slowly progressive clinical course dominated by neurological deterioration with hardly any involvement of other organs. All available specimens from affected subjects showed reduced amounts of CoQ10 and often displayed a decrease in CoQ10-dependent ETC complex activities. The pathogenic role of all identified mutations was experimentally validated in a recombinant yeast model; oxidative growth, strongly impaired in strains lacking COQ4, was corrected by expression of human wild-type COQ4 cDNA but failed to be corrected by expression of COQ4 cDNAs with any of the mutations identified in affected subjects. COQ4 mutations are responsible for early-onset mitochondrial diseases with heterogeneous clinical presentations and associated with CoQ10 deficiency.
Main Text
Coenzyme Q (CoQ), or ubiquinone, is a lipophilic component of the electron transport chain (ETC), where it shuttles electrons derived from NADH and FADH2 to ETC complex III (cIII) or ubiquinone-cytochrome c reductase. The main electron donors to CoQ are ETC complexes I (cI) and II (cII) but also include other mitochondrial flavoproteins, for instance, electron transfer flavoprotein-ubiquinone oxidoreductase, mitochondrial (ETF-dehydrogenase [ETFDH]), which is the terminal component of fatty acid β-oxidation and branched-chain amino acid oxidation pathways. CoQ can also act as an antioxidant and a membrane stabilizer, is a cofactor of additional mitochondrial enzymes (e.g., uncoupling protein UCP1),1,2 and plays an indispensable role in the de novo pyrimidine biosynthesis as the electron acceptor from dihydroorotate dehydrogenase.3–5
CoQ is a 1,4-benzoquinone with a tail of 10 isoprenyl units in humans (CoQ10) but of variable length in other species (e.g., CoQ6 in yeast). The synthesis of the isoprenoid moieties proceeds via either mevalonate or 2-C-methyl-D-erythritol 4-phosphate pathways, whereas the aromatic precursor of the CoQ benzoquinone ring is p-hydroxybenzoate, derived from tyrosine.6 After the isoprenoid “tail” is bound to the aromatic “head,” the ring undergoes sequential modification. At least ten enzymes participate in CoQ biosynthesis; in yeast, and possibly mammals as well, these enzymes are all localized in mitochondria.
Primary CoQ10 deficiency is the biochemical signature of a group of rare, clinically heterogeneous autosomal-recessive disorders caused by mutations in several genes encoding proteins involved in CoQ10 biosynthesis.7 Mutations in COQ2 (MIM 609825), COQ6 (MIM 614647), ADCK3 (COQ8 [MIM 606980]), ADCK4 (MIM 615573), COQ9 (MIM 612837), PDSS1 (MIM 607429), and PDSS2 (MIM 610564) have been reported in subjects with severe infantile mitochondrial syndromes associated with severe tissue CoQ10 deficiency, whereas the genetic bases underpinning adult-onset CoQ10 deficiency remain mostly undefined.8,9 COQ4 (MIM 612898) codes for a ubiquitously expressed 265-amino-acid protein that is peripherally associated with the mitochondrial inner membrane on the matrix side;10 the precise function of human COQ4 is not known, but the yeast ortholog seems to play a structural role crucial in the stabilization of a multiheteromeric complex including several, if not all, of the CoQ biosynthetic enzymes.11
We report here the identification of pathogenic biallelic COQ4 mutations in a total of five individuals from four families; these subjects were part of a cohort of severe mitochondrial cases where the CoQ10 defect was not anticipated. The family pedigrees are shown in Figure 1A.
Figure 1.
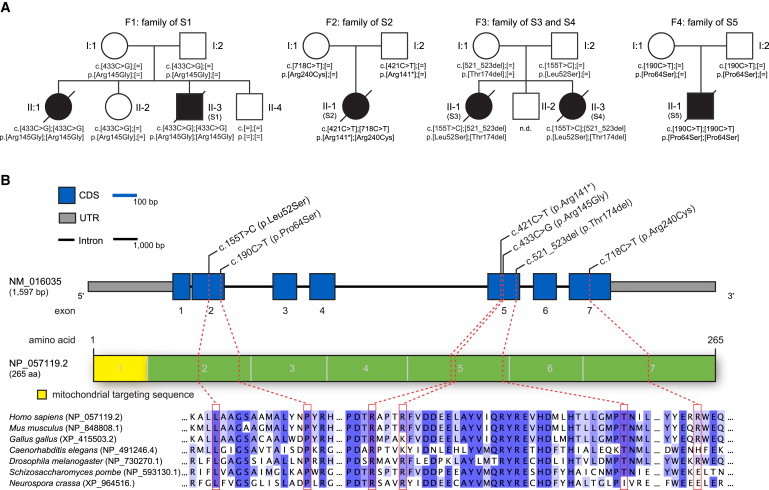
Pedigrees of Investigated Families and COQ4 Structure and Conservation of Identified Mutations
(A) Pedigrees of four families affected by mutations in COQ4. The mutation status of affected and unaffected family members is indicated by closed and open symbols, respectively.
(B) COQ4 structure showing the identified mutations. The structure of the gene product, COQ4, is also shown with known domains and localization and conservation of amino acid residues affected by the mutations. Intronic regions are not drawn to scale.
Subject 1 (S1; II-3, family 1), a boy, was the third of four siblings and was born to healthy, non-consanguineous Italian parents after an uncomplicated pregnancy and elective cesarean delivery. His oldest sister (II-1), who presented with bradycardia and hypotonia, died at birth, and his 16-year-old second sister and his 5-year-old brother are alive and well. At birth, S1 had a weight of 3,410 g, a length of 49.5 cm, and a head circumference of 34.5 cm. Apgar scores were 7 and 10 at 1 and 5 min after birth, respectively. At birth, his condition appeared critical, given that he showed severe hypotonia, areflexia, acrocyanosis, bradycardia, and respiratory insufficiency. Ultrasound examination revealed markedly decreased motility of the left ventricle with an ejection fraction of 20%–25%. No evidence of hepatic or renal impairment was observed. Dobutamine infusion via an umbilical venous catheter was ineffective, and the baby died 4 hr after birth. His blood glucose level was normal, as were renal and hepatic parameters; plasma creatine kinase was moderately elevated (861 U/l; normal value [n.v.] < 400), and blood lactate was extremely high (20.1 mM; n.v. < 2). Analysis of urinary organic acids showed elevated levels of 2-OH glutaric acid, whereas plasma and urinary amino acids were within normal ranges. The autopsy examination revealed left ventricular hypoplasia with septum hypertrophy and a patent ductus arteriosus. No brain examination was performed.
The activities of the ETC complexes in autoptic skeletal-muscle homogenate showed severe defects of both coupled cI+cIII and cII+cIII reactions, normalized to citrate synthase (CS), and a decrease in CS-normalized cI (Table 1). In both liver and cultured fibroblasts, the CS-normalized activities of each of the individual ETC complexes were in the control range. Although the coupled cI+cIII activity cannot be reliably assayed in cultured cells,12 the coupled cII+cIII activity was clearly decreased in S1 fibroblasts (65% of the control mean).
Table 1.
Mitochondrial ETC Activities in Muscle
| Subject | cI/CSa | cI+III/CSa | cII/CSa | cII+III/CSa | cIII/CSa | cIV/CSa | CSb | |
|---|---|---|---|---|---|---|---|---|
| Muscle biopsy | S1c | 36 | 24 | N | 34 | N | N | 64 |
| S2c | 6 | ND | 42 | 43 | 10 | 30 | 57 | |
| S3 | N | N | N | 55 | N | 50 | 54 | |
| S4 | 145 | N | N | N | 222 | 189 | 109 | |
| S5 | <5 | ND | N | 30 | 50 | N | 65 |
Abbreviations are as follows: N, value in the control range; ND, not done; cI, complex I; cII, complex II; cIII, complex III; cIV, complex IV; cI+III, coupled activity of complexes I and III; and cII+III, coupled activity of complexes II and III. The analyses were performed in different laboratories, and the reference values are diverse (they usually range between 60% and 140% of the mean control value). The values of ETC complex activities out of the control range (specific to each enzymatic activity and to each laboratory) are reported.
Mean control value (%) of CS-normalized ETC complex activities.
Percentage of mean control value.
Sample from autopsy.
S2 (II-1, family 2) was born at the 34th week of gestation and was the female first child of non-consanguineous Japanese parents. Her birth weight was 1,120 g (−2.2 SDs). Apgar scores were 7 and 8 at 1 and 5 minutes after birth, respectively. There was no family history of neurological or cardiac disease. The pregnancy was complicated by severe intrauterine growth delay and ultrasound-documented hypertrophic cardiomyopathy. On S2’s first day of life, she became apnoeic and was intubated as a result of respiratory failure. She initially displayed moderate lactic acidosis, but soon after her admission to Neonatal Medical Center, her lactic acidosis rapidly worsened (blood lactate = 11.2–18.8 mM; n.v. < 2); her hypertrophic cardiomyopathy evolved into severe heart failure, leading to death at the age of 1 day.
The metabolic profile (urinary and plasmatic amino acids, organic acids, and acylcarnitines) showed no significant findings. A liver autoptic specimen showed a severe deficiency of cI (cI/CS ratio = 2.9%); autoptic skeletal-muscle homogenate also showed a cI deficiency together with less pronounced reductions of other ETC complexes (Table 1).
Sisters S3 (II-1, family 3) and S4 (II-3, family 3) are the first and third, respectively, of three siblings and were born to healthy, non-consanguineous Austrian parents. Their brother (II-2) is a healthy, unaffected boy. S3 and S4 were born prematurely at gestational ages of 32 weeks (birth weight = 1,550 g) and 34 weeks (birth weight = 2,170 g), respectively.
Performed at the 20th week of gestation, prenatal organ screening of S3 revealed a suspected malformation of the cerebellum. A postnatal cranial ultrasound showed cerebellar hypoplasia. After birth, she showed distal arthrogryposis, but no other dysmorphic features. At birth, she suffered from respiratory-distress syndrome, and a few hours later, a severe myoclonic epileptic encephalopathy ensued; blood lactic acid at 36 hr of age was 6.4 mM and rose to 14 mM prior to her death by multiorgan failure on the third day of life. Echocardiography showed a normal heart. Metabolic investigations (amino acids in plasma, acylcarnitine profile, and standard newborn screening) were essentially normal. Analysis of organic acids in urine showed excretion of glycerol and 2-OH-glutarate. In frozen postmortem muscle (obtained within 30 min after death), ETC enzyme activities were slightly decreased (Table 1). An autopsy of the brain revealed severe olivopontocerebellar and thalamic hypoplasia and scattered cavitations in the white matter; the visceral organs appeared normal for the gestational age.
Six years later, prenatal organ screening of the sister, S4, showed cerebellar hypoplasia, suggesting the same disease as in S3. Similar to her sister, S4 suffered from neonatal respiratory distress. No dysmorphic features were present. Echocardiography was normal. A cranial ultrasound confirmed cerebellar hypoplasia. Six hours after birth, epileptic encephalopathy ensued; blood lactic acid was 3.5 mM at 2 hr of age and rose to 9 mM at death on the second day of life. Metabolic investigations showed normal newborn-screening results and a normal acylcarnitine profile. Amino acids in plasma were grossly elevated but showed no specific pattern. Analysis of urinary organic acids showed excretion of a “mitochondrial dysfunctional pattern” with malate, fumarate, and 2-OH-glutarate, as well as vitamin B6 metabolites and N-acetyl-tyrosine. Analysis of frozen postmortem muscle showed elevated levels of ETC activities (Table 1). In both girls, blood glucose concentration and renal and hepatic parameters were in the normal range.
S5 (II-1, family 4) is an 18-year-old young man and is the only offspring of healthy Italian parents who deny consanguinity and originate from a medium-size town in southern Italy. Pregnancy was normal, and delivery was via cesarean section because of a podalic presentation. He was born at term, and his weight at birth was 4,100 g. Weight and motor development were reportedly normal in his first year of life, but he started to show slowly progressive motor deterioration after the age of 10 months, when he manifested unsteadiness in maintaining acquired sitting position. He achieved the ability to walk with a spastic ataxic gait at 3 years of age but lost ambulation by 6 years of age and has been wheelchair bound since then. At 12 years of age, he started manifesting epileptic seizures in the form of prolonged right-side hemiclonic seizures. MRI showed bilateral increased signal intensity in fluid-attenuated-inversion-recovery and T2-weighted sequences in both occipital-cortical and juxtacortical areas (Figures S1A–S1D). Around the same period, he started to have swallowing difficulties. He was admitted for extensive investigation. Thorough blood tests excluded liver and kidney involvement and did not show lactic acidosis. A specific pattern of organic aciduria was excluded. Electrophysiological examination showed a sensory motor polyneuropathy with slowed conduction velocities. During a 5-year follow-up, he showed a slowly progressive downhill course with recurrent treatment-resistant seizures, worsened swallowing impairment, progressive scoliosis, and cognitive deterioration. A muscle biopsy was performed when he was 12 years old. Spectrophotometric assays of the ETC complexes in muscle homogenate showed virtually undetectable cI/CS ratios and reduced cII+cIII/CS and cIII/CS ratios. The other ETC complex activities were within control limits (Table 1). Since the age of 15 years, he has used a percutaneous-endoscopic-gastronomy tube and has developed severe scoliosis with a Cobb angle of 75°. Control MRI performed when he was 17 years old showed cerebellar atrophy, widening of ventricular brain spaces, and scars from cortical necrotic lesions in both occipital areas (Figures S1E–S1H).
In agreement with the Declaration of Helsinki, informed consent for genetic and biochemical studies was signed by the parents of all subjects, and the ethics committee of the Technische Universität München approved the study.
We performed whole-exome sequencing (WES) to investigate the molecular bases of the mitochondrial disease presentations of S1, S4, and S5, as described previously.13 Coding DNA sequences were enriched with a SureSelect Human All Exon 50 Mb V4 or V5 Kit (Agilent) and subsequently sequenced on a HiSeq2500 system (Illumina). Read alignment to the human reference assembly (UCSC Genome Browser hg19) was done with the Burrows-Wheeler Aligner (version 0.7.5), and single-nucleotide variants and small insertions and deletions were identified with SAMtools (version 0.1.19). On the basis of the rare disease phenotype and a pattern concordant with autosomal-recessive inheritance, we sought genes carrying rare (minor allele frequency [MAF] < 0.1% in 4,500 control exomes) variants predicted to be compound heterozygous or homozygous. We then prioritized variants in genes coding for proteins with known or predicted mitochondrial localization.14 This filtering strategy led to the identification of recessive variants in COQ4, coding for a mitochondrial protein involved in CoQ10 biosynthesis,10 in all three subjects. In S2, we used the SeqCap EZ Library (version 1.0; Roche NimbleGen). Details on the bioinformatics pipeline and variant filtering have been reported recently.15 Sequencing statistics are provided in Table S1.
We identified COQ4 mutations (RefSeq accession number NM_016035.3) in four individuals (Figure 1). In S1, we identified a homozygous missense variant, c.433C>G (p.Arg145Gly). Both parents and a healthy sister are heterozygous carriers, and a healthy brother has two reference alleles. No material was available from the deceased sister. S2 was found to be compound heterozygous for a nonsense variant on the paternal allele and a missense variant on the maternal allele: c.[421C>T];[718C>T], p.[Arg141∗];[Arg240Cys]. S4 was found to be compound heterozygous for a missense mutation and an exon 5 in-frame deletion: c.[155T>C];[521_523delCCA], p.[Leu52Ser];[Thr174del]. Both variants were also confirmed in the DNA of S3, whereas the parents are heterozygous for only one variant each (the father carries the missense mutation, and the mother carries the deletion). In S5, we identified a homozygous mutation, c.190C>T (p.Pro64Ser). Both parents are heterozygous for this mutation.
None of the identified variants are present in our exome database, which contains 4,500 samples, or in public SNP databases, including dbSNP, the NHLBI Exome Sequencing Project Exome Variant Server, and the Exome Aggregation Consortium (ExAC) Browser. The only exception is the c.718C>T variant (rs143441644), which is reported to have an extremely low frequency (MAF = 0.00023; 28/12,0330 alleles) in the ExAC Browser. Moreover, all missense changes are predicted to be deleterious by several bioinformatics tools (Table S2).
Because of the identified genetic defects, we tested CoQ10 levels in available specimens from the subjects. In a muscle biopsy from S1, we detected a clear reduction of CoQ10 (32.9 nmol CoQ10/g protein; n.v. = 101–183; 1.16 nmol CoQ10/CS; n.v. = 1.75–3.46). In fibroblasts from S1, the levels of CoQ10 were also lower than CoQ10 levels in neonatal control fibroblasts (54% of control mean). In frozen muscle from S3, CoQ10 was reduced (13.5 nmol CoQ10/g protein; n.v. = 160–1,200; 0.3 nmol CoQ10/CS; n.v. = 2.7–7); in muscle from S4, CoQ10 was profoundly reduced (25.7 nmol CoQ10/g protein; n.v. = 160–1,200; 0.1 nmol CoQ10/CS; n.v. = 2.7–7), whereas in S5 muscle, the amount of CoQ10 was slightly decreased (88.9 μg CoQ10/g protein; n.v. = 101–183; 1.70 μg nmol CoQ10/CS; n.v. = 1.75–3.46) (Figure 2A). No residual sample from the muscle biopsy of S2 was available. Together, these findings are consistent with a deleterious role of the mutations identified in COQ4.
Figure 2.
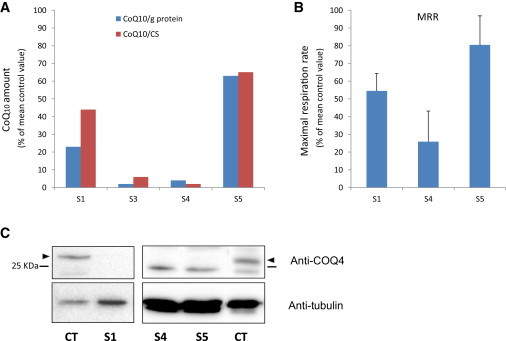
Biochemical Studies in COQ4 Mutant Muscle and Fibroblasts
(A) CoQ10 in muscle from affected subjects S1 and S3–S5 is reported as a percentage of the mean of control values (the analyses were performed in different laboratories, and the reference values are diverse; see text). Data are reported after normalization to protein content or CS activity.
(B) Maximal respiration rate (MRR) measured in fibroblasts from subjects S1, S4, and S5; MRR values are expressed as percentages of MRR values obtained in control fibroblasts. The graphs represent the mean values from two independent experiments, each with six to eight replicates. Error bars represent the SD.
(C) Immunoblot analysis of COQ4 in fibroblasts from subjects S1, S4, and S5 and control individuals (Ct). Arrowheads indicate the band corresponding to COQ4. An antibody against tubulin was used as a loading control.
By Seahorse micro-oxygraphy,16 we detected that maximal respiratory rates were lower in S1, S4, and S5 fibroblasts than in control cells (Figure 2B). Moreover, a drastic decrease in the amount of COQ4 was detected by immunoblot analysis in S1, S4, and S5 fibroblasts (Figure 2C), confirming that the identified COQ4 nucleotide variants are deleterious.
The Saccharomyces cerevisiae ortholog of human COQ4 is yCOQ4; yCOQ4-null strains have been reported to be effectively complemented by human COQ4.10 In order to functionally test the effect of all the mutations found in our cohort, we transformed a COQ4-null strain (Δcoq4) by inserting the following hCOQ4 variants into the multicopy pYES2.1 vector: pYES:hCOQ4WT (human wild-type [WT]), pYES2.1 (empty vector), pYES:yCOQ4WT (positive control), pYES:hcoq4p.Arg145Gly (mutation c.433C>G), pYES:hcoq4p.Arg141∗ (c.421C>T), pYES:hcoq4p.Arg240Cys (c.718C>T), pYES:hcoq4p.Leu52Ser (c.155T>C), pYES:hcoq4p.Thr174del (c.521_523delCCA), and pYES:hcoq4p.Pro64Ser (c.190C>T). In addition, to replicate the compound-heterozygous condition found in probands of families 2 and 3, we transformed the Δcoq4 strain via a pYES construct harboring both the c.155T>C and the c.521_523delCCA mutations (pYES:hcoq4p.Leu52Ser/p.Thr174del) and a pYES construct expressing the c.421C>T and c.718C>T mutations (pYES:hcoq4p.Arg141∗/p.Arg240Cys). A WT strain transformed with the pYES2.1 empty vector was also included as an additional control. In order to reveal a possible respiratory defect, we compared the growth of our transformant strains cultured in either glucose (a fermentable carbon source) or glycerol (a non-fermentable carbon source) after inducing gene expression with galactose for 4 hr. Notably, whereas the growth of the pYES2.1:hCOQ4WT transformant strain was comparable to that of the pYES2.1:yCOQ4WT transformant strain, the strains transformed with the hCOQ4 mutant vectors grew as slowly as that transformed with pYES2.1 (Figure 3A). This result clearly indicates that each mutation reported in our probands leads to a virtually complete loss of function of the corresponding protein, COQ4. Next, we found that the CoQ6 content in one Δcoq4 mutant strain, hcoq4p.Arg145Gly, was markedly decreased, whereas Δcoq4 strains transformed with either pYES2.1:yCOQ4 or pYES2.1:hCOQ4 had CoQ6 levels similar to those in the WT strain (Figure 3B). This result indicates that mutant hcoq4p.Arg145Gly impairs CoQ biosynthesis.
Figure 3.

Yeast Studies
(A) Glycerol (YPG) growth of transformed ΔCOQ4 yeast with the different mutated versions of human COQ4 (pYES2.1, empty vector; hCOQ4, pYES:hCOQ4WT; yCOQ4, pYES:yCOQ4WT; c.433C>G, pYES:hcoq4p.Arg145Gly; c.421C>T, pYES:hcoq4p.Arg141∗; c.718C>T, pYES:hcoq4p.Arg240Cys; c.155T>C, pYES:hcoq4p.Leu52Ser; c.521_523delCCA, pYES:hcoq4p.Thr174del; c.190C>T, pYES:hcoq4p.Pro64Ser; c.155T>C and c.521_523delCCA, pYES:hcoq4p.Leu52Ser/p.Thr174del; and c.421C>T and c.718C>T, pYES:hcoq4p.Arg141∗/p.Arg240Cys). WT indicates the wild-type yeast transformed with the YES2.1 empty vector. Cells were grown in selective medium for 16 hr, induced in galactose for 4 hr, and inoculated in YPG at 0.1 U of optical density (OD) at 600 nm. Growth at 30°C was monitored over 5 days by measurement of OD cultures at 600 nm.
(B) Yeast mitochondrial CoQ6 levels. Purified mitochondria lipid extraction and high-performance-liquid-chromatography quantification of CoQ6 was performed in the ΔCOQ4 strain transformed with the empty vector (pYES2.1), WT yeast (yCOQ4), or human (hCOQ4) or hcoq4p.Arg145Gly (c.433C>G) COQ4 genes. A WT strain transformed with the empty vector was included as a positive control. Error bars represent the SD.
Primary CoQ10 deficiency, caused by genetic defects in CoQ10 biosynthesis, is a clinically heterogeneous condition associated with a spectrum of different phenotypes, including encephalomyopathic forms with seizures and/or ataxia,17–19 multisystem infantile forms with encephalomyopathy and renal failure,20 nephrotic syndrome with sensorineural deafness,21,22 adult Leigh syndrome,23 and isolated myopathic forms.24 Mutations in seven genes encoding proteins involved in CoQ10 biosynthesis have been reported in single families or in a few singleton cases;25 the genetic defect has not been determined in most of the cases of CoQ10 deficiency, and only a few data are available regarding specific genotype-phenotype correlations. Secondary CoQ10 deficiency has been reported in association with glutaric aciduria type IIC (MIM 231680), caused by mutations in ETFDH (MIM 231675; encoding electron-transfer dehydrogenase); ataxia-oculomotor apraxia syndrome (MIM 208920), caused by mutations in APTX (MIM 606350; encoding aprataxin); a cardio-facio-cutaneous syndrome caused by a mutation in BRAF (MIM 115150; encoding serine/threonine-protein kinase B-Raf)26; and glucose transporter GLUT1 deficiency.27 Interestingly, although the mechanisms linking these heterogeneous genetic conditions to a decrease in CoQ10 remain obscure, most of these individuals benefitted from CoQ10 supplementation.28,29
We found six COQ4 mutations in five affected subjects from four unrelated families. All these individuals carried homozygous or compound-heterozygous mutations, clearly indicating that the resulting disease is an autosomal-recessive trait. Two alleles carried nonsense mutations, which are both transmitted by descent in combination with missense COQ4 mutations to different individuals (S2 and sisters S3 and S4) and are predicted to lead to a truncated and aberrant COQ4. Given that the heterozygous parents carrying the nonsense mutations are alive and well, it is unlikely that COQ4 haploinsufficiency is pathogenic, even though a previous study reported on a boy carrying a de novo heterozygous deletion, including COQ4, in chromosomal region 9q34.30 Because the biosynthetic pathway of CoQ is conserved throughout evolution from human to Saccharomyces cerevisiae, we modeled in yeast the mutations found in our subjects. Using this system, we demonstrated that each mutation, or the allelic combinations found in S2 and siblings S3 and S4, was associated with a severe defect of oxidative growth. In parallel, we also showed that COQ4 was strongly reduced in mutant fibroblast cell lines from S1, S4, and S5. In the skeletal muscle of S1 and S3–S5, the CoQ10 content was reduced as well. Taken together, these results demonstrate the pathogenic role of the COQ4 mutations found in our cohort.
In keeping with the essential role of COQ4, four of our five subjects had a prenatal or perinatal onset with a fatal outcome in the first days of life. S1 and S2 presented with severe hypotonia, bradycardia, and respiratory insufficiency at birth; in S2, hypertrophic cardiomyopathy had been evident since fetal development. A markedly different, albeit equally severe, clinical presentation dominated by premature delivery, antenatal cerebellar hypoplasia, neonatal respiratory-distress syndrome, and epileptic encephalopathy characterized sisters S3 and S4. Rapidly progressive, severe lactic acidosis was a common feature in all four affected newborn subjects and is likely to have determined their fatal outcome. Involvement of the heart has been very rarely documented in CoQ10-deficient subjects, often as part of multisystem phenotypes, where cardiomyopathy develops later than brain, muscle, or kidney impairment.20 For instance, a homozygous nonsense mutation in COQ9 was described in a baby who presented with neonatal lactic acidosis and later developed hypertrophic cardiomyopathy as part of a multisystem disease including intractable seizures, global developmental delay, and renal tubular dysfunction.9 In spite of his early onset, the clinical course of S5 was slowly progressive and dominated by neurological deterioration with hardly any involvement of other organs, including the heart and kidneys.
Although the link between specific genetic defects and phenotypes is often unclear in mitochondrial disorders, organs with the highest energy requirements, such as the heart, kidneys, and brain, have the highest CoQ10 concentrations31 and are the most frequently affected by CoQ10 deficiency. The level of expression of COQ genes in different cells seems to correlate poorly with the primarily affected tissue or organ; for instance, COQ2, mutations of which typically cause renal impairment, has expression levels that are relatively higher in skeletal muscle and the heart than in other organs,32 whereas COQ4, mutated in our subjects with cardiac or brain failure, is ubiquitously expressed and has relatively higher levels in the liver, lungs, and pancreas.
Because cardiomyocytes have a remarkably high energy requirement, and cardiomyopathy is quite common in individuals with various inherited mitochondrial disorders, the cardiac involvement in subjects with mutations in COQ genes can be overlooked. Indeed, the crucial role of CoQ10 in cardiomyocyte function has been recognized for a very long time; for instance, myocardial biopsies from individuals with congestive heart failure33 or cardiomyopathy34,35 show low CoQ10 levels, which correlate with the severity of heart damage.36 Moreover, statins, cholesterol-lowering drugs that inhibit HMG-CoA reductase (the key enzyme common to the biosynthesis of both cholesterol and CoQ10) can cause CoQ10 deficiency, ultimately leading to cardiomyopathy;37 interestingly, this harmful side effect can be overcome by oral CoQ10 supplementation.38 Moreover, long-term CoQ10 treatment of individuals with chronic heart failure is safe, improves symptoms, and reduces major adverse cardiovascular events.39 These observations all converge on a strict association between CoQ10 deficiency and cardiomyopathy.
Notably, S3–S5 showed no sign of heart involvement, whereas the clinical phenotype was dominated by encephalopathy with seizures and a more progressive, but mainly neurological, syndrome is the clinical hallmark of S5, indicating the heterogeneity of the clinical presentations associated with COQ4 defects. The variable specificity of organ failure (e.g., heart versus brain) in the neonatal cases of our cohort could be due to the fulminant course of the disease, which prevented the deployment of multisystem involvement. In support of this view, although cardiomyopathy dominated the clinical picture, the presence of severe hypotonia and hyporeflexia suggests concomitant involvement of the nervous system in S1 and S2 as well. Clinical heterogeneity was accompanied by an equally striking variability of the biochemical findings, which ranged from multiple (S1 and S5) to isolated (S2 and S3) ETC defects in muscle and fibroblasts to hardly any detectable defect at all (S4). This biochemical diversity could be due to differences in individual adaptive responses to reduced CoQ10 availability or could reflect the striking tissue specificity observed in the clinical presentations, but at the moment, a mechanistic explanation for these observations is lacking. Poor correlation with the clinical and biochemical phenotypes has also been reported for other genes related to CoQ10 biosynthesis. For instance, mutations in COQ2, the first mutated gene identified in affected individuals with primary CoQ10 deficiency, have been associated with a wide range of clinical presentations, often including nephrotic syndrome but also including fatal neonatal multisystemic disorder, Leigh syndrome, myoclonic epilepsy, hypertrophic cardiomyopathy, deafness, and adult-onset multisystem atrophy.25,40 In any case, the identification of COQ4 mutations in subjects with such a wide spectrum of clinical and biochemical abnormalities is a further indication of the advantage of unbiased screening such as WES for the identification of genes newly associated with mitochondrial disorders.
Unfortunately, the fulminant fatal outcome in S1–S4 was so rapid that it prevented both the diagnosis of CoQ10 deficiency and the start of CoQ10 supplementation. Prompt diagnosis is a main challenge for syndromes of primary CoQ10 deficiency but is very important given that co-factor deficiencies are virtually the only group of mitochondrial disorders for which beneficial pharmacological treatment is currently available. Treatment of the long-surviving subject, S5, has now started and will hopefully provide some useful indication of its efficacy in the near future.
Acknowledgments
We would like to thank the families for their collaboration. We thank Roberto Bellavia, Yoshihito Kishita, Yoshimi Tokuzawa, and Ana Sánchez-Cuesta for their technical support; Consolato Sergi for his help; and K. Muroya and M. Adachi for referral of subject materials. This work was supported by Fondazione Telethon (GGP11011), the Italian Ministry of Health (GR2010–2316392 [D.G.]; “Ricerca corrente” [E.B.]), Fondazione CARIPLO (2011/0526), the Mariani Foundation, the Italian Association of Mitochondrial Disease Patients and Families (Mitocon), the European Research Council Advanced Grant FP7-322424, the German Ministry of Education and Research through the E-Rare project GENOMIT (01GM1207 [T.M.; H.P.], FWF I 920-B13 [J.A.M.], J41J11000420001 [D.G.]), the German Network for mitochondrial disorders (mitoNET; 01GM1113C [T.M.; H.P.]), the German Center for Heart Research (Z76010017300; Z56010015300 [T.M.]) by the German Research Foundation within the Munich Cluster for Systems Neurology (EXC-1010-SyNergy), the UK Medical Research Council, the Spanish Instituto de Salud Carlos III (FIS-PI11-00078), the Research Program of Innovative Cell Biology by Innovative Technology (Cell Innovation) from the Japanese Ministry of Education, Culture, Sports, Science, and Technology [Y.O.], Grants-in-Aid for the Research on Intractable Diseases (Mitochondrial Disease) from the Ministry of Health, Labour, and Welfare of Japan [A.O.; K.M.], and the Kawano Masanori Memorial Public Interest Incorporated Foundation for Promotion of Pediatrics [K.M.]. We also acknowledge the Cell Lines and DNA Bank of Paediatric Movement Disorders and Mitochondrial Diseases and the Bank of Muscle Tissue, Peripheral Nerve, DNA, and Cell Culture of the Telethon Network of Genetic Biobanks (grant GTB12001J) and the EurobiobanK Network.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Contributor Information
Holger Prokisch, Email: prokisch@helmholtz-muenchen.de.
Daniele Ghezzi, Email: dghezzi@istituto-besta.it.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Exome Aggregation Consortium (ExAC) Browser, http://exac.broadinstitute.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Turunen M., Olsson J., Dallner G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta. 2004;1660:171–199. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Echtay K.S., Winkler E., Klingenberg M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature. 2000;408:609–613. doi: 10.1038/35046114. [DOI] [PubMed] [Google Scholar]
- 3.Miller R.W., Curry J.R. Mammalian dihydroorotate—ubiquinone reducatse complex. II. Correlation with cytochrome oxidase, mode of linkage with the cytochrome chain, and general properties. Can. J. Biochem. 1969;47:725–734. doi: 10.1139/o69-110. [DOI] [PubMed] [Google Scholar]
- 4.Schmelzer C., Lorenz G., Rimbach G., Döring F. Influence of Coenzyme Q_10 on release of pro-inflammatory chemokines in the human monocytic cell line THP-1. Biofactors. 2007;31:211–217. doi: 10.1002/biof.5520310308. [DOI] [PubMed] [Google Scholar]
- 5.Bentinger M., Tekle M., Dallner G. Coenzyme Q—biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010;396:74–79. doi: 10.1016/j.bbrc.2010.02.147. [DOI] [PubMed] [Google Scholar]
- 6.Kawamukai M. Biosynthesis and bioproduction of coenzyme Q10 by yeasts and other organisms. Biotechnol. Appl. Biochem. 2009;53:217–226. doi: 10.1042/BA20090035. [DOI] [PubMed] [Google Scholar]
- 7.Quinzii C.M., Emmanuele V., Hirano M. Clinical presentations of coenzyme q10 deficiency syndrome. Mol Syndromol. 2014;5:141–146. doi: 10.1159/000360490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeviani M., Carelli V. Mitochondrial disorders. Curr. Opin. Neurol. 2007;20:564–571. doi: 10.1097/WCO.0b013e3282ef58cd. [DOI] [PubMed] [Google Scholar]
- 9.Duncan A.J., Bitner-Glindzicz M., Meunier B., Costello H., Hargreaves I.P., López L.C., Hirano M., Quinzii C.M., Sadowski M.I., Hardy J. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am. J. Hum. Genet. 2009;84:558–566. doi: 10.1016/j.ajhg.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casarin A., Jimenez-Ortega J.C., Trevisson E., Pertegato V., Doimo M., Ferrero-Gomez M.L., Abbadi S., Artuch R., Quinzii C., Hirano M. Functional characterization of human COQ4, a gene required for Coenzyme Q10 biosynthesis. Biochem. Biophys. Res. Commun. 2008;372:35–39. doi: 10.1016/j.bbrc.2008.04.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marbois B., Gin P., Gulmezian M., Clarke C.F. The yeast Coq4 polypeptide organizes a mitochondrial protein complex essential for coenzyme Q biosynthesis. Biochim. Biophys. Acta. 2009;1791:69–75. doi: 10.1016/j.bbalip.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spinazzi M., Casarin A., Pertegato V., Salviati L., Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012;7:1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 13.Haack T.B., Haberberger B., Frisch E.M., Wieland T., Iuso A., Gorza M., Strecker V., Graf E., Mayr J.A., Herberg U. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J. Med. Genet. 2012;49:277–283. doi: 10.1136/jmedgenet-2012-100846. [DOI] [PubMed] [Google Scholar]
- 14.Elstner M., Andreoli C., Klopstock T., Meitinger T., Prokisch H. The mitochondrial proteome database: MitoP2. Methods Enzymol. 2009;457:3–20. doi: 10.1016/S0076-6879(09)05001-0. [DOI] [PubMed] [Google Scholar]
- 15.Ohtake A., Murayama K., Mori M., Harashima H., Yamazaki T., Tamaru S., Yamashita Y., Kishita Y., Nakachi Y., Kohda M. Diagnosis and molecular basis of mitochondrial respiratory chain disorders: exome sequencing for disease gene identification. Biochim. Biophys. Acta. 2014;1840:1355–1359. doi: 10.1016/j.bbagen.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 16.Invernizzi F., D’Amato I., Jensen P.B., Ravaglia S., Zeviani M., Tiranti V. Microscale oxygraphy reveals OXPHOS impairment in MRC mutant cells. Mitochondrion. 2012;12:328–335. doi: 10.1016/j.mito.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogasahara S., Engel A.G., Frens D., Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalomyopathy. Proc. Natl. Acad. Sci. USA. 1989;86:2379–2382. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamperti C., Naini A., Hirano M., De Vivo D.C., Bertini E., Servidei S., Valeriani M., Lynch D., Banwell B., Berg M. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology. 2003;60:1206–1208. doi: 10.1212/01.wnl.0000055089.39373.fc. [DOI] [PubMed] [Google Scholar]
- 19.Mignot C., Apartis E., Durr A., Marques Lourenço C., Charles P., Devos D., Moreau C., de Lonlay P., Drouot N., Burglen L. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J. Rare Dis. 2013;8:173. doi: 10.1186/1750-1172-8-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rötig A., Appelkvist E.-L., Geromel V., Chretien D., Kadhom N., Edery P., Lebideau M., Dallner G., Munnich A., Ernster L., Rustin P. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356:391–395. doi: 10.1016/S0140-6736(00)02531-9. [DOI] [PubMed] [Google Scholar]
- 21.Heeringa S.F., Chernin G., Chaki M., Zhou W., Sloan A.J., Ji Z., Xie L.X., Salviati L., Hurd T.W., Vega-Warner V. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Invest. 2011;121:2013–2024. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashraf S., Gee H.Y., Woerner S., Xie L.X., Vega-Warner V., Lovric S., Fang H., Song X., Cattran D.C., Avila-Casado C. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Invest. 2013;123:5179–5189. doi: 10.1172/JCI69000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Maldergem L., Trijbels F., DiMauro S., Sindelar P.J., Musumeci O., Janssen A., Delberghe X., Martin J.J., Gillerot Y. Coenzyme Q-responsive Leigh’s encephalopathy in two sisters. Ann. Neurol. 2002;52:750–754. doi: 10.1002/ana.10371. [DOI] [PubMed] [Google Scholar]
- 24.Lalani S.R., Vladutiu G.D., Plunkett K., Lotze T.E., Adesina A.M., Scaglia F. Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch. Neurol. 2005;62:317–320. doi: 10.1001/archneur.62.2.317. [DOI] [PubMed] [Google Scholar]
- 25.Doimo M., Desbats M.A., Cerqua C., Cassina M., Trevisson E., Salviati L. Genetics of coenzyme q10 deficiency. Mol Syndromol. 2014;5:156–162. doi: 10.1159/000362826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aeby A., Sznajer Y., Cavé H., Rebuffat E., Van Coster R., Rigal O., Van Bogaert P. Cardiofaciocutaneous (CFC) syndrome associated with muscular coenzyme Q10 deficiency. J. Inherit. Metab. Dis. 2007;30:827. doi: 10.1007/s10545-007-0612-0. [DOI] [PubMed] [Google Scholar]
- 27.Yubero D., O Callaghan M., Montero R., Ormazabal A., Armstrong J., Espinos C., Rodríguez M.A., Jou C., Castejon E., Aracil M.A. Association between coenzyme Q 10 and glucose transporter (GLUT1) deficiency. BMC Pediatr. 2014;14:284. doi: 10.1186/s12887-014-0284-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Musumeci O., Naini A., Slonim A.E., Skavin N., Hadjigeorgiou G.L., Krawiecki N., Weissman B.M., Tsao C.Y., Mendell J.R., Shanske S. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology. 2001;56:849–855. doi: 10.1212/wnl.56.7.849. [DOI] [PubMed] [Google Scholar]
- 29.DiMauro S., Schon E.A., Carelli V., Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013;9:429–444. doi: 10.1038/nrneurol.2013.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salviati L., Trevisson E., Rodriguez Hernandez M.A., Casarin A., Pertegato V., Doimo M., Cassina M., Agosto C., Desbats M.A., Sartori G. Haploinsufficiency of COQ4 causes coenzyme Q10 deficiency. J. Med. Genet. 2012;49:187–191. doi: 10.1136/jmedgenet-2011-100394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aberg F., Appelkvist E.L., Dallner G., Ernster L. Distribution and redox state of ubiquinones in rat and human tissues. Arch. Biochem. Biophys. 1992;295:230–234. doi: 10.1016/0003-9861(92)90511-t. [DOI] [PubMed] [Google Scholar]
- 32.Forsgren M., Attersand A., Lake S., Grünler J., Swiezewska E., Dallner G., Climent I. Isolation and functional expression of human COQ2, a gene encoding a polyprenyl transferase involved in the synthesis of CoQ. Biochem. J. 2004;382:519–526. doi: 10.1042/BJ20040261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanaki Y., Sugiyama S., Ozawa T., Ohno M. Ratio of low-density lipoprotein cholesterol to ubiquinone as a coronary risk factor. N. Engl. J. Med. 1991;325:814–815. doi: 10.1056/nejm199109123251116. [DOI] [PubMed] [Google Scholar]
- 34.Langsjoen P.H., Langsjoen P.H., Folkers K. Long-term efficacy and safety of coenzyme Q10 therapy for idiopathic dilated cardiomyopathy. Am. J. Cardiol. 1990;65:521–523. doi: 10.1016/0002-9149(90)90824-k. [DOI] [PubMed] [Google Scholar]
- 35.Manzoli U., Rossi E., Littarru G.P., Frustaci A., Lippa S., Oradei A., Aureli V. Coenzyme Q10 in dilated cardiomyopathy. Int. J. Tissue React. 1990;12:173–178. [PubMed] [Google Scholar]
- 36.Mortensen S.A., Vadhanavikit S., Folkers K. Deficiency of coenzyme Q10 in myocardial failure. Drugs Exp. Clin. Res. 1984;10:497–502. [PubMed] [Google Scholar]
- 37.Silver M.A., Langsjoen P.H., Szabo S., Patil H., Zelinger A. Statin cardiomyopathy? A potential role for Co-Enzyme Q10 therapy for statin-induced changes in diastolic LV performance: description of a clinical protocol. Biofactors. 2003;18:125–127. doi: 10.1002/biof.5520180214. [DOI] [PubMed] [Google Scholar]
- 38.Ghirlanda G., Oradei A., Manto A., Lippa S., Uccioli L., Caputo S., Greco A.V., Littarru G.P. Evidence of plasma CoQ10-lowering effect by HMG-CoA reductase inhibitors: a double-blind, placebo-controlled study. J. Clin. Pharmacol. 1993;33:226–229. doi: 10.1002/j.1552-4604.1993.tb03948.x. [DOI] [PubMed] [Google Scholar]
- 39.Mortensen S.A., Rosenfeldt F., Kumar A., Dolliner P., Filipiak K.J., Pella D., Alehagen U., Steurer G., Littarru G.P., Q-SYMBIO Study Investigators The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC Heart Fail. 2014;2:641–649. doi: 10.1016/j.jchf.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 40.Scalais E., Chafai R., Van Coster R., Bindl L., Nuttin C., Panagiotaraki C., Seneca S., Lissens W., Ribes A., Geers C. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2) Eur. J. Paediatr. Neurol. 2013;17:625–630. doi: 10.1016/j.ejpn.2013.05.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.