

Abstract
The association between a geographical region and an mtDNA haplogroup(s) has provided the basis for using mtDNA haplogroups to infer an individual’s place of origin and genetic ancestry. Although it is well known that ancestry inferences using mtDNA haplogroups and those using genome-wide markers are frequently discrepant, little empirical information exists on the magnitude and scope of such discrepancies between multiple mtDNA haplogroups and worldwide populations. We compared genetic-ancestry inferences made by mtDNA-haplogroup membership to those made by autosomal SNPs in ∼940 samples of the Human Genome Diversity Panel and recently admixed populations from the 1000 Genomes Project. Continental-ancestry proportions often varied widely among individuals sharing the same mtDNA haplogroup. For only half of mtDNA haplogroups did the highest average continental-ancestry proportion match the highest continental-ancestry proportion of a majority of individuals with that haplogroup. Prediction of an individual’s mtDNA haplogroup from his or her continental-ancestry proportions was often incorrect. Collectively, these results indicate that for most individuals in the worldwide populations sampled, mtDNA-haplogroup membership provides limited information about either continental ancestry or continental region of origin.
Introduction
The high level of polymorphism, lack of recombination, and high copy number of mtDNA have made it a useful tool for studying human demographic history.1 Early studies classified branches of the human mtDNA phylogenetic tree into groups of closely related haplotypes,1,2 defined by lineage-specific polymorphisms in continental-scale populations such as Native Americans,3,4 sub-Saharan Africans,5 and Europeans.6,7 Populations with recent shared ancestry and/or living in geographical proximity displayed similar haplotypes that were grouped by relatedness into haplogroups.1,2 These standardized haplotypes and haplogroups8 facilitated detailed studies of population origins, genetic structure, gene flow,9,10 and detection of sex-biased demography.11–13
For a little over a decade, commercial genetic-testing laboratories have leveraged the information captured by the analysis of mtDNA haplogroups with widespread public interest in genealogical research and human origins to provide direct-to-consumer (DTC) ancestry tests. Specifically, the association between a geographical region and an mtDNA haplogroup(s) provided the basis for using mtDNA haplogroups to infer an individual’s place of origin and genetic ancestry. However, such lineage-based analyses overlook the contribution of the vast majority of an individual’s ancestors to his or her genome.14 Moreover, DTC ancestry tests have proven controversial because they use proprietary methods that lack transparency, present conflicts between cultural and scientific conceptions of ancestry, and lack federal regulation.14–22
The major alternative to lineage-based ancestry tests is model-based ancestry inference using either genome-wide, multi-locus, SNP genotype data23–25 or ancestry-informative markers (AIMs), which are autosomal SNPs with differing allele frequencies between populations.26 Both genome-wide SNPs and AIMs can be used for estimating an individual’s proportion of ancestry from inferred populations that are assumed to correspond to unique ancestral populations. Several recent population-specific studies have recently assessed the relationship between ancestry inferences using mtDNA haplogroups versus autosomal SNPs and have found frequent discrepancies, particularly in recently admixed populations.27–30 However, little empirical information exists on the magnitude and scope of such discrepancies across multiple mtDNA haplogroups and worldwide populations.
In 2008 and again in 2012, the American Society of Human Genetics acknowledged the popularity of commercial ancestry testing and provided a series of recommendations for academic scientists and for companies that perform DTC ancestry testing.18,21 These recommendations expressed concern that commercial testing provides little information about how the accuracy of lineage-based ancestry estimation compares to that of multi-locus ancestry estimation from autosomal markers.18 In particular, the extent to which ancestry information is, in general, captured by mtDNA haplogroups is unknown. To begin to address some of these concerns, we quantified and compared the variation in continental-ancestry proportions among individuals with the same mtDNA haplogroup in 938 individuals from 52 worldwide populations and 327 individuals from recently admixed populations in the 1000 Genomes Project (1KGP) dataset.
Material and Methods
HGDP Dataset
We downloaded Illumina 650Y SNP array genotype data for the HGDP-CEPH Human Genome Diversity Cell Line Panel,31 which consists of 1,043 individuals from 52 worldwide populations (Figure 1A), as previously reported.33 After removing previously identified relatives and duplicate samples,34 as well as samples with low-quality SNP genotype data, we were left with 938 samples in our HGDP dataset. Next, we obtained hypervariable region 1 (HVR1) sequence data for 891 of these 938 samples from the NCBI (PopSet accession number 189174470). For the 47 individuals without publicly available sequence data, we Sanger sequenced HVR1 by using DNA obtained from the CEPH.
Figure 1.
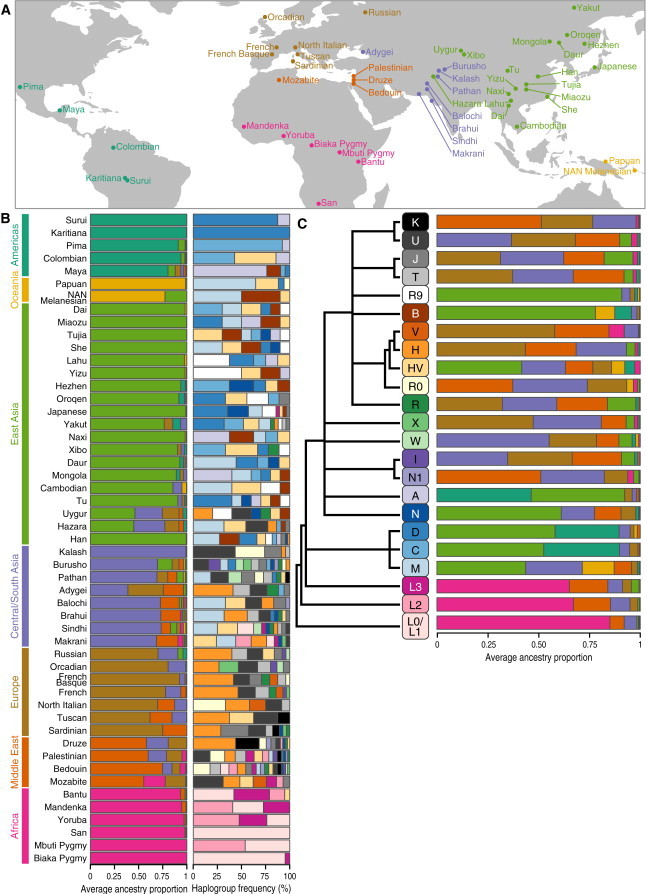
Geographic Location, mtDNA-Haplogroup Frequencies, and Average Ancestry Proportions in the HGDP Populations
(A) World map showing sample locations (points) for each of the populations included in the HGDP (labels).
(B) Left column: barplots of continental-ancestry proportions averaged within each HGDP population. Barplots are colored by continental region (labeled by colored bars on the left) and sorted by continental ancestry. Right column: barplots of haplogroup frequencies within each population. Barplots are colored by mtDNA haplogroup (labeled by the haplogroup tree in C).
(C) Barplots of continental-ancestry proportions averaged within each mtDNA haplogroup within the HGDP dataset. Barplots are colored by continental region (labeled by colored bars on the left of A). The unscaled phylogeny on the left shows the relationships between the mtDNA haplogroups.32
1KGP Dataset
We downloaded 1KGP35 variant call format (.vcf) files for phase 1 low-coverage whole-genome sequence data (release 20101123). We selected five populations from world regions with high levels of recent admixture: ASW (Americans of African ancestry in southwest USA), CLM (Colombians from Medellin, Colombia), GBR (British in England and Scotland), MXL (Mexican ancestry from Los Angeles, CA, USA), and PUR (Puerto Ricans from Puerto Rico). Our total sample consisted of data from 327 people. Using Tabix36 (v.0.2.6) and VCFtools37 (v.0.1.10), we removed indels, extracted variant sites with rs numbers matching SNPs from the HGDP 650Y SNP data, and converted the data to PLINK’s map/ped file format. In PLINK38 (v.1.07), we merged the 1KGP data with the HGDP data and removed 77 SNPs with unresolvable strand mismatches and 141 SNPs that could not be converted from hg18 to hg19 coordinates (UCSC Genome Browser). Our final dataset consisted of 646,356 SNPs. We also downloaded .vcf files for all mitochondrial variants in each 1KGP population.
mtDNA-Haplogroup Typing
We obtained DNA for 965 of the HGDP individuals from the CEPH for use in mtDNA-haplogroup typing. To begin, we first reviewed the literature for SNPs that uniquely identify each of the 23 major mtDNA haplogroups (diagnostic SNPs). Initially, we selected 24 candidate SNPs from Mitomap39 and the Genographic Project32 (one haplogroup required two diagnostic SNPs). We checked each of these candidate diagnostic SNPs in the PhyloTree8 comprehensive mtDNA phylogeny to confirm that the SNP was diagnostic. If a candidate diagnostic SNP was not supported by information from PhyloTree, we selected an additional diagnostic SNP on the basis of the PhyloTree phylogeny. Using a total of 28 diagnostic SNPs, we classified samples into the 23 mtDNA haplogroups (Figure S1).
Next, we used the Genographic Project’s nearest-neighbor haplogroup prediction tool to assign a predicted haplogroup to each sample on the basis of its HVR1 sequence. The prediction tool uses a sample’s haplotype affinity to HVR1 sequences in the Genographic Project’s extensive reference database to predict the sample’s haplogroup.32 (The Genographic Project Haplogroup Prediction Tool appears to no longer be available on the website we accessed.) Next, we experimentally confirmed these haplogroup predictions by genotyping each HGDP sample (either by Sanger sequencing or restriction digest) for the diagnostic SNP(s) of its predicted haplogroup (see Table S1 for reaction conditions for all 28 diagnostic SNPs). Finally, if our initial prediction was incorrect, we genotyped the sample by using our 28 diagnostic SNPs; we began with the SNP diagnostic of ancestral haplogroup L0/L1 and traversed the mtDNA phylogeny from trunk to tips.
For samples from the 1KGP, we used mitochondrial variant calls from phase 1 low-coverage genome data. We extracted all of the variants corresponding to our 28 diagnostic SNPs and used these diagnostic-SNP variant calls to assign a mitochondrial haplogroup to samples from all 327 people.
Continental-Ancestry Estimation from Autosomal SNPs
We used ADMIXTURE24 (v.1.22) to estimate ancestry proportions from each of seven continental regions in each sample from the HGDP Illumina 650Y genotype data. Because ADMIXTURE does not account for linkage disequilibrium (LD), we pruned the genotype markers according to observed correlation coefficients in the data by using a threshold of R2 ≥ 0.1 and a 50-SNP window advancing by ten SNPs in PLINK. We used this dataset in ADMIXTURE with k = 7 according to previously established population-structure parameters in the HGDP.33 The seven inferred populations correspond to continental regions: Africa, the Americas, Central and South Asia, East Asia, Europe, the Middle East, and Oceania. Continental-ancestry proportions for each sample in each HGDP population are shown in Figure S2, and estimated individual ancestry proportions are reported in Table S2.
To estimate continental ancestry in samples from the 1KGP dataset, we first selected HGDP pseudo-ancestors.25 For each of the seven continental groups, we selected the 20 HGDP individuals with the highest fraction of ancestry from their respective continents, resulting in 140 pseudo-ancestors. By including these proxies for ancestral populations, we ensured that the seven continental-ancestry components identified in the 1KGP data would match those identified in the HGDP dataset. Combining SNP data from the pseudo-ancestors and the 1KGP populations, we used PLINK to prune the SNPs according to the same pruning settings described above and estimated ancestry from this pruned dataset. We estimated ancestry proportions for the pseudo-ancestors twice (once with the HGDP data and once with the 1KGP data). Each sample’s two sets of estimated ancestry proportions were highly correlated (Pearson’s R2 > 0.99, p < 0.0001; Figure S3). Continental-ancestry proportions for each sample in each 1KGP population are shown in Figure S4, and estimated individual ancestry proportions are reported in Table S3.
Analysis of Continental-Ancestry Estimates within Each mtDNA Haplogroup
We first examined the average continental-ancestry proportions within each mtDNA haplogroup (Table S4). This produced an h × 7 matrix of means, where h is the number of haplogroups and 7 is the number of continental regions. To determine whether each average continental-ancestry component was significantly higher within a haplogroup than we would expect by chance, we performed a permutation test. For each replicate of the permutation test, we shuffled the haplogroup labels for the sample and recalculated the h × 7 matrix of means. Then we used 999 replicates, plus the original data, to calculate p values for each of the means in the original matrix.
To examine inter-individual variation in the composition of each individual’s continental-ancestry proportions within mtDNA haplogroups, we calculated SDs for each of the continental regions within each haplogroup. To measure this variability in more detail, we calculated the mean pairwise Euclidean distance (d) within each mtDNA haplogroup. Specifically, we considered each individual Pi = (p1, p2, … p7) to be a point in seven-dimensional (7D) space (defined by his or her ancestry proportions). Then we calculated the 7D Euclidean distance between each pair of individuals (Pi, Qj) within the haplogroup. Finally, we averaged these distances for all unique pairs within haplogroups according to the equation below:
To determine how well an mtDNA haplogroup could be linked to a particular continental region, we estimated a consistency score, the proportion of individuals within an mtDNA haplogroup whose highest continental-ancestry proportion was the same as the mtDNA haplogroup’s highest average continental ancestry (Table S5). For example, the highest average continental ancestry within mtDNA haplogroup L2 in the HGDP dataset was Africa. The consistency score of mtDNA haplogroup L2 in the HGDP dataset was 0.67, given that the highest continental-ancestry proportion was Africa in 67% of people with this mtDNA haplogroup.
Multinomial Logistic Regression Model
To explore whether there was a significant predictive relationship between mtDNA haplogroups and continental-ancestry composition, we used a training set of HGDP and 1KGP samples combined to fit a multinomial logistic regression (logit) model. First, we excluded all haplogroups with fewer than ten total samples or fewer than six samples in either dataset. The remaining ten mtDNA haplogroups included L0/L1, L2, L3, C, D, A, H, B, T, and U. The training set consisted of a randomly selected third of the samples from each dataset. The remaining two-thirds of samples were reserved for the test set.
We first used the R package nnet40 to fit a preliminary logit model in which the mtDNA haplogroup was the dependent variable and continental-ancestry proportions were the independent variables. To prevent collinearity, we excluded one continental region—the least common ancestry (Oceania) within our dataset—as a variable. Each of the six continental-ancestry variables improved the fit of the model to the data (likelihood-ratio test [LRT], p < 0.001). We observed possible nonlinear patterns in the data, so we tested logarithmic and exponential relationships for each of the continental-ancestry variables and included any nonlinear relationships that improved the model fit by a LRT (p < 0.001). With six independent variables, 57 possible interaction terms could be included in the model. We used LRTs to determine which of these interaction terms contributed significantly to a better fit of the model and identified 39 significant interaction terms to include; the inclusion of these interaction terms significantly improved the fit of the model (LRT, p < 0.001).
Our final model produced a set of nine logit equations describing the relative odds of belonging to each mtDNA haplogroup instead of the reference mtDNA haplogroup, L0/L1. We used these relative odds to determine each sample’s classification probability for each of our ten mtDNA haplogroups. Additionally, we used the logit equations to calculate the fitted classification probabilities of each sample in our test set for each mtDNA haplogroup (Table S9). For each individual, the mtDNA haplogroup with the highest classification probability was the mtDNA haplogroup predicted by the model. We repeated the model-fitting procedure with three different randomly selected training subsets from our data. Although particular details did change depending on the training set used, the general performance of the model was consistent for multiple training sets.
Results
Composition of mtDNA Haplogroups Varies among Populations
We first explored the relationship between mtDNA haplogroups and continental ancestry within populations of the HGDP. For each of the samples in the HGDP dataset, we classified the mtDNA haplogroup and estimated the proportion of autosomal ancestry from each of the seven continental regions (sub-Saharan Africa, the Middle East, Europe, Central and South Asia, East Asia, Oceania, and the Americas). We then averaged continental-ancestry proportions among samples in each population (Figure 1B, left column). Additionally, we tabulated the frequency of each mtDNA haplogroup in each population (Figure 1B, right column).
Each mtDNA haplogroup was found in several populations, most populations (50/52) contained more than one mtDNA haplogroup, and 46/52 populations contained three or more mtDNA haplogroups (the median was six mtDNA haplogroups per population; Figure 1B). Populations from sub-Saharan Africa (e.g., Biaka Pygmy, Bantu, and Yoruba) consisted almost exclusively of samples assigned to mtDNA haplogroups L0/L1, L2, or L3. Haplogroup H appeared at relatively high frequencies in populations from Europe and the Middle East. Haplogroups M, C, D, and A were most frequent in populations from East Asia and the Americas. Haplogroup M was frequent in populations of mostly East Asian, Central and South Asian, or Oceanian ancestry. Overall, the relationship between a population’s continental origin and its haplogroup composition was markedly heterogeneous.
In each of the five 1KGP populations (ASW, CLM, GBR, MXL, and PUR), we identified eight different mtDNA haplogroups, significantly higher than the median value of six mtDNA haplogroups per population found in HGDP populations (one-tailed t test, p < 0.001). The high frequency of haplogroup H in GBR and of haplogroup L3 in ASW was consistent with the coincidence of these haplogroups with European ancestry and African ancestry, respectively, in the HGDP populations. Haplogroup A was the most common haplogroup in the PUR, MXL, and CLM populations, even though it was not observed in any European HGDP populations, and in fact was most frequent in the Maya. These observations suggest that the relationship between mtDNA haplogroups and geographic origin breaks down in recently admixed populations.
Estimates of Continental Ancestry within mtDNA Haplogroups
To assess variation in continental ancestry among individuals with the same mtDNA haplogroup, we averaged the individual ancestry proportions among samples within each mtDNA haplogroup in populations from the HGDP (Figure 1C) and the 1KGP (Figure 3B). The maximum proportion of ancestry from any single continental region for each haplogroup was, on average, higher in the 1KGP (one-tailed t test, p < 0.01). This might be attributed to higher diversity in continental-ancestry proportions in the HGDP populations than in the 1KGP populations and to the larger contribution of continental ancestry from Europe in 1KGP populations.
Figure 3.
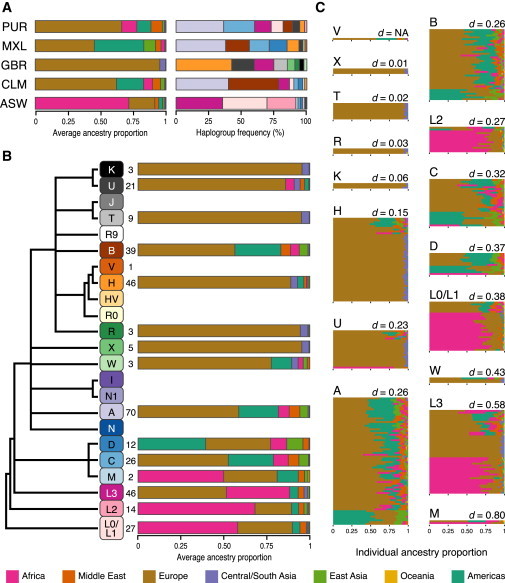
mtDNA-Haplogroup Frequencies, Population-Averaged and Haplogroup-Averaged Continental-Ancestry Proportions, and Individual Continental-Ancestry Proportions in the 1KGP Samples
(A) Left column: barplots of continental-ancestry proportions averaged within each 1KGP population. Barplots are colored by continental region (labeled by colored bars in Figure 1A) and sorted by continental ancestry. Right column: barplots of mtDNA-haplogroup frequencies within each population. Barplots are colored by haplogroup (labeled by the haplogroup boxes in B).
(B) Barplots of continental-ancestry proportions averaged within each mtDNA haplogroup within the 1KGP. Barplots are colored by continental region (see key at bottom). The unscaled phylogeny on the left is the same as in Figure 1C. Numbers to the right of haplogroup labels are the sample size for each haplogroup. Some haplogroups were not observed in the 1KGP samples.
(C) Individual continental-ancestry proportions within each haplogroup. Each horizontal line is a barplot for a single 1KGP sample and indicates individual continental-ancestry proportions. Continental regions are colored according to the key at bottom. Individual barplots are grouped into mtDNA haplogroups as indicated on the top left. Each haplogroup is labeled with the mean pairwise Euclidean distance (see Material and Methods), and haplogroups are sorted by increasing mean pairwise Euclidean distance from top to bottom and left to right. NA indicates not available.
Next, we performed a permutation test to determine whether, in any mtDNA haplogroup, one or more of the seven continental-ancestry proportions were higher than expected by chance. In each haplogroup, one or two continental-ancestry components were significantly higher than expected (Figure S5). These results suggest that some haplogroups are associated with a higher average ancestry proportion from a specific continental region than expected by chance. This affirms the ad hoc visual relationships suggested by the co-occurrence of high average continental ancestry and high haplogroup frequency within the HGDP populations (Figure 1B).
Except for ASW, the 1KGP populations had higher average proportions of ancestry from Europe than from other geographical regions, presumably because of recent admixture, and this decreased our ability to detect associations between continental ancestry regions and mtDNA haplogroups by using a permutation test (Figure S5). Accordingly, many of the associations we observed within the HGDP populations were also present in the 1KGP dataset, but fewer of these associations were significant.
Heterogeneity of Individual Continental-Ancestry Proportions within mtDNA Haplogroups
Even if certain mtDNA haplogroups are significantly associated with a higher proportion of ancestry from a specific geographical region, the extent to which estimates of individual continental-ancestry proportions can be accurately inferred is unclear. To address this issue, we first examined the inter-individual variation in continental-ancestry composition within mtDNA haplogroups. We found that individual continental-ancestry proportions, as measured by the SD of ancestry proportions within each mtDNA haplogroup, in the HGDP dataset varied considerably among individuals (Table S6). For example, the SD of East Asian ancestry was >0.40 in mtDNA haplogroups M, C, D, N, A, and HV, which limited the conclusions we could make regarding East Asian ancestry in individuals with these haplogroups. SDs of continental-ancestry proportions within each mtDNA haplogroup were generally lower in the 1KGP populations, perhaps because the geographical origin of individuals in these populations is less diverse than that in the HGDP.
Next, we calculated the mean pairwise Euclidean distance between continental-ancestry proportions among individuals within each mtDNA haplogroup (Figure 2; Figure 3C). This distance is a quantitative measure of the inter-individual variability in continental-ancestry proportions within a haplogroup. The mean pairwise Euclidean distance was relatively low (i.e., <0.5) in a few mtDNA haplogroups (e.g., R9 in HGDP and T and H in the 1KGP), suggesting a stronger association between individual continental-ancestry proportions and an mtDNA haplogroup. However, the mean pairwise Euclidean distance of most mtDNA haplogroups (17/23) was high (e.g., J, HV, and M in HGDP and L3 in 1KGP), indicating that these mtDNA haplogroups are less informative for inferring individual genetic ancestry.
Figure 2.
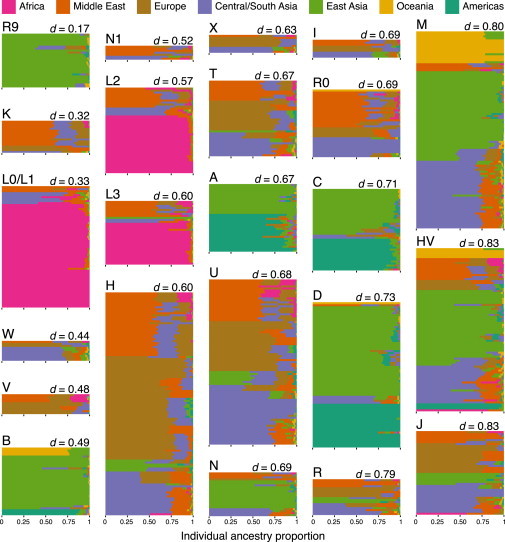
Individual Continental-Ancestry Proportions within Each Haplogroup in the HGDP
Each horizontal line is a barplot for a single HGDP sample and indicates individual continental-ancestry proportions. Continental regions are colored according to the key at the top. Individual barplots are grouped into the 23 mtDNA haplogroups as indicated on the top left. Each haplogroup is labeled with the mean pairwise Euclidean distance (see Material and Methods), and haplogroups are sorted by increasing mean pairwise Euclidean distance from top to bottom and left to right.
Overall, the mean pairwise Euclidean distance within each mtDNA haplogroup was much lower in the 1KGP populations (Figure S6) than in the HGDP. Additionally, mean pairwise Euclidean distances were not necessarily similar for the same mtDNA haplogroup in 1KGP and HGDP (Figure 2; Figure 3C). For example, haplogroup H had a much lower mean pairwise Euclidean distance in 1KGP populations (d = 0.15) than in HGDP populations (d = 0.60). This indicates that inferences drawn from the HGDP dataset are not necessarily portable to the 1KGP populations and vice versa.
To quantitatively assess how informative each mtDNA haplogroup in the HGDP populations was for predicting individual continental-ancestry proportions, we calculated the consistency score within each mtDNA haplogroup in the HGDP (Table S7). The consistency score measures the frequency with which an individual’s highest continental-ancestry proportion matches the highest average continental-ancestry proportion in that individual’s mtDNA haplogroup. In HGDP, consistency ranged from 0.28 to 0.93 with a mean of 0.56. However, only 14/23 mtDNA haplogroups had a consistency score > 50%, meaning that in slightly more than half of mtDNA haplogroups in the HGDP, individuals’ highest continental-ancestry proportions matched their corresponding mtDNA haplogroup’s highest average continental-ancestry proportion (Figure S7). HGDP mtDNA haplogroups with high consistency scores had low mean pairwise Euclidean distances (Pearson’s product-moment correlation, R2 = −0.86, p < 0.001). For example, haplogroup R9, found only in East Asians, had the lowest mean pairwise Euclidean distance (d = 0.17) and was also the most consistent (consistency = 0.92), suggesting that individuals from R9 could reasonably be described as having recent East Asian genetic ancestry.
For the 1KGP, we measured consistency by using the HGDP as a reference panel to determine the maximum continental-ancestry component for each mtDNA haplogroup. About half of the mtDNA haplogroups in 1KGP had consistency scores > 50%, and in many cases, the scores were higher than in the HGDP (Figure S7; Table S7). The correlation between consistency and mean pairwise Euclidean distances was both weaker and not statistically significant in the 1KGP data (R2 = −0.49, p = 0.07), and several haplogroups (M, C, D, W, B, U, and K) even had consistency scores of 0. The consistency scores of some haplogroups varied substantially between the HGDP and 1KGP datasets (e.g., haplogroup A had d = 0.67 in HGDP and d = 0.26 in 1KGP).
Measuring the Association between mtDNA-Haplogroup Membership and Autosomal Estimates of Continental Ancestry
To determine the association between individual continental autosomal ancestry and mtDNA-haplogroup membership, we fit a multinomial logit model to predict each individual’s mtDNA haplogroup from his or her specific combination of continental-ancestry proportions. Our logit model was a significantly better fit to the data than a null model (LRT, p < 0.001). McFadden’s pseudo-R2 for this final model was 0.84, indicating a very good fit to the data. When we used the logit model to predict mtDNA-haplogroup membership for each of the individuals in our test set, 24% of the predictions were correct in the HGDP populations; for comparison, a random assignment of mtDNA haplogroup would be expected to be correct 10% of the time. Furthermore, the classification probabilities for the correct mtDNA haplogroup of each sample were significantly higher (t test, p < 0.001) than the classification probabilities for incorrect mtDNA haplogroups. Classification probabilities for the correct mtDNA haplogroup were also significantly higher (t test, p = 0.02) than classification probabilities for incorrect haplogroups in 1KGP populations (Figure S8). However, only 7% of the predictions were correct in the 1KGP populations. Prediction accuracy was significantly higher in the HGDP (χ2 test, p = 1 × 10−14), even though both datasets were used for building the model. For both datasets, there were a considerable number of samples for which the prediction’s classification probability was very high, but the prediction was nonetheless incorrect (points in the upper left corner of each panel in Figure S9). The effect sizes of the estimated coefficients revealed interesting relationships between autosomal continental-ancestry proportions and mtDNA haplogroups (Table S8). For example, the combination of Middle Eastern and Central and South Asian ancestry drastically decreased an individual’s probability of belonging to haplogroup C—the highest coefficient effect size (β = −1,166.12).
We examined the classification probabilities in more detail to identify which mtDNA haplogroups were most likely to be classified incorrectly by our logit model (Figure 4). In the HGDP samples, classification accuracy ranged from 9% (L3) to 57% (L0/L1) (Figure 4A), and except for haplogroup L0/L1, all mtDNA haplogroups were more likely to be classified incorrectly. For many haplogroups, the highest classification probability did not match the correct haplogroup. For example, L2 was classified as L0/L1 42% of the time and classified correctly only 9%, and T was classified incorrectly as U 39% of the time and classified correctly 13% of the time. Samples from haplogroups A–D were often incorrectly classified as one other (Figure 4A). Classification probabilities for a given haplogroup in the 1KGP dataset usually differed from their counterparts in the HGDP populations, and overall, classification of mtDNA haplogroups showed even poorer performance in the 1KGP dataset. All mtDNA haplogroups in 1KGP populations had higher probabilities of misclassification than did HGDP populations, and none were more likely to be classified correctly (Figure 4B). One haplogroup (D) was never classified correctly.
Figure 4.
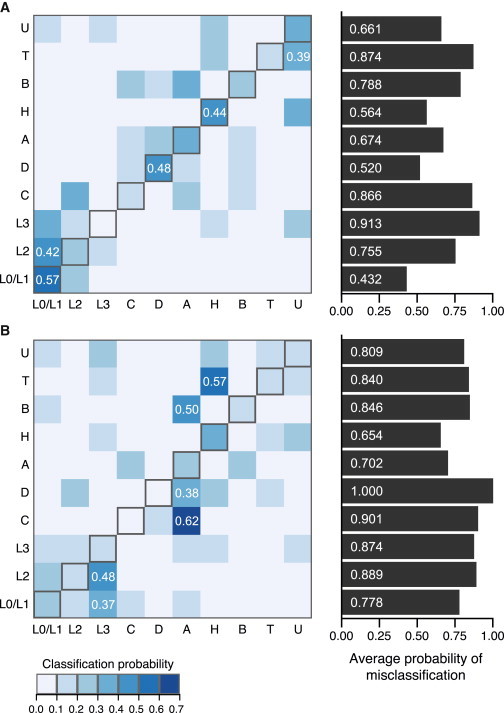
Misclassification Probabilities in the HGDP and 1KGP
Each cell (row i, column j) denotes the probability that a sample experimentally determined as haplogroup i is classified as haplogroup j on the basis of a fitted logit model. Cells are colored by increasing classification probabilities from white to blue (see key at bottom). Diagonal entries (gray outlines) are the probability of being classified correctly. Barplots on the right show the average probability of misclassification for each haplogroup, which is the total of all non-diagonal values in each row. The top 10% of non-zero classification probabilities are labeled (white text).
(A) Misclassification in the HGDP.
(B) Misclassification in the 1KGP.
Discussion
We compared genetic-ancestry inferences made by mtDNA-haplogroup membership to those made by autosomal SNPs in worldwide populations. Continental-ancestry proportions often varied widely among individuals sharing the same mtDNA haplogroup (e.g., Figure 5). For only half of the mtDNA haplogroups did the majority of individuals have their highest continental-ancestry proportion match the haplogroup’s highest average continental-ancestry proportion. Predicting an individual’s mtDNA haplogroup from his or her continental-ancestry proportions was usually incorrect. Collectively, these results indicate that for most individuals in our sample, mtDNA-haplogroup membership provides limited information about either continental ancestry or geographical origin.
Figure 5.
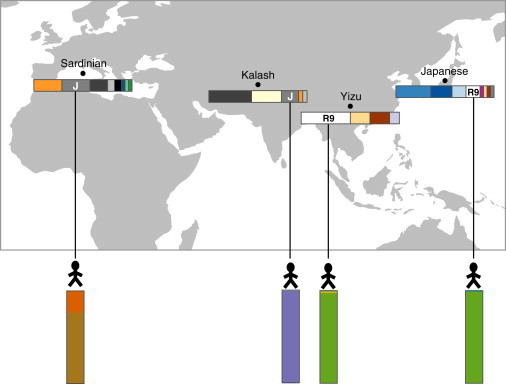
mtDNA-Haplogroup Membership Might Not Be Associated with Autosomal Ancestry Proportions
Each point on the map marks a sampled population, and the population’s mtDNA-haplogroup frequencies are shown in the horizontal barplots below (color key corresponds to haplogroups in Figures 1C and 3B). One individual from each population, along with a vertical barplot of the individual’s autosomal-ancestry proportions, is shown below the map. The two individuals from haplogroup R9 have highly similar autosomal-ancestry proportions, whereas those from haplogroup J are very different.
Mean individual ancestry proportions varied substantially in all but a few of the major mtDNA haplogroups. Moreover, high inter-individual variation in continental-ancestry proportions, as measured by mean pairwise Euclidean distance (Figure 2), indicates that many mtDNA haplogroups consist of individuals with diverse ancestry backgrounds. Thus, even for haplogroups that have high average continental-ancestry proportions, many individuals within that haplogroup do not have more than 50% ancestry from that same continent. For example, 24% of the Brahui population belonged to haplogroup H, but the average European ancestry in the Brahui was only 0.01. mtDNA-haplogroup distributions in the HGDP agree with previously observed descriptions of geographic distributions (e.g., L0/L1, L2, and L3 are frequent in Africa and H is frequent in Europe, etc.). However, there are some significant departures from these generalizations.
Our results show that most mtDNA haplogroups are associated with higher average continental ancestry from a particular continental region than predicted by chance (Figure S5). This result is consistent with our observation that the highest continental-ancestry proportion for most individuals is the same as that of the haplogroup to which they belong. In other words, mtDNA-haplogroup membership does capture some information about continental origins. Although this information appears to support the usefulness of mtDNA haplogroups for estimating genetic ancestry, approximately one-third of mtDNA haplogroups do not exhibit this pattern (Table S7). For example, haplogroups C, D, and R9 are found predominantly in East Asian populations in the HGDP dataset (Figure 1C); however, a substantial number of individuals with haplogroups C and D have negligible East Asian autosomal ancestry and predominately Native American autosomal ancestry (Figure 2).
The second-highest and third-highest continental-ancestry proportions within an individual’s overall ancestry profile often varied significantly within a haplogroup (Figure S5). Several mtDNA haplogroups also showed significant associations with higher-than-average continental ancestry from a second continental region, but most did not. Thus, although an mtDNA-haplogroup classification might provide accurate information about an individual’s highest continental-ancestry component, information regarding an individual’s other ancestry components is limited.
It is also important to note that sex-biased admixture can result in a mismatch between an individual’s highest autosomal-ancestry proportion and mtDNA haplogroup. For example, Hispanic and Latino populations have high European autosomal-ancestry proportions but have almost exclusively Native American mtDNA haplogroups as a result of disproportionate contributions from European men and Native American women.41,42 We observed a similar pattern in the PUR, MXL, and CLM 1KGP populations.
If the association between an individual’s mtDNA haplogroup and his or her combination of continental-ancestry proportions is high, then we should be able to use an individual’s continental-ancestry proportions to predict that individual’s mtDNA haplogroup. However, our logit-based mtDNA-haplogroup predictions were often incorrect (76% incorrect in the HGDP), despite the good fit of our model. Some haplogroups were difficult to distinguish from one another, and many samples were misclassified as a phylogenetically distant mtDNA haplogroup (e.g., C misclassified as L2; Figure 4A). Our failed predictions were not necessarily “close” to being correct. Often the failed predictions had very high classification probabilities, so the strength of the prediction did not necessarily indicate its confidence level (Figure S9). These inaccurate predictions underscore the observation that a substantial amount of ancestry information is not captured by mtDNA-haplogroup classifications.
A highly informative mtDNA haplogroup would have a strong association with ancestry proportion(s) from specific continental region(s), a low mean pairwise Euclidean distance among individuals within the haplogroup, and a high consistency score. Only three haplogroups satisfy these criteria: R9, K, and L0/L1. Ideally, a haplogroup should also have a high consistency score in both the HDGP reference panel and in admixed populations. This is the case for L0/L1, but not for K (R9 was not present in 1KGP).
Our results demonstrate that the majority of mtDNA haplogroups convey information about one, or possibly two, top ancestry components, whereas other ancestry information is lost. Accordingly, most mtDNA haplogroups that are assigned to a continental group (e.g., an “African haplogroup” or a “European haplogroup”) offer an incomplete picture of the complexity of continental ancestry within an mtDNA haplogroup. Effectively communicating this complexity to a consumer or the public poses a substantial challenge,22 and failure to communicate this information could perpetuate misinterpretations.
Overall, our results question the validity of making anything but fairly crude inferences of continental ancestry on the basis of most mtDNA lineage tests. The limitations of lineage-based ancestry inference should be acknowledged by researchers and made explicit to consumers of commercial ancestry-testing products. Although this might merely bolster DTC justifications for independent ancestry inference using autosomal markers, we think it highlights the continued development and refinement of guidelines for genetic-ancestry inference. Finally, it also suggests that additional consumer education is required for more fully understanding the relationship between lineage- and autosomal-based ancestry testing.
Acknowledgments
We acknowledge Arthur Baines and Matthew P. Conomos for statistical consulting through the University of Washington’s Biostatistics Consulting program. This research was supported in part by NIH grant R01GM110068 and an NIH National Human Genome Research Institute Genome Training Grant.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org
HGDP SNP data, http://www.hagsc.org/hgdp/files.html
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Underhill P.A., Kivisild T. Use of y chromosome and mitochondrial DNA population structure in tracing human migrations. Annu. Rev. Genet. 2007;41:539–564. doi: 10.1146/annurev.genet.41.110306.130407. [DOI] [PubMed] [Google Scholar]
- 2.Cavalli-Sforza L.L., Feldman M.W. The application of molecular genetic approaches to the study of human evolution. Nat. Genet. 2003;33:266–275. doi: 10.1038/ng1113. [DOI] [PubMed] [Google Scholar]
- 3.Torroni A., Schurr T.G., Cabell M.F., Brown M.D., Neel J.V., Larsen M., Smith D.G., Vullo C.M., Wallace D.C. Asian affinities and continental radiation of the four founding Native American mtDNAs. Am. J. Hum. Genet. 1993;53:563–590. [PMC free article] [PubMed] [Google Scholar]
- 4.Torroni A., Sukernik R.I., Schurr T.G., Starikorskaya Y.B., Cabell M.F., Crawford M.H., Comuzzie A.G., Wallace D.C. mtDNA variation of aboriginal Siberians reveals distinct genetic affinities with Native Americans. Am. J. Hum. Genet. 1993;53:591–608. [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y.S., Torroni A., Excoffier L., Santachiara-Benerecetti A.S., Wallace D.C. Analysis of mtDNA variation in African populations reveals the most ancient of all human continent-specific haplogroups. Am. J. Hum. Genet. 1995;57:133–149. [PMC free article] [PubMed] [Google Scholar]
- 6.Torroni A., Huoponen K., Francalacci P., Petrozzi M., Morelli L., Scozzari R., Obinu D., Savontaus M.L., Wallace D.C. Classification of European mtDNAs from an analysis of three European populations. Genetics. 1996;144:1835–1850. doi: 10.1093/genetics/144.4.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrnstadt C., Elson J.L., Fahy E., Preston G., Turnbull D.M., Anderson C., Ghosh S.S., Olefsky J.M., Beal M.F., Davis R.E., Howell N. Reduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian, and European haplogroups. Am. J. Hum. Genet. 2002;70:1152–1171. doi: 10.1086/339933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Oven M., Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009;30:E386–E394. doi: 10.1002/humu.20921. [DOI] [PubMed] [Google Scholar]
- 9.Kivisild T., Rootsi S., Metspalu M., Mastana S., Kaldma K., Parik J., Metspalu E., Adojaan M., Tolk H.-V., Stepanov V. The genetic heritage of the earliest settlers persists both in Indian tribal and caste populations. Am. J. Hum. Genet. 2003;72:313–332. doi: 10.1086/346068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong Q.-P., Yao Y.-G., Liu M., Shen S.-P., Chen C., Zhu C.-L., Palanichamy M.G., Zhang Y.-P. Mitochondrial DNA sequence polymorphisms of five ethnic populations from northern China. Hum. Genet. 2003;113:391–405. doi: 10.1007/s00439-003-1004-7. [DOI] [PubMed] [Google Scholar]
- 11.Boattini A., Martínez-Cruz B., Sarno S., Harmant C., Useli A., Sanz P., Yang-Yao D., Manry J., Ciani G., Luiselli D., Genographic Consortium Uniparental markers in Italy reveal a sex-biased genetic structure and different historical strata. PLoS ONE. 2013;8:e65441. doi: 10.1371/journal.pone.0065441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wen B., Xie X., Gao S., Li H., Shi H., Song X., Qian T., Xiao C., Jin J., Su B. Analyses of genetic structure of Tibeto-Burman populations reveals sex-biased admixture in southern Tibeto-Burmans. Am. J. Hum. Genet. 2004;74:856–865. doi: 10.1086/386292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood E.T., Stover D.A., Ehret C., Destro-Bisol G., Spedini G., McLeod H., Louie L., Bamshad M., Strassmann B.I., Soodyall H., Hammer M.F. Contrasting patterns of Y chromosome and mtDNA variation in Africa: evidence for sex-biased demographic processes. Eur. J. Hum. Genet. 2005;13:867–876. doi: 10.1038/sj.ejhg.5201408. [DOI] [PubMed] [Google Scholar]
- 14.Shriver M.D., Kittles R.A. Genetic ancestry and the search for personalized genetic histories. Nat. Rev. Genet. 2004;5:611–618. doi: 10.1038/nrg1405. [DOI] [PubMed] [Google Scholar]
- 15.Bolnick D.A., Fullwiley D., Duster T., Cooper R.S., Fujimura J.H., Kahn J., Kaufman J.S., Marks J., Morning A., Nelson A. Genetics. The science and business of genetic ancestry testing. Science. 2007;318:399–400. doi: 10.1126/science.1150098. [DOI] [PubMed] [Google Scholar]
- 16.Frudakis T. The legitimacy of genetic ancestry tests. Science. 2008;319:1039–1040. doi: 10.1126/science.319.5866.1039b. author reply 1039–1040. [DOI] [PubMed] [Google Scholar]
- 17.Sarata, A.K. (2008). Genetic Ancestry Testing: CRS Report for Congress. http://assets.opencrs.com/rpts/RS22830_20080312.pdf
- 18.The American Society of Human Genetics (2008). Ancestry testing statement. http://www.ashg.org/pdf/ASHGAncestryTestingStatement_FINAL.pdf
- 19.Lee D.Y., Hayes J.J., Pruss D., Wolffe A.P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- 20.Via M., Ziv E., Burchard E.G. Recent advances of genetic ancestry testing in biomedical research and direct to consumer testing. Clin. Genet. 2009;76:225–235. doi: 10.1111/j.1399-0004.2009.01263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Royal C.D., Novembre J., Fullerton S.M., Goldstein D.B., Long J.C., Bamshad M.J., Clark A.G. Inferring genetic ancestry: opportunities, challenges, and implications. Am. J. Hum. Genet. 2010;86:661–673. doi: 10.1016/j.ajhg.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner J.K., Cooper J.D., Sterling R., Royal C.D. Tilting at windmills no longer: a data-driven discussion of DTC DNA ancestry tests. Genet. Med. 2012;14:586–593. doi: 10.1038/gim.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pritchard J.K., Stephens M., Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang H., Peng J., Wang P., Risch N.J. Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol. 2005;28:289–301. doi: 10.1002/gepi.20064. [DOI] [PubMed] [Google Scholar]
- 26.Shriver M.D., Smith M.W., Jin L., Marcini A., Akey J.M., Deka R., Ferrell R.E. Ethnic-affiliation estimation by use of population-specific DNA markers. Am. J. Hum. Genet. 1997;60:957–964. [PMC free article] [PubMed] [Google Scholar]
- 27.Salas A., Acosta A., Alvarez-Iglesias V., Cerezo M., Phillips C., Lareu M.V., Carracedo A. The mtDNA ancestry of admixed Colombian populations. Am. J. Hum. Biol. 2008;20:584–591. doi: 10.1002/ajhb.20783. [DOI] [PubMed] [Google Scholar]
- 28.Watkins W.S., Xing J., Huff C., Witherspoon D.J., Zhang Y., Perego U.A., Woodward S.R., Jorde L.B. Genetic analysis of ancestry, admixture and selection in Bolivian and Totonac populations of the New World. BMC Genet. 2012;13:39. doi: 10.1186/1471-2156-13-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardena M.M.S.G., Ribeiro-Dos-Santos A., Santos S., Mansur A.J., Pereira A.C., Fridman C. Assessment of the relationship between self-declared ethnicity, mitochondrial haplogroups and genomic ancestry in Brazilian individuals. PLoS ONE. 2013;8:e62005. doi: 10.1371/journal.pone.0062005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poetsch M., Wiegand A., Harder M., Blöhm R., Rakotomavo N., Freitag-Wolf S., von Wurmb-Schwark N. Determination of population origin: a comparison of autosomal SNPs, Y-chromosomal and mtDNA haplogroups using a Malagasy population as example. Eur. J. Hum. Genet. 2013;21:1423–1428. doi: 10.1038/ejhg.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cann H.M., de Toma C., Cazes L., Legrand M.-F., Morel V., Piouffre L., Bodmer J., Bodmer W.F., Bonne-Tamir B., Cambon-Thomsen A. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- 32.Behar D.M., Rosset S., Blue-Smith J., Balanovsky O., Tzur S., Comas D., Mitchell R.J., Quintana-Murci L., Tyler-Smith C., Wells R.S., Genographic Consortium The Genographic Project public participation mitochondrial DNA database. PLoS Genet. 2007;3:e104. doi: 10.1371/journal.pgen.0030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L., Myers R.M. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- 34.Rosenberg N.A. Standardized subsets of the HGDP-CEPH Human Genome Diversity Cell Line Panel, accounting for atypical and duplicated samples and pairs of close relatives. Ann. Hum. Genet. 2006;70:841–847. doi: 10.1111/j.1469-1809.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- 35.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics. 2011;27:718–719. doi: 10.1093/bioinformatics/btq671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., 1000 Genomes Project Analysis Group The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruiz-Pesini E., Lott M.T., Procaccio V., Poole J.C., Brandon M.C., Mishmar D., Yi C., Kreuziger J., Baldi P., Wallace D.C. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007;35:D823–D828. doi: 10.1093/nar/gkl927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venables W.N., Ripley B.D. Springer; New York: 2002. Modern Applied Statistics with S. [Google Scholar]
- 41.Rojas W., Parra M.V., Campo O., Caro M.A., Lopera J.G., Arias W., Duque C., Naranjo A., García J., Vergara C. Genetic make up and structure of Colombian populations by means of uniparental and biparental DNA markers. Am. J. Phys. Anthropol. 2010;143:13–20. doi: 10.1002/ajpa.21270. [DOI] [PubMed] [Google Scholar]
- 42.Bryc K., Auton A., Nelson M.R., Oksenberg J.R., Hauser S.L., Williams S., Froment A., Bodo J.-M., Wambebe C., Tishkoff S.A., Bustamante C.D. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc. Natl. Acad. Sci. USA. 2010;107(2):786–791. doi: 10.1073/pnas.0909559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.