

Abstract
We studied a group of individuals with elevated urinary excretion of 3-methylglutaconic acid, neutropenia that can develop into leukemia, a neurological phenotype ranging from nonprogressive intellectual disability to a prenatal encephalopathy with progressive brain atrophy, movement disorder, cataracts, and early death. Exome sequencing of two unrelated individuals and subsequent Sanger sequencing of 16 individuals with an overlapping phenotype identified a total of 14 rare, predicted deleterious alleles in CLPB in 14 individuals from 9 unrelated families. CLPB encodes caseinolytic peptidase B homolog ClpB, a member of the AAA+ protein family. To evaluate the relevance of CLPB in the pathogenesis of this syndrome, we developed a zebrafish model and an in vitro assay to measure ATPase activity. Suppression of clpb in zebrafish embryos induced a central nervous system phenotype that was consistent with cerebellar and cerebral atrophy that could be rescued by wild-type, but not mutant, human CLPB mRNA. Consistent with these data, the loss-of-function effect of one of the identified variants (c.1222A>G [p.Arg408Gly]) was supported further by in vitro evidence with the mutant peptides abolishing ATPase function. Additionally, we show that CLPB interacts biochemically with ATP2A2, known to be involved in apoptotic processes in severe congenital neutropenia (SCN) 3 (Kostmann disease [caused by HAX1 mutations]). Taken together, mutations in CLPB define a syndrome with intellectual disability, congenital neutropenia, progressive brain atrophy, movement disorder, cataracts, and 3-methylglutaconic aciduria.
Introduction
The implementation of exome sequencing (ES) and genome sequencing has expanded our knowledge of genes that cause pediatric syndromic phenotypes.1 For a subset of these individuals, the diagnostic approach can be assisted by findings from metabolic markers in blood and urine that might point toward hitherto unappreciated inborn errors of metabolism (IEMs).2
3-methylglutaconic acid (3-MGA) detected in urine samples is such a marker, showing consistently and significantly elevated levels in a rapidly growing group of IEMs with a syndromic phenotype.3 This group encompasses several disorders in which phospholipid remodeling and other mitochondrial membrane-related processes are defective. Clinical features are heterogeneous but distinctive. For example, Barth syndrome (MIM 302060), caused by mutations in TAZ (MIM 300394), is associated with (cardio)myopathy, neutropenia, and delayed motor milestones; Sengers syndrome (MIM 212350), driven by mutations in AGK (MIM 610345), manifests cardiomyopathy and cataracts; and MEGDEL syndrome (MIM 614739), caused by mutations in SERAC1 (MIM 614725), is associated with deafness and dystonia.4 In addition, congenital neutropenia and central nervous system involvement have also been reported in disorders without 3-MGA-uria, such as Kostmann syndrome (MIM 610738; HAX1 [MIM 605998]), Shwachman-Bodian-Diamond disease (MIM 260400; SBDS [MIM 607444]), and Cohen syndrome (MIM 216550; VPS13B [MIM 607817]).5
Here we present a constellation of pathologies that cannot be reconciled with any known clinical entity. This phenotypic spectrum encompasses intellectual disability (ID)/developmental delay (DD), congenital neutropenia, progressive brain atrophy, movement disorder, and bilateral cataracts. Though all individuals share 3-MGA-uria as a characteristic biomarker, the severity of the other signs and symptoms shows interindividual variability. Nonetheless, under the hypothesis that the consistently observed metabolic signature underpins a discrete molecular genetic disorder, we performed ES in two unrelated individuals, followed by subsequent candidate gene testing in a further 16 affected individuals. Through these studies, the implementation of an in vivo model that recapitulated key neuroanatomical aspects of the disorder, and biochemical in vitro testing, we report the identification of loss-of-function mutations in CLPB (RefSeq accession number NM_030813.4).
Subjects and Methods
Clinical Cohort
Individuals #6 and #9 manifested an overlapping phenotype of ID, neutropenia, cataracts, and 3-MGA-uria and were independently evaluated by ES at two different centers (RadboudUMC, Nijmegen, the Netherlands; Helmholtz Zentrum Munich, Germany). After identification of the genetic defect in these cases, we selected 16 additional individuals with an overlapping clinical presentation from the internal database at RadboudUMC. This study adhered to the Declaration of Helsinki and written informed consent was obtained from each individual.
Exome Sequencing and Variant Identification
ES for individual 6 was performed as previously described.6 In brief, we used a SureSelect Human All Exon 50 Mb Kit (Agilent) for enrichment and a HiSeq2500 (Illumina). Reads were aligned to the UCSC human reference assembly (hg19) with BWA v.0.5.8. More than 90% of the exome was covered at least 20×. Single-nucleotide variants (SNVs) and small insertions and deletions were detected with SAMtools v.0.1.7. Variant prioritization was performed based on an autosomal-recessive pattern of inheritance. ES for individual 9 was performed as previously described with minor adjustments.2 In brief, we used the SureSelectXT Human All Exon 50Mb Kit (Agilent) for enrichment and a SOLiD 5500XL (Life Technologies). We excluded all nongenic, intronic (other than canonical splice sites), and synonymous variants and all known variants with a frequency >1% in dbSNP v.132 and our in-house variant database consisting of 672 exomes. Next, because a recessive disease model was expected and given the assumption of a common ancestral allele (based on parental consanguinity), we prioritized variants according the percentage of variant reads. For this, we used the threshold of >75% variant reads as an indicator for homozygous variants.
Mutation Analysis by Sanger Sequencing
Primer sequences for amplification of all protein coding exons of CLPB (RefSeq NM_030813.4) are shown in Table S1 available online. PCR conditions are available upon request. PCR products were sequenced with the ABI PRISM BigDye Terminator Cycle Sequencing v.2.0 Ready Reaction Kit and analyzed with the ABI PRISM 3730 DNA analyzer (Applied Biosystems).
In Vivo Functional Modeling of CLPB Mutations in Zebrafish
Zebrafish embryos and adults were maintained and mated as described and all experiments were carried out with the approval of the Institutional Animal Care and Use Committee (IACUC).7 For the in vivo complementation experiments, a translational-blocking morpholino (MO) against the sole zebrafish clpb ortholog was designed (Figure S1) and obtained from Gene Tools. We injected 1 nl of diluted MO (2.5 ng) and/or RNA (200 pg for WT or mutant clpb) into wild-type zebrafish embryos at the 1- to 4-cell stage. For acetylated tubulin staining marking the neuronal axon processes, injected embryos were treated as described.8 For RNA rescue and overexpression experiments, the human wild-type mRNA of the canonical isoform (RefSeq NM_030813.4) of CLPB was cloned into the pCS2+ vector and transcribed in vitro with the SP6 Message Machine kit (Ambion). The four variants tested—p.Arg408Gly, p.Met411Ile, p.Tyr617Cys, and p.Gly646Val—were introduced with Phusion high-fidelity DNA polymerase (New England Biolabs) and custom-designed primers. Image acquisition and analysis was performed with Nikon NIS-Elements Advanced Research software. All experiments were repeated in triplicate and significance of the morphant phenotype was judged by Student’s χ2 test.
Purification and Biochemical Characterization of Human CLPB
The CLPBΔN92 construct (deletion of predicted signal peptide) was PCR generated with a pENTR223-hCLPB plasmid as a template. The amplified DNA fragment was cloned into the pET15b vector (Novagen) at the NdeI/BamHI sites, resulting in a construct pET15b-CLPBΔN92 with an introduced N-terminal His-tag. The construct was verified by DNA sequencing. The p.Arg408Gly change of CLPBΔN92 was introduced into pET15b-CLPBΔN92 by site-directed mutagenesis and confirmed by DNA sequencing.
The CLPBΔN92 and its p.Arg408Gly variant were expressed in E. coli BL21(DE3). Pelleted cells were diluted 1/1 (v/v) with buffer (NaCl 300 mM, imidazole 20 mM, glycerol 20%, 2-mercaptoethanol 5 mM, HEPES 20 mM [pH 7.4]), French press lysed, and centrifuged at 75,000 × g for 1 hr. The lysate was incubated with 2 ml Ni-NTA resin in batch mode. After washing with buffer (NaCl 150 mM, imidazole 20 mM, glycerol 20%, 2-mercaptoethanol 5 mM, HEPES 40 mM [pH 7.4]) and then with the same buffer supplemented with 48 mM imidazole, proteins were eluted with 300 mM pure imidazole and applied on a PD10 desalting column equilibrated with buffer without imidazole (the other components of washing buffers, elution buffers, and desalting buffers remained the same). Protein identity was confirmed by immunoblot, via an anti-CLPB antibody (Abcam, ab87253) (Figure S2A). Figure S2B shows purified CLPB_WT and the purified CLPB_p.Arg408Gly on SDS-PAGE. Protein concentrations were estimated by Coomassie-stained SDS-PAGE gel densitometry with BSA as a standard. The ATPase activity of purified human wild-type CLPB (CLPB_WT) and its p.Arg408Gly mutant (CLPB_p.Arg408Gly) was analyzed with the coupled pyruvate kinase/lactate dehydrogenase assay, as previously described.9 2 μM hCLPB was incubated with 20 mM ATP at 36°C and the absorbance change was recorded at 1 s intervals. The rate was calculated from the linear part of the curve (steady-state rate). For WT hCLPB, the measurement was additionally performed in the presence of casein (0.2 mg/ml).
Results
The Phenotypic Spectrum of Individuals with Mutations in CLPB
The clinical presentation and course of the 14 affected individuals (eight females, six males) varied substantially from a mild phenotype (individuals #1 and #2) associated with cataracts and neutropenia but no neurological involvement or infections to the most severe phenotype (individuals #9–#14) associated with neonatal or even prenatal onset of neurological symptoms (progressive brain atrophy, absence of development, movement disorder, seizures), severe neutropenia with progression into leukemia, and death in the first months of life. The detailed clinical histories of the cohort can be found in the Supplemental Data and are summarized in Table 1 and Figures 1 and 2. Common features include ID/DD (12/14 individuals investigated), congenital neutropenia (10/14), brain atrophy (7/9), microcephaly (7/12), movement disorder (7/13), cataracts (5/10), and 3-MGA-uria (12/12 individuals). The oldest affected participant alive is 18 years old and the youngest is 2 years old. Six individuals passed away between the ages of 24 days and 46 months.
Table 1.
Clinical, Biochemical, and Neuroradiological Findings in Individuals with Mutations in CLPB
| Individual, Gender | Family | ID | Change (aa) | Neutropenia | Generalized Brain Atrophy | Movement Disorder | Muscle Tone | Cataracts | 3-MGA-Uria | Current Age | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| #1, m | 1 | no | p.Met411Ile and p.Tyr617Cys | CS | ND | − | normal | - | + | 10 years | |
| #2, f | 1 | no | p.Met411Ile and p.Tyr617Cys | CM | − | − | normal | + | + | 8 years | |
| #3, m | 2 | moderate | p.Arg408Gly and p.Arg417∗ | IS | ND | ataxia | floppy infant, mild truncal hypotonia | − | + | 5 years 9 months | neonatal hypoglycaemia, microcephaly |
| #4, m | 2 | moderate | p.Arg408Gly and p.Arg417∗ | − | ND | − | floppy infant, mild truncal hypotonia | + | + | 2 years 2 months | neonatal hypoglycaemia, microcephaly |
| #5, f | 2 | moderate | p.Arg408Gly and p.Arg417∗ | − | ND | − | floppy infant, mild truncal hypotonia | − | + | 2 years 2 months | neonatal hypoglycaemia, microcephaly |
| #6, f | 4 | mild | p.Glu435_Gly436delinsAspPro and p.Gly646Val | CM | +a | ataxia, dysarthria, tremor | mild tetraspasticity | + | + | 18 years | microcephaly, hypothyreoidea, hypergonadotropic hypogonadism |
| #7, f | 5 | severe | p.Cys486Arg homozygous | − | + | dystonia | floppy infant, progressive tetraspasticity | + | + | 17 years | microcephaly IUGR, epilepsy, hypothyreoidea, mild cardiac septal hypertrophy, nystagmus |
| #8, f | 5 | severe | p.Cys486Arg homozygous | − | + | dystonia, athetosis | floppy infant, progressive tetraspasticity | − | + | 9 years | microcephaly, epilepsy, nystagmus |
| #9, f | 6 | severe | p.Ala591Val homozygous | CS | + | hyperkinesia, dystonia | floppy infant, progressive tetraspasticity | + | + | 3 years 10 monthsb | microcephaly, IUGR, neonatal hypoglycaemia, life-threatening drooling |
| #10, m | 7 | severe | p.Tyr272Cys and p.Tyr567Cys | CS | + | − | floppy infant, progressive tetraspasticity | ND | ND | 3 monthsb | hepatosplenomegaly, leukemia, facial dysmorphism |
| #11, f | 7 | severe | p.Tyr272Cys and p.Tyr567Cys | CS | + | − | floppy infant, generalized hypotonia | ND | + | 3 monthsb | hepatosplenomegaly, myelodysplastic and preleukemic syndrome, facial dysmorphism |
| #12, f | 8 | severe | p.Cys647Leufs∗26 and p.Ile682Asn | CM | + | ataxia, tremor | stiff baby | − | + | 5 monthsb | microcephaly, IUGR, epilepsy |
| #13, m | 9 | severe | p.Arg250∗, p.Arg417∗, and p.Glu501Lys | CS | ND | jittery | stiff baby | ND | + | 24 daysb | epilepsy |
| #14, m | 9 | severe | p.Arg250∗, p.Arg417∗, and p.Glu501Lys | CS | ND | ND | stiff baby | ND | ND | 54 daysb | IUGR, mild dilated cardiomyopathy |
All individuals are of European descent. Abbreviations are as follows: CM, chronic moderate; CS, chronic severe; ID, intellectual disability; IS, intermittent severe; IUGR, intrauterine growth retardation; ND, no data; 3-MGA-uria, 3-methylglutaconic aciduria.
Isolated cerebellar atrophy.
Age deceased.
Figure 1.
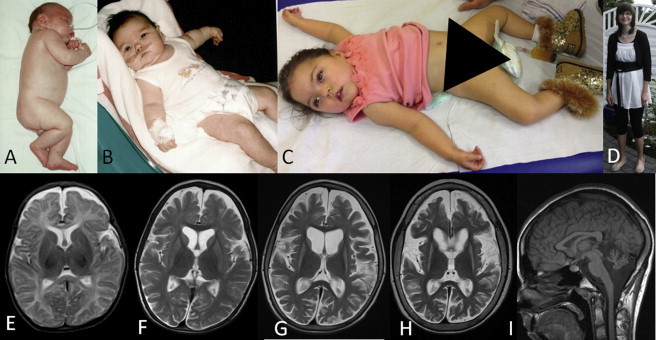
Individual Photographs and MRI Findings
(A) Individual #10 was born with increased muscle tone.
(B and C) Study participants #11 (B) and #9 (C) with tented mouth, hypertelorism, and truncal hypotonia.
(D) Individual #6 displayed no facial dysmorphisms and is able to stand freely.
(E–H) Consecutive T2-weighted MR images of individual #8, axial view, at the age of 2.5 months (E), 16 months (F), 3.5 years (G), and 7 years (H), demonstrating progressive brain atrophy with both cortical and white matter volume decrease over time. Progressive, symmetrical basal ganglia atrophy was supported by abnormally increased T2 signal intensity in the caudate nucleus and putamen starting at the age of 16 months.
(I) Isolated cerebellar atrophy was observed in study participant #6 as determined T1-weighted sagittal MRI.
Figure 2.
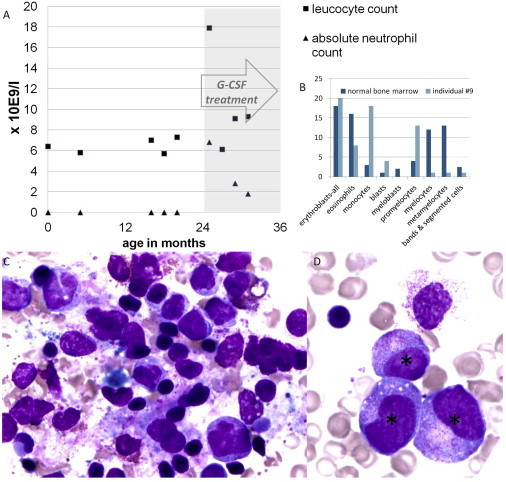
Leucocyte Counts, Images, and Maturation Arrest
(A) Leucocyte (reference range 4.5–10 g/l) and absolute neutrophil count (reference range at birth, 12–15 g/l; 2–12 months, >2 g/l; >12 months, >1.5 g/l) of individual #9 before and during successful treatment with G-CSF.
(B) The total bone marrow composition at 20 months of life of the same individual: blasts 4%, promyelocytes 13%, myelocytes 1%, metamyelocytes 1%, bands and segmented cells 1%, neutrophils 2%, basophils 0%, eosinophils 8%, lymphocytes 32%, monocytes 18%, plasma-cells 0%, normoblasts 20% in comparison with a normal bone marrow.
(C and D) Crista biopsy of individual #9 at 20 months of age shown in two distinct magnifications. The bone marrow contains many promyelocytes (∗), but no mature neutrophils (maturation arrest at promyelocyte stage), many macrophages, hemophagocytosis, and atypical lymphocytes.
Neurological Phenotype
Two individuals (#1 and #2), currently aged 8 and 10 years, showed no neurological involvement at all as determined by a normal neurological examination, normal IQ test, and, in one person, normal brain imaging. However, individual #1 had ADHD, dyslexia, and dysgraphia and individual #2 had a tendency to impulsivity. All other persons (#3–#14) showed DD/ID; the most severe cases (#10–#14) did not develop at all, did not make any eye or other contact, and suffered from (episodic to permanent) unconsciousness from birth until their early death. Most of them (individuals #3–#14) showed pyramidal tract involvement, progressing from severe hypotonia during the first months of life to severe bilateral spasticity thereafter. In the most severe cases (individuals #12–#14), these degenerative processes probably started during fetal life, because all were born as “stiff babies” with generalized increased muscle tension including contractures and jaw lock. Additionally, 11 individuals suffered swallowing difficulties, potentially of both muscular and central nervous origin, necessitating tube feeding. In four individuals, epilepsy was reported. Furthermore, there is a spectrum of MRI abnormalities ranging from isolated cerebellar atrophy (#6, Figure 1J) in less severely affected individuals to atrophy of both cerebral hemispheres, the basal ganglia, and the cerebellum in the most severely affected persons (Figures 1E–1H). Additionally, white matter involvement was seen in individuals #7–#9 and 11. The brain atrophy corresponds with the clinical finding of microcephaly in 7 of 12 investigated persons. In the individuals with basal ganglia involvement (#7–#9), clinically established dystonia was observed.
Hematological/Immunological Phenotype
Neutropenia was noted in 10 of the 14 individuals, as severe (<0.5 g/l) in six (#1, #9–#11, #13, #14) and moderate (0.5–1.0 g/l) in three (#2, #6, #12). One person (#3) was found to be neutropenic only subsequent to infections, the severity of which appear concomitant with the severity of the neurological phenotype. Whereas individuals without neurological symptoms (#1, #2) did not suffer from recurrent infections, individuals #3–#8 suffered from more frequent infections than peers but without serious complications. The remaining individuals from the study cohort (#9–#14) suffered regularly from serious, often life-threatening infections and were treated with G-CSF (#9 and #12, during infections also #3), continuous antibiotics, and antimycotics (see Figure 2A for details on neutropenia of #9). The bone marrow examination of individual #9 showed a maturation arrest at the stage of the promyelocyte (Figures 2B and 2C). In addition, we observed an absence of mature neutrophils in the bone marrow of individuals #10 and #11. Of note, two siblings, who were not treated with G-CSF, progressed to (1) an acute myeloid leukemia (M5, acute monocytic, with the typical finding of a somatic monosomy of chromosome 7; individual #10) or a (2) myelodysplastic syndrome/preleukemia of myelomonocytic type (individual #11), respectively. Chemotherapeutic treatment was initiated in individual #10, who died shortly after. No treatment was initiated in individual #11, who also died shortly after the diagnosis. Bone marrow aspiration in the third available individual (#13) showed a vacuolar degeneration of the phagocytic mononuclear system without typical signs of neutropenia.
Other Signs and Symptoms
For ten of the study participants, information about ophthalmological findings was available. Five of these individuals had bilateral cataracts, and one (#7) was diagnosed with a suspected pigmentary retinopathy. Two individuals showed cardiac involvement, namely mild septal hypertrophy (#7) and mild dilated cardiomyopathy (#14). Two study participants had endocrine abnormalities (#6 and #7). At least three individuals (#9–#11) showed similar facial dysmorphisms (e.g., low nasal bridge, hypertelorism, tented mouth; Figures 1B and 1C). From a biochemical standpoint, no consistently elevated serum lactate or alanine levels were observed in any of the investigated individuals, nor did they have an elevated urinary excretion of Krebs cycle intermediates. All individuals had consistent and significant excretion of 3-MGA in their urine, amounting to 2–15 times over the limit of the reference range, pointing to a possible mitochondrial dysfunction. It was observed that the concentration of this biomarker could fluctuate in an individual (#2, 29–109 [reference < 12 μmol/mmol creatinine]; #6, 34–160; and #12, 52–93 [reference < 20]), seemingly without having a direct relation to the clinical condition.
Identification of Mutations in CLPB and Mutational Spectrum
For each individual, ES resulted in a list of four to six candidate genes. For individual #6, these were OBSCN (MIM 608616), SCN4A (MIM 603967), CEACAM20, and CLPB; and for individual #9, CLVS2, PDZD7 (MIM 612971), TMEM63C, COG7 (MIM 606978), BAHCC1, and CLPB. Besides being the only overlapping gene, CLPB was considered a candidate gene because of the mitochondrial targeting sequence predicting a mitochondrial localization and the previous association of 3-MGA-uria with mitochondrial dysfunction.10 We detected two heterozygous variants, c.1305_1307inv (p.Glu435_Gly436delinsAspPro) and c.1937G>T (p.Gly646Val) in the coding region of CLPB (RefSeq NM_030813.3) for individual #6 while both parents were heterozygous for one of the two variant alleles. In individual #9, a homozygous variant, c.1772C>T (p.Ala591Val), was identified and confirmed to be heterozygous in each of the parents. Given these findings, we performed Sanger sequencing of the entire coding region of CLPB in 16 additional subjects that shared phenotypes with our discovery cohort; we identified likely pathogenic CLPB mutations in 12 of them, all of which segregated (wherever testing was possible) with the disorder under an autosomal-recessive paradigm.
In total, we identified 14 different CLPB mutations (2 nonsense, 1 frameshift, 11 missense; Table 2, Figure 3, Figure S3) in 14 affected individuals from nine independent families of primarily northern European descent (Canada, Australia, Germany, Turkey, Italy, Poland, Estonia). Consistent with a disease-causal role, all variants are either absent or were rare (<0.1% MAF) in the in-house database of 5,036 exomes (Department of Human Genetics, Helmholtz Zentrum, Munich, Germany) and in public databases. The most frequent change, c.1222A>G (p.Arg408Gly), had a MAF of 0.011% in the ExAC browser (detected in 22/122,848 alleles, seen only in heterozygosity and never in homozygosity). The nonsense and frameshift mutations are predicted to result in nonsense-mediated mRNA decay (NMD). Of note, each of the most severely affected individuals (#13 and #14) is compound heterozygous for a nonsense and a missense mutation. Because the parents are unavailable for further research, we could not determine whether these nonsense mutations map on the same haplotype. However, it is tempting to speculate that compound heterozygous nonsense mutations result in a more severe phenotype than missense variants. All missense mutations affect amino acids that are evolutionarily conserved across vertebrates and mostly in bacteria (Figure S4). Moreover, all missense mutations are predicted to have a deleterious impact on protein function by different prediction programs (SIFT, PolyPhen, MutationTaster)11–13 and map to functional domains and the C-tail of CLPB. There was no clear correlation between the severity of the disease and the position and nature of the specific missense mutations. Pathogenic mutations in CLPB from this study have been uploaded to the Leiden Open Variation Database (LOVD-CLPB page).
Table 2.
Mutations in CLPB and Their Predicted Effects at the Protein Level
| Individual | Mutation (nt) | Change (aa) | Exon | Mutation Type | Domains Affected | (Predicted) Effect | Occurence in the ExAC Browser |
|---|---|---|---|---|---|---|---|
| #13, #14 | c.748C>T | p.Arg250∗ | 6 | nonsense | all | truncated protein, NMD | not detected |
| #10, #11 | c.815A>G | p.Tyr272Cys | 6 | missense | ANK | – | 2 het / 122918 alleles |
| #3, #4, #5 | c.1222A>G | p.Arg408Gly | 11 | missense | AAA+ | probably affects ATP binding | 22 het / 122848 alleles |
| #1, #2 | c.1233G>A | p.Met411Ile | 11 | missense | AAA+ | probably affects ATP binding | not detected |
| #3, #4, #5, #13, #14 | c.1249C>T | p.Arg417∗ | 11 | nonsense | AAA+, D2 | truncated protein, NMD | 4 het / 122824 alleles |
| #6 | c.1305_1307inv | p.Glu435_Gly436delinsAspPro | 12 | missense | AAA+ | likely to affect substrate interaction30 | not detected |
| #7, #8 | c.1456T>C | p.Cys486Arg | 13 | missense | AAA+ | – | not detected |
| #13, #14 | c.1501G>A | p.Glu501Lys | 13 | missense | AAA+ | – | 1 het / 121514 alleles |
| #10, #11 | c.1700A>G | p.Tyr567Cys | 15 | missense | D2 boundary | – | 5 het /122590 alleles |
| #9 | c.1772C>T | p.Ala591Val | 16 | missense | D2 | probably affects stabilization of D2 domain | not detected |
| #1, #2 | c.1850A>G | p.Tyr617Cys | 16 | missense | D2 | probably affects oligomer stabilization | not detected |
| #6 | c.1937G>T | p.Gly646Val | 17 | missense | D2 | – | 1 het / 122598 alleles |
| #12 | c.1937dupG | p.Cys647Leufs∗26 | 17 | frameshift | D2 | truncated protein, NMD | not detected |
| #12 | c.2045T>A | p.Ile682Asn | 17 | missense | C-tail | – | not detected |
Overview of all mutations found in CLPB in the different individuals. Abbreviations are as follows: aa, amino acid; het, heterozygous; NMD, nonsense-mediated mRNA decay; nt, nucleotide.
Figure 3.
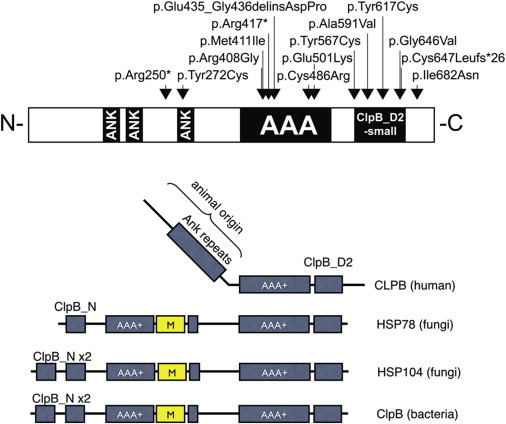
Schematic Representation of CLBP in Human, Fungi, and Bacteria
(A) The positions of all mutations identified in human. Black boxes represent the two main functional domains: the ankyrin domain (ANK) consisting of a short 34 residue repeat implicated in a wide range of protein-protein interactions and the ATPase domain (AAA+).31,32
(B) Evolutionary changes in domain composition of CLPB proteins in human, fungi (mitochondrial HSP78 and cytosolic HSP104), and bacteria. All CLPB-family homologs possess at least a single AAA+ domain and the specific CLPB-D2 domain. In contrast to human CLPB, fungi and bacteria possess an “M domain,” a ClpB N domain, and an additional AAA+ domain. Ankyrin repeats are present only in the human homolog and are probably species specific.
CLPB was shown to be expressed in a broad range of human fetal and adult tissues with a significantly higher expression in adult brain tissues (Figure S5). Of note, the expression in fetal brain is approximately five times lower than in adult brain; we also observed low expression in granulocytes.
Evaluation of Mitochondrial Function in Fibroblasts from Affected Individuals
The first 92 amino acid residues of human CLPB are predicted to encode a mitochondrial targeting sequence (MITOPROT).14 The predicted mitochondrial localization for human CLPB was confirmed by immunofluorescence of CLPB in U2OS cells (Figure S6). Given that other IEMs with 3-MGA-uria as a discriminative feature exhibit mitochondrial dysfunction (e.g., Barth, Sengers, and MEGDEL syndromes), we wondered whether mutations in CLPB could affect oxidative phosphorylation.3,10 We did not detect histological abnormalities, with the exception of a modest dominance of type I fibers, in muscle of individuals #6 and #9. Evaluation of the oxidative phosphorylation in the same individuals in fresh muscle and cultured fibroblasts did not show any abnormalities (Table S3). Finally, autophagy and mitophagy were assessed in cell lines from four individuals, yielding results that were indistinguishable from controls cells (Figure S7). Taken together, these data suggest that CLPB mutations are unlikely to affect mitochondrial function in general, or respiratory chain oxidative phosphorylation defects or other dysfunction in particular.
Evaluation of Phospholipid Metabolism
Several of the IEMs with 3-MGA-uria as discriminative feature have been associated with defective phospholipid metabolism. For instance, typical abnormalities in the quantity and acyl-chain composition of cardiolipin (CL), phosphatidylglycerol (PG), and bis(monoacylglycerol)phosphate (BMP) species are characteristic biomarkers in fibroblasts from individuals with Barth and MEGDEL syndromes.2,4 As such, we investigated the different phospholipid species in fibroblasts from our cohort. Phospholipid analysis of fibroblasts of individuals #1, #3, #6, and #9 showed normal quantity and acyl-chain composition of PG and BMP compared to seven control cell lines (Figure S8). The total amount of CL was lower in affected individuals than in controls (Figure S8). However, the significance level (p = 0.037) as well as the difference in total CL amounts was marginal, which might reflect the limited number of data points. No abnormalities were observed in other phospholipid species in the fibroblasts from the four individuals tested (phosphatidic acids, phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, phosphatidylinositols, cardiolipins, sphingomyelines, and their lyso-analogs; data not shown). Taken together, we found no unambiguous evidence for a general role of CLPB in phospholipid metabolism.
Functional Analysis of Nonsynonymous Variants in CLPB by In Vivo Complementation in Zebrafish Embryos
To investigate the pathogenic potential of the nonsynonymous missense CLPB alleles identified in our study cohort, we turned to the developing zebrafish as a surrogate model. We first evaluated the effect of the MO-induced knockdown on the cerebellar integrity and the ability of human CLPB mRNA to rescue that phenotype in the developing embryos. Among the observed pathologies in individuals with CLPB mutations, CNS defects are the most penetrant structural phenotypes (>90% of individuals; Table 1). As such, given that previous studies have demonstrated that cerebellar defects can be modeled in the developing D. rerio,8,15 we focus on this phenotype. We first identified the sole ortholog of CLPB in the zebrafish genome by reciprocal BLAST. Because all six exons of the gene were divisible by three, hampering our ability to generate bona fide loss-of-function alleles by inhibiting splicing, we designed a translational blocking morpholino (tbMO). Injection of 2.5 ng of the clpb MO resulted in approximately 50% of the injected embryos developing cerebellar defects that ranged in severity from depletion of the axonal connections across the midline of the cerebellum to complete atrophy (Figures 4 and S9). The phenotype was dosage sensitive: progressive increases led to concomitant increase of the penetrance of the cerebellar phenotype to 100% of embryos at 6 ng (Figure S9). The MO-induced phenotype was rescued significantly and reproducibly (p < 0.0001; performed in triplicate, scored blind to injection cocktail) by coinjection with 200 pg of human capped CLPB mRNA (Figures 4 and S9). By contrast, coinjection of the clpb MO with human mRNA encoding each of the four candidate pathogenic variants tested (p.Arg408Gly, p.Met411Ile, p.Tyr617Cys, and p.Gly646Val) were indistinguishable to MO alone (p.Arg408Gly [p = 0.79], p.Met411Ile [p = 0.54], p.Tyr617Cys [p = 0.47], and p.Gly646Val [p = 0.92]), suggesting that these variants have little or no residual activity (Figure 4). Overexpression of CLPB WT mRNA, or mRNA harboring each of the four variants, had no effect on the cerebellar integrity (Figure S10).
Figure 4.
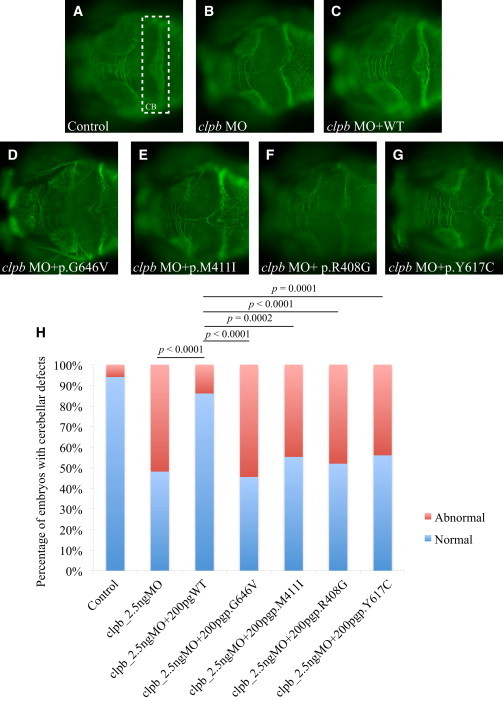
The CLPB Zebrafish Model
In vivo complementation of four CLPB variants (p.Arg408Gly, p.Met411Ile, p.Tyr617Cys, and p.Gly646Val) identified in evaluated study participants, in zebrafish.
(A–G) Dorsal view of the developing zebrafish brain stained with acetylated tubulin at 3 days postfertilization (dpf). The cerebellum (CB) is highlighted by a white dashed rectangle in (A) showing a control embryo.
(A–C) Control (A), morpholino injection (B), and rescue (C) by human WT mRNA.
(D–G) Illustration of the effects of the tested alleles.
(H) Graphic representation of the scoring for CB defects in control and injected embryos. Coinjection of MO with WT human CLPB mRNA results is a statistically significant reduction in the number of embryos with cerebellar defects (p < 0.0001). By contrast, coinjection of MO with each of the human CLPB mRNAs carrying the four mutations tested score as pathogenic being indistinguishable from the MO-injected embryos.
Evaluation of ATPase Function of Human CLPB
We were able to express human CLPB in E. coli after removing the mitochondrial targeting sequence and confirm that human CLPB retains ATPase activity (Figure S11). Unlike the bacterial ClpB chaperone, the ATPase activity was not found to be stimulated by the presence of casein, a natively unfolded soluble substrate. We also expressed the mutant CLPB_p.Arg408Gly (found in individuals #3–#5) in E. coli. This change in the AAA+ domain of CLPB is at the interface between the CLPB oligomers and in the vicinity of the ATP binding site and hence is predicted to influence the ATPase activity through impaired ATP binding. Consistent with this prediction, the ATPase activity level of the mutant protein is 26% of that of wild-type human CLPB in E. coli (Figure S11).
Protein Interaction Network for CLPB and HAX1
A database search resulted in 17 proteins with an established physical interaction with CLPB. The live-cell screen of CLPB against a library of 100 proteins by means of bioluminescence resonance energy transfer (BRET) identified 19 additional direct interactions (Tables S4 and S5). For HAX1, a total of 38 protein interactions was listed in the BioGRID database. Within the networks of first-order protein interactions for both proteins, a link between CLPB and HAX1 is established by mutual interaction with the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (ATP2A2 [MIM 108740]).
Discussion
Here we present our clinical and molecular genetic analyses of a cohort of individuals, the phenotype(s) of which cannot be reconciled with any known clinical entity. The phenotypic spectrum encompasses ID/DD, congenital neutropenia, progressive brain atrophy, movement disorder, and bilateral cataracts. Though all individuals share 3-MGA-uria as a characteristic biomarker, the severity of the other signs and symptoms described shows interindividual variability. On aggregate, however, we propose that our cohort represents a distinct clinical entity that leads to an encephalopathy predominantly involving gray matter, which can start as early as in fetal life, as well as chronic moderate to severe neutropenia due to a maturation arrest at the promyelocyte stage. We do not know whether these pathologies are related. However, we note that individuals with the more attenuated neurological phenotypes also had the more severe hematologic disease, which proceeded to leukemia in two siblings. The broad phenotypic spectrum cannot be reconciled readily by the underlying genotype. However, we note that the individuals with the most severe phenotypes (#12–#14) also carry disease-causing variants predicted to lead to the complete absence of functional protein. Given the observed clinical variability, we speculate that other genetic as well as environmental factors (e.g., infections) could exacerbate the clinical presentation.
Under the hypothesis that the consistently observed metabolic signature underpins a discrete molecular genetic disorder, we performed ES in two unrelated individuals, followed by subsequent candidate gene testing in an additional 16 affected individuals. Through these studies we identified 14 independent mutations in CLPB in 14 individuals from 9 unrelated families. Taken together, our data suggest that CLPB is the major locus for this phenotype, although additional genes are likely to exist. We further supported these claims by developing a zebrafish model of the disorder, in which we recapitulated key aspects of the neuroanatomical phenotypes of the affected individuals. Specifically, embryos bereft of endogenous clpb show microcephaly, reduction of the size of the optic tectum (OT; a structure equivalent to the superior culicullus in humans), and degeneration of the axons forming the cerebellum, reminiscent of the clinical features in the individuals with CLPB mutations. Additionally, coinjection of the clpb MO with human mRNA bearing each of the four detected in our cohort alleles tested were indistinguishable to MO alone, suggesting that these alleles have little or no residual activity and that the observed syndromic phenotypes in humans are driven by null or near null mutations in all cases studied. It will be important to generate genetic clpb mutants to study the progression and molecular pathology of these phenotypes, as well as the possible manifestation of other phenotypes, such as cataracts, that are found in some but not all individuals with CLPB mutations. Likewise, it will be critical to accumulate additional individuals with CLPB mutations and ascertain whether mutations that retain partial protein function correlate with an attenuated phenotype, or indeed any phenotype at all.
CLPB belongs to the large AAA+ (ATP-ases associated with diverse cellular activities) superfamily. AAA+ proteins usually form ring-shaped homo-hexamers.16 Members of this superfamily typically have one or two highly conserved ATPase domains and are involved in various processes, such as DNA replication and repair and protein disaggregation and refolding, and operate as part of dynein motors, as chelatases or proteases.17 The unifying characteristic of this family of proteins consists in the hydrolysis of ATP through the AAA+ domain to produce energy required to exert mechanic force onto their substrates. The human CLPB protein is characterized further by the presence of a specific C-terminal D2 domain (PFAM identifier PF10431), which is typical for AAA+ proteins involved in polypeptide chain threading through the hexamer central channel. Proteins with this domain form the subfamily of Caseinolytic peptidase (Clp) proteins, also called HSP100 (Heat shock proteins 100 kDa).18 The human CLPB protein was named after its high homology to the C-terminal part of bacterial ClpB protein which, in cooperation with Hsp70, is involved in the process of disaggregation of protein aggregates and hence is called a disaggregase. Bacterial ClpB proteins dissolve protein aggregates and rescue aggregated proteins by assisting them to fold back into a native, biologically active form.19 What distinguishes the human CLPB protein from its microbial and plant paralogs is primarily the domain composition. These microbial orthologs contain an additional AAA+ domain and a small N-terminal domain (Figure 3) and have an “M domain,” necessary for disaggregation. Another feature characteristic of the human CLPB is the presence of ankyrin repeats instead of the first of two ATPase domains found in bacteria and fungi, which are used commonly as protein-protein interaction platforms (Figure 3).20,21 The species-specific presence of the ankyrin repeats in the N-terminal part of the human protein might have evolved to ensure a more elaborate or more refined substrate recognition, to mediate the interaction with as yet unknown protein partners, or even to support a putative chaperone function. Despite the fact that only one of the two ATPase domains is retained in the human CLPB, its presence is postulated to be sufficient to mediate the use of ATP hydrolysis energy for threading unfolded polypeptide through the central channel of the hexamer ring.22
Consistent with this hypothesis, we were able to confirm in vitro the ATPase activity of human CLPB. Furthermore, we were able to show that upon the presence of the p.Arg408Gly variant (detected in individuals #3–#5) that is predicted to affect the ATP binding capability of human CLPB, the ATPase activity measured was diminished. Despite the convergence of human and bacterial CLPB proteins on several aspects of molecular function, unlike the bacterial counterpart, the human CLPB cannot be stimulated by casein.
Comparison of the clinical and genetic pathology of individuals with CLPB mutations is likewise informative. Mutations in CLPB lead to a congenital neutropenia syndrome comparable to Kostmann disease, driven by mutations in HAX1, which encodes HCLS1-associated protein X-1 (HAX1).23 Both disorders can show neurological involvement, which seems, however, to be more severe in individuals with CLPB mutations. In individuals with HAX1 mutations, neurological involvement (epilepsy, ID) can be appreciated only when both HAX1 isoforms A and B are affected.24 Despite the variability of neurological symptoms in the two syndromes, remarkable mimicry exists from a hematological standpoint, with individuals affected by either one of the two disorders displaying maturation arrest of the neutrophils at the promyelocyte stage upon bone marrow examination.25 Furthermore, individuals with either CLPB or HAX1 mutations also exhibit disease progression from neutropenia into leukemic pictures in the absence of GCSF.26
Biochemical analyses have established a role of HAX1 in stabilizing the mitochondrial membrane potential and preventing apoptosis via interaction with the mitochondrial proteases presenilin-associated rhomboid-like (PARL), HtrA serine peptidase 2 (HTRA2), and BCL2-associated X protein (Bax).23,26,27 Further evidence for an antiapoptotic role of HAX1 stems from the observation that it interacts with and downregulates the protein level of the sarco/endoplasmic reticulum Ca2+-ATPase (SR Ca(2+)-ATPase 2 encoded by ATP2A2), which regulates the endoplasmic reticulum Ca2+ concentration.28 Evaluation of the protein interaction networks of CLPB predict a biochemical interaction between CLPB and SR Ca(2+)-ATPase 2 (Figure 5; for method, see Table S5), which allows us to speculate that the effect of CLPB mutations on hematopoesis could be driven by excessive apoptosis, as is the case in Kostmann syndrome.
Figure 5.
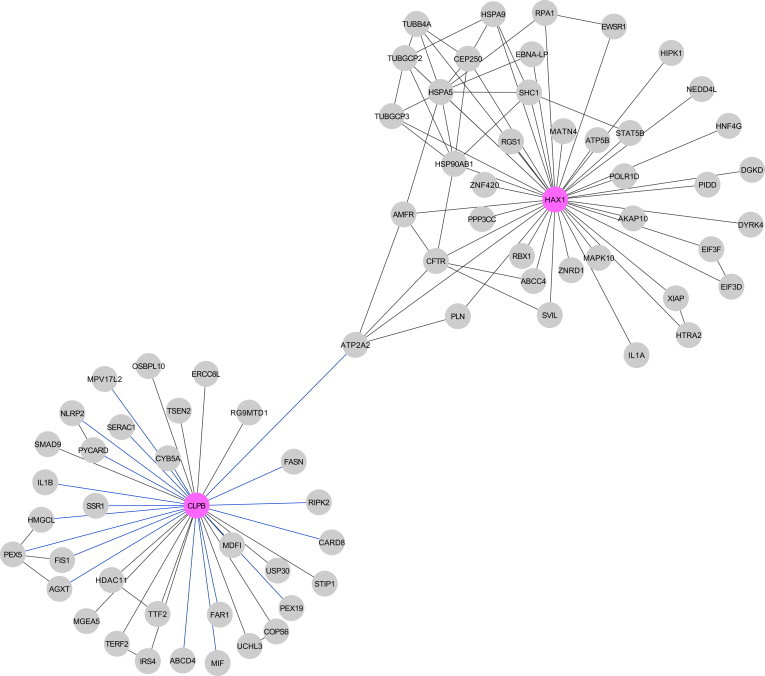
Protein Interaction Network for CLPB and HAX1
A database search resulted in 17 proteins with an established physical interaction with CLPB. The live-cell screen of CLPB against a library of 100 proteins by means of bioluminescence resonance energy transfer (BRET) identified 19 additional direct interactions (Tables S4 and S5). For HAX1, a total of 38 protein interactions were listed in the BioGRID database. Within the networks of first-order protein interactions for both proteins, a link between CLPB and HAX1 is established by mutual interaction with the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SR Ca2+-ATPase 2 encoded by ATP2A2).
To enrich our understanding of the CLPB-mutation-mediated disease pathomechanisms, we reasoned that CLPB defects might have an effect on lipid biosynthesis and metabolism, similar to what is observed in Barth syndrome, one of the IEMs with 3-MGA-uria as a discriminative feature, characterized by neutropenia as well as (cardio)myopathy and delayed motor milestones.29 Despite extensive analysis of the levels of various phospholipids, we detected no involvement of lipid biosynthesis, turnaround, and metabolism in the pathogenesis mediated by mutations in CLPB. We also were unable to detect overt abnormalities in mitophagy or autophagy in cells from individuals with CLPB mutations. In aggregate, our data argue that despite the clinical mimicry between CLPB disease and Barth syndrome or other IEMs with 3-MGA-uria as a discriminative feature, the mechanisms underlying each condition are diverse.
In conclusion, we describe an as yet unknown inborn error of metabolism with 3-MGA-uria as discriminative feature (CLPB defect). Underlying mutations were found in CLPB and lead to a broad phenotypic spectrum encompassing ID/DD, congenital neutropenia, progressive brain atrophy, movement disorder, and bilateral cataracts. The function of human CLPB is currently poorly described but our data show that it might be related to apoptosis.
Acknowledgments
We thank the individuals and their parents for participation in this study. The study was financially supported by Van Leersumfonds, Koninklijke Nederlandse Akademie van Wetenschappen (project VLF2013277 to S.B.W.), by the Dutch society for the study of inborn errors of metabolism (ESN stimulatie beurs to S.B.W.), the NARSAD Young Investigator Grant from BBRF (to C.G.), the Canadian Institutes of Health Research (#301221 grant to C.v.K.), NIH P50MH094268 (to N.K.), the German Bundesministerium für Bildung und Forschung (BMBF) through the German Network for mitochondrial disorders (mitoNET; 01GM1113C to T.M. and H.P.), and through the E-Rare project GENOMIT (01GM1207 for T.M. and H.P.). N.K. is a Distinguished George W. Brumley Professor. C.K. is the recipient of a career development award from the Hermann and Lilly Schilling Foundation and is supported by a grant from the Deutsche Forschungsgemeinschaft (DFG). C.v.K. is the recipient of a scholar award from the Michael Smith Foundation for Health Research. R.S. is financially supported by the Metakids Foundation. We thank Edwin van Kaauwen and Liesbeth Wintjes (Radboudumc Nijmegen) for excellent technical assistance, Dr. Graham Sinclair (University of British Columbia) for providing biochemical data, and Dirk Klee (Heinrich-Heine University) for the MRI images of individual #8. We thank Han Brunner (Radboudumc Nijmegen), K. Liberek (Department of Molecular and Cellular Biology, University of Gdańsk, Poland), Angela Luyf and Antoine van Kampen (AMC, Amsterdam), and Mathias Woidy and Philipp Guder (Ludwig-Maximilians-University, Munich, Germany), as well as Harry Kampinga (University Medical Center Groningen, Groningen, Germany) for fruitful discussions.
Accession Numbers
The databank accession number for the data reported in this paper is http://www.lovd.nl/clpb.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BioGRID, http://thebiogrid.org/
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
ExAC Browser, http://exac.broadinstitute.org/
HPRD, http://www.hsls.pitt.edu/obrc/index.php?page=URL1055173331
MitoProt, http://ihg.gsf.de/ihg/mitoprot.html
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, www.genetics.bwh.harvard.edu/pph2/
SAMtools, http://samtools.sourceforge.net/
STRING 9.0, http://www.string-db.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Acehan D., Xu Y., Stokes D.L., Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab. Invest. 2007;87:40–48. doi: 10.1038/labinvest.3700480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wortmann S.B., Vaz F.M., Gardeitchik T., Vissers L.E., Renkema G.H., Schuurs-Hoeijmakers J.H., Kulik W., Lammens M., Christin C., Kluijtmans L.A. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat. Genet. 2012;44:797–802. doi: 10.1038/ng.2325. [DOI] [PubMed] [Google Scholar]
- 3.Wortmann S.B., Duran M., Anikster Y., Barth P.G., Sperl W., Zschocke J., Morava E., Wevers R.A. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J. Inherit. Metab. Dis. 2013;36:923–928. doi: 10.1007/s10545-012-9580-0. [DOI] [PubMed] [Google Scholar]
- 4.Wortmann S.B., Espeel M., Almeida L., Reimer A., Bosboom D., Roels F., de Brouwer A.P., Wevers R.A. Inborn errors of metabolism in the biosynthesis and remodelling of phospholipids. J. Inherit. Metab. Dis. 2014 doi: 10.1007/s10545-014-9759-7. Published online September 2, 2014. [DOI] [PubMed] [Google Scholar]
- 5.Donadieu J., Fenneteau O., Beaupain B., Mahlaoui N., Chantelot C.B. Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J. Rare Dis. 2011;6:26. doi: 10.1186/1750-1172-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haack T.B., Gorza M., Danhauser K., Mayr J.A., Haberberger B., Wieland T., Kremer L., Strecker V., Graf E., Memari Y. Phenotypic spectrum of eleven patients and five novel MTFMT mutations identified by exome sequencing and candidate gene screening. Mol. Genet. Metab. 2014;111:342–352. doi: 10.1016/j.ymgme.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 7.Niederriter A.R., Davis E.E., Golzio C., Oh E.C., Tsai I.C., Katsanis N. In vivo modeling of the morbid human genome using Danio rerio. J. Vis. Exp. 2013;78:e50338. doi: 10.3791/50338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Margolin D.H., Kousi M., Chan Y.M., Lim E.T., Schmahmann J.D., Hadjivassiliou M., Hall J.E., Adam I., Dwyer A., Plummer L. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med. 2013;368:1992–2003. doi: 10.1056/NEJMoa1215993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nørby J.G. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 1988;156:116–119. doi: 10.1016/0076-6879(88)56014-7. [DOI] [PubMed] [Google Scholar]
- 10.Wortmann S.B., Kluijtmans L.A., Rodenburg R.J., Sass J.O., Nouws J., van Kaauwen E.P., Kleefstra T., Tranebjaerg L., de Vries M.C., Isohanni P. 3-Methylglutaconic aciduria—lessons from 50 genes and 977 patients. J. Inherit. Metab. Dis. 2013;36:913–921. doi: 10.1007/s10545-012-9579-6. [DOI] [PubMed] [Google Scholar]
- 11.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 14.Claros M.G., Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 15.Matsui H., Namikawa K., Babaryka A., Köster R.W. Functional regionalization of the teleost cerebellum analyzed in vivo. Proc. Natl. Acad. Sci. USA. 2014;111:11846–11851. doi: 10.1073/pnas.1403105111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neuwald A.F., Aravind L., Spouge J.L., Koonin E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 17.Snider J., Thibault G., Houry W.A. The AAA+ superfamily of functionally diverse proteins. Genome Biol. 2008;9:216. doi: 10.1186/gb-2008-9-4-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zolkiewski M. A camel passes through the eye of a needle: protein unfolding activity of Clp ATPases. Mol. Microbiol. 2006;61:1094–1100. doi: 10.1111/j.1365-2958.2006.05309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenzweig R., Moradi S., Zarrine-Afsar A., Glover J.R., Kay L.E. Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction. Science. 2013;339:1080–1083. doi: 10.1126/science.1233066. [DOI] [PubMed] [Google Scholar]
- 20.Mosavi L.K., Cammett T.J., Desrosiers D.C., Peng Z.Y. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004;13:1435–1448. doi: 10.1110/ps.03554604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J., Mahajan A., Tsai M.D. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry. 2006;45:15168–15178. doi: 10.1021/bi062188q. [DOI] [PubMed] [Google Scholar]
- 22.Weibezahn J., Tessarz P., Schlieker C., Zahn R., Maglica Z., Lee S., Zentgraf H., Weber-Ban E.U., Dougan D.A., Tsai F.T. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119:653–665. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 23.Klein C., Grudzien M., Appaswamy G., Germeshausen M., Sandrock I., Schäffer A.A., Rathinam C., Boztug K., Schwinzer B., Rezaei N. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease) Nat. Genet. 2007;39:86–92. doi: 10.1038/ng1940. [DOI] [PubMed] [Google Scholar]
- 24.Germeshausen M., Grudzien M., Zeidler C., Abdollahpour H., Yetgin S., Rezaei N., Ballmaier M., Grimbacher B., Welte K., Klein C. Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood. 2008;111:4954–4957. doi: 10.1182/blood-2007-11-120667. [DOI] [PubMed] [Google Scholar]
- 25.Kostmann R. Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria. Acta Paediatr. Suppl. 1956;45(105):1–78. [PubMed] [Google Scholar]
- 26.Yetgin S., Olcay L., Koç A., Germeshausen M. Transformation of severe congenital neutropenia to early acute lymphoblastic leukemia in a patient with HAX1 mutation and without G-CSF administration or receptor mutation. Leukemia. 2008;22:1797. doi: 10.1038/leu.2008.64. [DOI] [PubMed] [Google Scholar]
- 27.Chao J.R., Parganas E., Boyd K., Hong C.Y., Opferman J.T., Ihle J.N. Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature. 2008;452:98–102. doi: 10.1038/nature06604. [DOI] [PubMed] [Google Scholar]
- 28.Vafiadaki E., Arvanitis D.A., Pagakis S.N., Papalouka V., Sanoudou D., Kontrogianni-Konstantopoulos A., Kranias E.G. The anti-apoptotic protein HAX-1 interacts with SERCA2 and regulates its protein levels to promote cell survival. Mol. Biol. Cell. 2009;20:306–318. doi: 10.1091/mbc.E08-06-0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Houtkooper R.H., Turkenburg M., Poll-The B.T., Karall D., Pérez-Cerdá C., Morrone A., Malvagia S., Wanders R.J., Kulik W., Vaz F.M. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta. 2009;1788:2003–2014. doi: 10.1016/j.bbamem.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 30.Leidhold C., von Janowsky B., Becker D., Bender T., Voos W. Structure and function of Hsp78, the mitochondrial ClpB homolog. J. Struct. Biol. 2006;156:149–164. doi: 10.1016/j.jsb.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 31.Letunic I., Doerks T., Bork P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku949. Published online October 9, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sedgwick S.G., Smerdon S.J. The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem. Sci. 1999;24:311–316. doi: 10.1016/s0968-0004(99)01426-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.