

Abstract
Keppen-Lubinsky syndrome (KPLBS) is a rare disease mainly characterized by severe developmental delay and intellectual disability, microcephaly, large prominent eyes, a narrow nasal bridge, a tented upper lip, a high palate, an open mouth, tightly adherent skin, an aged appearance, and severe generalized lipodystrophy. We sequenced the exomes of three unrelated individuals affected by KPLBS and found de novo heterozygous mutations in KCNJ6 (GIRK2), which encodes an inwardly rectifying potassium channel and maps to the Down syndrome critical region between DIRK1A and DSCR4. In particular, two individuals shared an in-frame heterozygous deletion of three nucleotides (c.455_457del) leading to the loss of one amino acid (p.Thr152del). The third individual was heterozygous for a missense mutation (c.460G>A) which introduces an amino acid change from glycine to serine (p.Gly154Ser). In agreement with animal models, the present data suggest that these mutations severely impair the correct functioning of this potassium channel. Overall, these results establish KPLBS as a channelopathy and suggest that KCNJ6 (GIRK2) could also be a candidate gene for other lipodystrophies. We hope that these results will prompt investigations in this unexplored class of inwardly rectifying K+ channels.
Main Text
Keppen-Lubinsky syndrome (KPLBS; MIM 614098) is a very rare disease originally described in 2001;1 two additional individuals were reported in subsequent years.2,3 Clinical features including severe developmental delay, intellectual disability, hypertonia, hyperreflexia, growth parameters above the 50th–75th centile at birth and below the fifth centile by age of 6–9 months, microcephaly, large prominent eyes, a narrow nasal bridge, a tented upper lip, a high palate, an open mouth, tightly adherent skin, and an aged appearance, are highly distinct and fit with a generalized lipodystrophy disorder. To identify the genetic cause of KPLBS, we sequenced the exomes of the three known individuals and their unaffected parents.
Individual 1 was the second child of American and Hispanic-descent parents, with an unremarkable family history. Pregnancy was normal, and the child was delivered at term. The mother was 22 years old when the child was born, and the father was 27. Birth weight was 3.9 kg. The individual had a striking appearance, including wrinkled forehead and chin skin and tightly adherent facial skin due to absence of subcutaneous tissue. Additional features included prominent eyes due to shallow orbits, a narrow nasal bridge, a protruding nasal tip, a narrow and high palate, and micrognathia. A gastrostomy tube was inserted when the child was 2 years old. Severe recurrent pneumonia and respiratory insufficiency were reported, prompting tracheostomy when the child was 4 years old and ventilation when he was 7. He had hypertonia, flexion contractures, and severe scoliosis. Eye closure was impaired. Seizures were first reported in infancy. Routine laboratory tests were unremarkable (Table S1).
Individual 2, of Italian ancestry,2 was the third child of non-consanguineous parents. Pregnancy was complicated by polyhydramnios. Delivery was by cesarean section. Birth weight was 3.4 kg (50th percentile), length 50 cm (50th percentile), and head circumference 34 cm (25th percentile); Apgar scores were 6 and 7, at 1 and 5 min, respectively. In the first month of life, generalized lipodystrophy, spastic tetraparesis, and severe developmental delay were recorded. At first evaluation at an age of 8.10 years, this child showed a complete absence of subcutaneous fat, prominent eyes, underdeveloped alae nasi, an open mouth, gingival overgrowth, scoliosis, prepubertal Tanner stage (Ph1Pg1), and testis in the scrotum with a volume of 2 ml according to Prader’s orchidometer. At 12.4 years he underwent a percutaneous endoscopic gastrostomy (PEG) application to improve his nutritional status. Routine laboratory tests were normal (Table S1).
Individual 3 was the second child of non-consanguineous Jewish parents of Yemenite origin.3 The mother was 20 years old when the child was born, and the father was 21. Pregnancy was unremarkable, and delivery was at term. At birth, the child showed severe developmental delay, consistently reduced facial subcutaneous fat resulting in a “mask face” with minimal facial movements during crying or smiling, and otherwise normal subcutaneous tissue throughout the body. At 2 weeks of age, he was noted to have an opisthotonic posture and breathing difficulties resulting from upper airway obstruction due to narrow nostrils. Between the ages of 2 and 4 years, several episodes of febrile seizures were recorded. In recent years, surgeries for hamstring-tendon release and hip-muscle release have been performed so that joint deformities could be corrected. He fractured his left femur shaft after minor trauma. Routine laboratory tests were normal (Table S1).
Exome and Sanger sequencing were performed on DNA from the three family trios (Table S2). Written informed consent for genetic analyses was collected from parents, and the genetic study was approved by the Bambino Gesù Children’s Hospital ethical committee.
The FASTQ reads were aligned to the hg19 reference genome with the Burrows-Wheeler Aligner (BWA v.0.7.5), and single-nucleotide variants (SNVs) and indels were identified with the Genome Analysis Toolkit (GATK v.2.8-1)4 following the GATK Best Practices. SNVs and indels were further filtered with KggSeq.5,6 Variants that had allele frequencies greater than 5% and that were present in dbSNP (v.138), the 1000 Genome Project, the Exome Variant Server ESP6500 dataset, or in an internal variant database that consists of data derived from exome experiments were excluded. Intergenic, intronic, and synonymous variants were excluded, but we kept variants that occurred in canonical splice sites. A total of 51 Mb for the probands of families 1 and 3 and 44 Mb for the proband of family 2 were targeted for sequencing. For each participant, at least 60,000,000 sequences of 90 bp have been generated, resulting in an average depth of >46-fold for the targeted bases, and in >95% of the target region’s, being covered at >10-fold. For each individual, approximately 28,000 variants were identified in coding regions; of these variants, 40% were missense. A detailed list of results obtained from exome sequencing (i.e., exon-capture statistics and number of SNPs and indels) is reported in Table S3.
On the basis of the unusual subcutaneous fat distribution in the individuals and their lipoatrophy, we investigated a list of candidate genes known to be mutated in lipodystrophy (see Table S4), but we found no pathogenic variant. We searched for the disease-causing mutation in the three individuals by assuming both autosomal-dominant and recessive models (Table S5). Exome-sequencing analysis revealed that the unique de novo common variants across the three individuals were located in exon 3 of KCNJ6 (GIRK2) (Figure 1A). This gene (RefSeq accession number NM_002240.3) maps to the Down syndrome critical region, between DYRK1A (Dual-specificity tyrosine phosphorylation-regulated kinase 1A [MIM 600855]) and DSCR4 (Down syndrome critical region gene 4 [MIM 604829]). Two individuals shared the same mutation, an in-frame heterozygous deletion of three nucleotides (c.455_457del) leading to loss of one amino acid (p.Thr152del). The third individual was heterozygous for a missense mutation (c.460G>A) that introduces an amino acid change from glycine to serine (p.Gly154Ser). No other homozygous or compound heterozygous variants of the protein-coding regions were shared in these three individuals (Figures 1B–C and Table S5).
Figure 1.
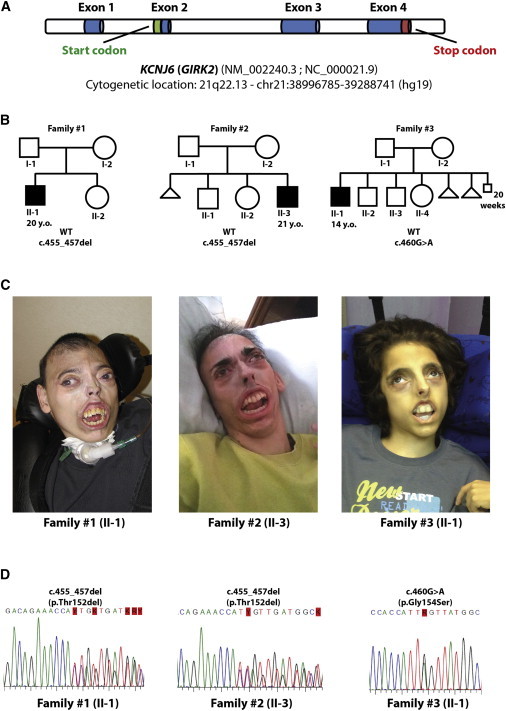
Pedigree of the Three Study Families and KCNJ6 (GIRK2) Details
(A) Cytogenetic localization of KCNJ6 (GIRK2).
(B) Pedigree of the three families examined.
(C) Photographs of the three individuals.
(D) Sanger sequencing of the mutated regions confirms the findings of next-generation sequencing in the three individuals.
To confirm the variants by Sanger sequencing, we designed two specific primers (forward, 5′-CCTCTTCTTGGGTTGAGAGATT-3′ and reverse, 5′-TCTGGTGGTTGATCGCATAC-3′) able to amplify an approximately 270 bp genomic region that was located in exon 3 of KCNJ6 (GIRK2) and that contained the variants identified by exome sequencing (Figure 1D). These variants have never been reported, nor have they been annotated in dbSNP138, in the 1000 Genome Project,7 in the Exome Variant Server,8 in the ExAc Browser (see Web Resources), or in 1,400 exomes from healthy controls.
To gain further insights into the functional consequences of KCNJ6 (GIRK2) variants, we performed 3D protein modeling of wild-type and mutated KCNJ6 (GIRK2). The crystal structure of the mammalian KCNJ6 (GIRK2) was reported by Whorton and MacKinnon in 20119 and 2013.10 KCNJ6 (GIRK2) is a transmembrane potassium-channel protein that crystallizes as a homo-tetramer in the presence of βγ G protein subunits, phosphatidylinositol-4,5-bisphosphate (PIP2), and intracellular sodium. This channel is principally made of a transmembrane domain (TMD) and a cytoplasmic domain (CTD) with different functional parts (i.e., a G loop and inner-helix gates and a selectivity filter for cations) (Figure 2A). To derive the functional consequences of the detected variants, we predicted the 3D structural models of the two mutant proteins (p.Thr152del and p.Gly154Ser), and compared them with the wild-type protein structure.
Figure 2.
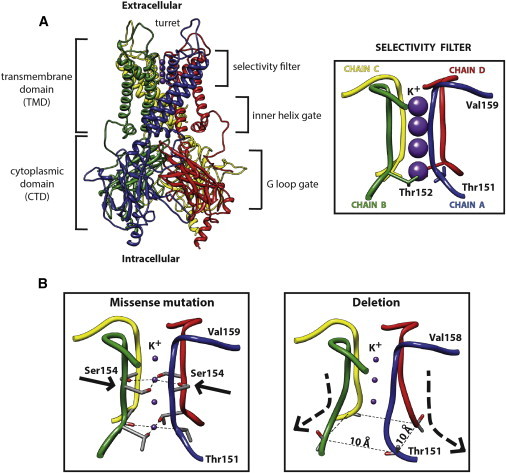
Protein Modeling
(A) Crystal structure of KCNJ6 (GIRK2) (left) and a magnification of the K+ selectivity filter (right). The protein (Protein Data Bank accession number 3SYO) is shown in ribbon form, whereas the four subunits are colored in blue (chain A), green (chain B), yellow (chain C), and red (chain D).
(B) The selectivity filter of the missense mutation and the deletion have been modeled and compared to the wild-type channel. The missense mutation determines the substitution of Ser154 instead of Gly154 residue within the channel, whereas the deletion leads to a shortening of the channel and a widening of the inner base of the K+ filter.
All substitutions were located in the selectivity filter domain of KCNJ6 (GIRK2). The missense mutation replaces a glycine (Gly154) with a serine (Ser154) in the middle of the filter channel (Figure 2B). Because of the more hydrophilic and polar character of the Ser residue compared to Gly, the potassium cations (K+) inside the channel might be differently stabilized by the -OH group, leading to flow inhibition or channel gating.11 Researchers proposed interactions between K+ ions and oxygen atoms more than 40 years ago to explain the selectivity of these channels toward K+.12 Furthermore, the substitution could also abolish the selectivity for K+, which could in turn cause an aberrant Na+ influx across the channel and trigger cell death.13
On the other side, the deletion of Thr152 within the protein backbone profoundly affects the structure of the protein channel, in particular by shortening the channel and widening the inner base of the selectivity filter (the chains of the four subunits are separated by a distance of 10.1 Å instead of 6.9 Å, as in the wild-type structure). Shortening of the channel is determined by the deletion of one Thr residue and a shift of the remaining one (Thr151) toward the channel bottom. This shift also changes the orientation of Thr residues that are no longer directed toward the inner channel but rather outside of it (Figure 2). This conformational change might jeopardize the stabilization of K+, probably impairing the channel functioning. Altogether, these results strongly suggest that KPLBS is a channelopathy originating from substitutions in the inwardly rectifying potassium channel KCNJ6 (GIRK2).
KCNJ6 (GIRK2) is an ATP-sensitive inwardly rectifying K+ channel that is controlled by G-proteins and closed by an increase of intracellular ATP levels. This function links cellular metabolism to electrical excitability of the plasma membrane. Inwardly rectifying channels are characterized by a greater tendency to allow cations to flow into the cell than out of it. The conduction properties characteristic of K+ channels are due to a specific catalytic domain, a highly conserved motif (see Table S1) comprising eight amino acids (Thr-X-X-Thr-X-Gly-Tyr-Gly) that define the “selectivity filter” region.14 Electrophysiology studies on oocytes isolated from Xenopus laevis have shown that specific substitutions in positions 1, 5, 6, or 8 of this region can significantly alter the selectivity toward K+. One of these substitutions, the replacement of Ser by Gly at position 6 of the channel, is identical to the substitution we found in one of our individuals.14 Other substitutions at position 3 or 7 showed no functional expression or failed to produce ionic current. Therefore, the specific sequence encompassing the region of the selectivity filter is an important determinant of K+ selectivity and channel functioning.
GIRK channels have recently acquired increasing recognition for their role in health and disease.13 According to the Human Protein Atlas15,16 (see Web Resources), KCNJ6 (GIRK2) is present at high levels in most mammalian tissues, where it participates in many physiological processes by associating with other channel proteins to form homo- or hetero-multimeric complexes. Neurons express GIRK1, KCNJ6 (GIRK2), and GIRK3. KCNJ6 (GIRK2) forms homotetramers and heterotetramers with GIRK1 and GIRK3 in the central nervous system17,18 and thus represents an important mechanism for the modulation of the expression level and function of neurotransmitter-coupled, inwardly rectifying K+ channels.19 The p.Gly154Ser substitution in KCNJ6 (GIRK2) subunits was reproduced in frog oocytes and led to an abnormal pattern for this K+ channel both alone and in combination with other GIRKs.20
An animal model related to KPLBS is the Weaver (wv) mouse.21 This well-studied model, carrying a Gly-to-Ser substitution (p.Gly156Ser, which aligns to the human p.Gly154Ser; Table S6) in the selectivity filter channel, arose spontaneously in the pedigreed expansion stocks of C57BL/6J at Jackson Laboratory in 1961 (see Web Resources). Mice homozygous for the wv mutation (Kcnj6wv/Kcnj6wv) have small size, gait instability, weakness, and hypotonia. Their cerebellum is small, simple, and almost devoid of granule cells, which degenerate during the second week. Heterozygote mice (Kcnj6wv/Kcnj6+) display some neurological abnormalities, including abnormal locomotor behavior; disruptions of Purkinje-cell alignment and morphology; cell-layer irregularities;18 and ectopic, stunted, and disorganized dendrites. Cultured granule cells from wild-type, wv/+, and wv/wv cerebella show that the wv mutation significantly reduces G-protein-activated potassium currents, mostly in homozygous neurons.22 Cerebellar granule cells fail to differentiate in affected mice, which die in the first three weeks of life. Neuronal death is mainly attributable to the loss of KCNJ6 (GIRK2)-mediated currents, resulting in excessive neuronal depolarization and excitability and epilepsy.
The importance of membrane polarization in neuronal functions has also been investigated in a KCNJ6 (GIRK2) triploid mouse model, where gain of function significantly reduces neural activity in similarity to what occurs in individuals affected by Down syndrome.23 These subjects show defects in hippocampal-dependent learning and memory, altered responses to reward, hampered depotentiation, excitatory synaptic plasticity (i.e., altered glutamatergic transmission), and accentuated long-term synaptic depression. However, fully elucidating the role of KCNJ6 (GIRK2) channel in Down-syndrome-affected individuals will require further studies.
In agreement with the pathogenic role of K+ channels in KPLBS, recent studies have shown that voltage-gated K+ channels (Kv) are implicated in the control of some cellular processes, including adipogenic and osteogenic differentiation of human mesenchymal stem cells (hMSCs) 24,25 and proliferation of immature neural progenitor cells.26 In particular, the blockade of Kv channels decreases the extent of adipogenic differentiation of hMSCs and white adipose cells from preadipocytes.24 In addition, in the developing embryo, immature cells exhibit strongly depolarized membrane potentials, whereas terminally differentiated quiescent cells exhibit strongly hyperpolarized membrane potentials.25 Membrane depolarization by high concentrations of extracellular K+ suppresses the adipogenic markers PPARG (peroxisome proliferator-activated receptor gamma), LPL (lipoprotein lipase), and the specific marker IBSP (integrin-binding sialoprotein) during osteogenic differentiation. Finally, depolarization exerts its effect in the early differentiation process.25 Altogether, these data suggest that membrane potential is mandatory for differentiation and cellular plasticity. Disruption of the membrane potential and impaired differentiation of adipocytes at the beginning of embryogenesis could be responsible for the abnormal distribution of subcutaneous fat in our three individuals.
The identification of two identical deletions and one missense mutation in three unrelated individuals is really a surprising, ultra-rare finding. Notwithstanding this, our finding further emphasizes the functional importance of the KCNJ6 (GIRK2) channel in the onset of KPLBS. In fact, owing to the high sequence conservation of the selectivity filter of KCNJ6 (GIRK2), the substitution of each of these amino acids is deemed crucial for the correct functioning of the channel. A suitable animal model for the in-frame deletion is currently not available, and this prevents us from drawing conclusive explanations. In any case, the two different substitutions we found in the selectivity-filter region support this hypothesis. Furthermore, we would suggest that these substitutions determine the formation of a protein with a dominant-negative function because all the individuals have a wild-type allele. In the central nervous system, this effect could be even more detrimental because KCNJ6 (GIRK2) is able to form heterotetramers with GIRK1 and GIRK3, and these heterotetramers are most likely involved in other important biological processes. However, fully elucidating the mutation mechanism and the involvement of this channel in other fundamental pathways that are so far unexplored will require further studies.
In conclusion, KPLBS is a channelopathy caused by KCNJ6 (GIRK2) mutations. Our study suggests that KCNJ6 (GIRK2) could be a candidate for other lipodystrophies and should prompt investigations in this so-far-unexplored class of inwardly rectifying K+ channels.
Acknowledgments
The authors are indebted to the families who contributed to this study. The financial support of the Italian Ministry of Health and the company BVLGARI is fully acknowledged.
Contributor Information
Andrea Masotti, Email: andrea.masotti@opbg.net.
Xun Xu, Email: xuxun@genomics.cn.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
The ExAC Browser, http://exac.broadinstitute.org/
The Human Protein Atlas, www.proteinatlas.org
OMIM, http://www.omim.org/
Weaver mouse at Jackson Laboratory, http://jaxmice.jax.org/strain/000247.html
References
- 1.Gorlin R.J., Cohen M.M., Hennekam R.C.M. Oxford University Press: Oxford; 2001. Syndromes of the Head and Neck. [Google Scholar]
- 2.De Brasi D., Brunetti-Pierri N., Di Micco P., Andria G., Sebastio G. New syndrome with generalized lipodystrophy and a distinctive facial appearance: confirmation of Keppen-Lubinski syndrome? Am. J. Med. Genet. A. 2003;117A:194–195. doi: 10.1002/ajmg.a.10936. [DOI] [PubMed] [Google Scholar]
- 3.Basel-Vanagaite L., Shaffer L., Chitayat D. Keppen-Lubinsky syndrome: Expanding the phenotype. Am. J. Med. Genet. A. 2009;149A:1827–1829. doi: 10.1002/ajmg.a.32975. [DOI] [PubMed] [Google Scholar]
- 4.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li M.X., Gui H.S., Kwan J.S., Bao S.Y., Sham P.C. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 2012;40:e53. doi: 10.1093/nar/gkr1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li M.X., Kwan J.S., Bao S.Y., Yang W., Ho S.L., Song Y.Q., Sham P.C. Predicting mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet. 2013;9:e1003143. doi: 10.1371/journal.pgen.1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu W., O’Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J., NHLBI Exome Sequencing Project Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whorton M.R., MacKinnon R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 2011;147:199–208. doi: 10.1016/j.cell.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whorton M.R., MacKinnon R. X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature. 2013;498:190–197. doi: 10.1038/nature12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hou P., Di A., Huang P., Hansen C.B., Nelson D.J. Impermeability of the GIRK2 weaver channel to divalent cations. Am. J. Physiol. Cell Physiol. 2000;278:C1038–C1046. doi: 10.1152/ajpcell.2000.278.5.C1038. [DOI] [PubMed] [Google Scholar]
- 12.Bezanilla F., Armstrong C.M. Negative conductance caused by entry of sodium and cesium ions into the potassium channels of squid axons. J. Gen. Physiol. 1972;60:588–608. doi: 10.1085/jgp.60.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lüscher C., Slesinger P.A. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 2010;11:301–315. doi: 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heginbotham L., Lu Z., Abramson T., MacKinnon R. Mutations in the K+ channel signature sequence. Biophys. J. 1994;66:1061–1067. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uhlén M., Björling E., Agaton C., Szigyarto C.A., Amini B., Andersen E., Andersson A.C., Angelidou P., Asplund A., Asplund C. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics. 2005;4:1920–1932. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 17.Thiriet M. Springer; New York: 2012. Signaling at the Cell Surface in the Circulatory and Ventilatory Systems. [Google Scholar]
- 18.Signorini S., Liao Y.J., Duncan S.A., Jan L.Y., Stoffel M. Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc. Natl. Acad. Sci. USA. 1997;94:923–927. doi: 10.1073/pnas.94.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kofuji P., Davidson N., Lester H.A. Evidence that neuronal G-protein-gated inwardly rectifying K+ channels are activated by G beta gamma subunits and function as heteromultimers. Proc. Natl. Acad. Sci. USA. 1995;92:6542–6546. doi: 10.1073/pnas.92.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slesinger P.A., Patil N., Liao Y.J., Jan Y.N., Jan L.Y., Cox D.R. Functional effects of the mouse weaver mutation on G protein-gated inwardly rectifying K+ channels. Neuron. 1996;16:321–331. doi: 10.1016/s0896-6273(00)80050-1. [DOI] [PubMed] [Google Scholar]
- 21.Herrup K. The weaver mouse: a most cantankerous rodent. Proc. Natl. Acad. Sci. USA. 1996;93:10541–10542. doi: 10.1073/pnas.93.20.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Surmeier D.J., Mermelstein P.G., Goldowitz D. The weaver mutation of GIRK2 results in a loss of inwardly rectifying K+ current in cerebellar granule cells. Proc. Natl. Acad. Sci. USA. 1996;93:11191–11195. doi: 10.1073/pnas.93.20.11191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooper A., Grigoryan G., Guy-David L., Tsoory M.M., Chen A., Reuveny E. Trisomy of the G protein-coupled K+ channel gene, Kcnj6, affects reward mechanisms, cognitive functions, and synaptic plasticity in mice. Proc. Natl. Acad. Sci. USA. 2012;109:2642–2647. doi: 10.1073/pnas.1109099109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.You M.H., Song M.S., Lee S.K., Ryu P.D., Lee S.Y., Kim D.Y. Voltage-gated K+ channels in adipogenic differentiation of bone marrow-derived human mesenchymal stem cells. Acta Pharmacol. Sin. 2013;34:129–136. doi: 10.1038/aps.2012.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sundelacruz S., Levin M., Kaplan D.L. Membrane potential controls adipogenic and osteogenic differentiation of mesenchymal stem cells. PLoS ONE. 2008;3:e3737. doi: 10.1371/journal.pone.0003737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaarschmidt G., Wegner F., Schwarz S.C., Schmidt H., Schwarz J. Characterization of voltage-gated potassium channels in human neural progenitor cells. PLoS ONE. 2009;4:e6168. doi: 10.1371/journal.pone.0006168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.