

Abstract
Singleton-Merten syndrome (SMS) is an infrequently described autosomal-dominant disorder characterized by early and extreme aortic and valvular calcification, dental anomalies (early-onset periodontitis and root resorption), osteopenia, and acro-osteolysis. To determine the molecular etiology of this disease, we performed whole-exome sequencing and targeted Sanger sequencing. We identified a common missense mutation, c.2465G>A (p.Arg822Gln), in interferon induced with helicase C domain 1 (IFIH1, encoding melanoma differentiation-associated protein 5 [MDA5]) in four SMS subjects from two families and a simplex case. IFIH1 has been linked to a number of autoimmune disorders, including Aicardi-Goutières syndrome. Immunohistochemistry demonstrated the localization of MDA5 in all affected target tissues. In vitro functional analysis revealed that the IFIH1 c.2465G>A mutation enhanced MDA5 function in interferon beta induction. Interferon signature genes were upregulated in SMS individuals’ blood and dental cells. Our data identify a gain-of-function IFIH1 mutation as causing SMS and leading to early arterial calcification and dental inflammation and resorption.
Main Text
Singleton-Merten syndrome (SMS [MIM 182250]) is an infrequently described entity characterized by abnormalities of blood vessels, teeth, and bone.1,2 Calcifications of the aorta and aortic and mitral valves occur in childhood or puberty (Figures 1A and 1B and Movies S1 and S2) and can lead to early demise. Dental findings include a delay in primary tooth exfoliation and permanent tooth eruption, truncated tooth root formation, early-onset periodontal disease, and severe root and alveolar bone resorption associated with dysregulated mineralization3 leading to tooth loss (Figure 1C). Osteoporosis (either generalized or limited to distal extremities), distal-limb osteolysis, widened medullary cavities, and easy tearing of tendons from bone are core osseous manifestations (Figure 1D). Less common features are an unusual face (high anterior hair line, broad forehead, smooth philtrum, and thin upper vermillion), generalized muscle weakness, psoriasis, early-onset glaucoma, and recurrent infections. SMS follows an autosomal-dominant pattern of inheritance and manifests with variable interfamilial and intrafamilial phenotypes.2 The cause is not known, although altered immunological functioning has been suggested.2
Figure 1.
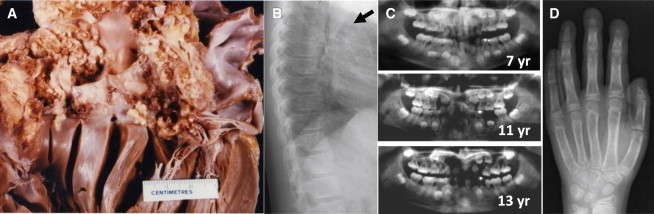
Core Phenotypic Features of SMS
(A) Postmortem heart specimen of subject IV-1 from family 1. Note the widespread, extremely marked calcifications in the outflow tract of the left ventricle, aortic valve, and ascending aorta.
(B) A chest X-ray of subject II-2 from family 3 at 12 years shows aortic calcification (for cardiac angiograms from subject II-1 from family 2, see Movies S1 and S2).
(C) Progressive dental abnormalities observed in subject II-3 from family 2 over a 6-year period. Normal formation of root structures and secondary dentition are shown at 7 years with rapid progressive development of periodontitis and root resorption.
(D) Hand X-ray of subject II-2 from family 3 at age 12 years. Note the marked distal osteolysis and wide medullary cavities.
Images in (A), (B), (D), and part of (C) were reproduced with permission from Feigenbaum et al.2
In an effort to elucidate the molecular basis for SMS, we undertook a broader approach by whole-exome sequencing (WES). We obtained blood samples and pedigrees after acquiring written informed consent from each participant. Approval for this human study was obtained from the ethical committees of the University of Amsterdam and from Muenster University Hospital. Five SMS subjects from three families and four unaffected relatives were included in the WES study (Figure 2A). The clinical features of these SMS-affected families were previously reported by Feigenbaum et al.2 DNA (1 μg) was fragmented by sonification (Covaris), end repaired, and adaptor ligated with the inclusion of sample index barcodes. After size selection, the library was subjected to enrichment with the SeqCap EZ Human Exome Library v.2.0 (Roche NimbleGen). The enriched library was subsequently sequenced on an Illumina HiSeq 2000 sequencing instrument with a paired-end 2 × 100 bp protocol. For the nine exomes, the sequence output was on average 90.3 million reads, and 85% were uniquely mapped. For the 20×, 30×, and mean coverage of target sequences, we obtained average values of 94%, 90%, and 102%, respectively. For data analysis, the Varbank pipeline v.2.3 and filter interface were used. Primary data were filtered according to signal purity by the Illumina Realtime Analysis software v.1.8. Subsequently, the reads were mapped to the human genome reference (UCSC Genome Browser hg19) with the Burrows-Wheeler Aligner.4 The Genome Analysis Toolkit v.1.65 was used to mark duplicated reads, perform local realignments around short indels, recalibrate the base and variant quality scores, and call SNPs and short indels. Scripts developed in house at the Cologne Center for Genomics were applied to detect protein changes, affected donor and acceptor splice sites, and overlaps with known variants. Acceptor and donor splice-site mutations were analyzed with a Maximum Entropy model.6 Filtering of variants was performed as described in Table S1.
Figure 2.
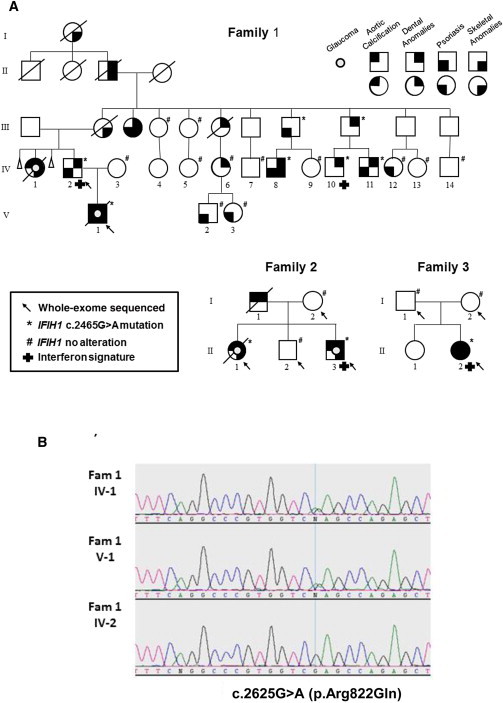
Pedigrees of Studied SMS-Affected Families and the Identified Disease-Causing Germline Mutation c.2625G>A in IFIH1
(A) Families 1 and 2 show an autosomal-dominant mode of inheritance. In family 3, the affected individual is a simplex case identified as having a de novo mutation. Details of the phenotype in these families are available in Feigenbaum et al.2
(B) Sanger sequencing electropherograms of two probands (IV-1 and V-1 from family 1) and one unaffected family member (IV-2) confirm the IFIH1 c.2625G>A (p.Arg822Gln) mutation.
Given the observed autosomal-dominant inheritance pattern, we assumed a de novo variant in the simplex case (family 3). WES data analysis identified putative deleterious mutations in two genes: SPECC1L (MIM 614140) and IFIH1 (interferon induced with helicase C domain 1 [MIM 606951]; RefSeq accession number NM_022168.3; Table S1). Additional analyses showed that the IFIH1 missense mutation, c.2465G>A (p.Arg822Gln), co-segregated with the disorder (Figure 2A). Sanger sequencing in 17 additional members of family 1 validated the IFIH1 variant in all affected subjects and demonstrated segregation in all members with dental anomalies, except in one person (Figures 2A and 2B). One individual with only psoriasis also carried the mutation, whereas three other subjects with only psoriasis harbored no mutation. No unaffected individual carried the variant. The IFIH1 c.2465G>A variant has been reported as a single-nucleotide variation (rs376048533) in 1 of 6,517 individuals sampled in the NHLBI Exome Sequencing Project (ESP) Exome Variant Server cohort populations. The variant is listed in the Exome Aggregation Consortium Browser with an allele frequency of 0.00002481.
Melanoma-differentiation-associated protein 5 (MDA5), encoded by IFIH1, is a member of the RIG-I-like receptor family and functions as a cytoplasmic pattern-recognition receptor recognizing virus double-stranded RNA (dsRNA) and secreted bacterial nucleic acids.7 To determine whether the localization pattern of MDA5 correlated with affected tissues in SMS individuals, we performed fluorescent immunohistochemistry on cryosections from human heart, skin, and cartilage tissues. MDA5 was detected in cardiac myocytes, epidermis, and cartilage chondrocytes (Figure 3A). For analysis of MDA5 localization in developing teeth, paraffin-embedded 28-day postnatal mouse mandibles were fixed and demineralized prior to immunohistochemistry. MDA5 was found to be localized in ameloblasts, odontoblasts, the periodontal ligament, and active osteoblasts at the surface of the alveolar bone (Figure 3B). These results indicate that MDA5 localized to all target tissues altered in SMS.
Figure 3.
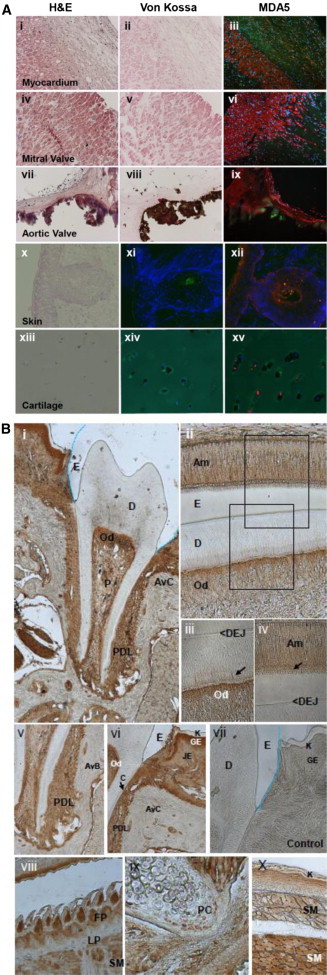
Immunohistochemistry of MDA5 in Human Heart and Skin and Murine Dental Tissues
(A) MDA5 localization in heart, skin, and cartilage. H&E staining shows general morphology (i, iv, vii, x, and xiii), and von Kossa staining demonstrates the presence or absence of hydroxyapatite deposits (ii, v, and viii). MDA5 immunolabeling (iii, vi, ix, xii, and xv) and control immunolabeling with no primary antibody (xi and xiv) are also shown. MDA5 is localized in cardiac myocytes (iii, vi, and ix), the epidermis (xii), and chondrocyctes of the cartilage (xv). Immunofluorescence signal of MDA5 was labeled with rabbit anti-MDA5 antibody (pAb AT113, Enzo LifeScience) at a 1:200 dilution and Cy3-conjugated secondary antibodies (red). DAPI was used to stain nuclei (blue). Autofluorescence of matrix structures is shown in green. The control individual is shown in (i)–(iii), the SMS individual’s mitral valve is shown in (iv)–(vi), and the SMS individual’s aortic valve is shown in (vii)–(ix).
(B) MDA5 localization in mouse 28-day and 7-day postnatal teeth. The rabbit anti-MDA5 antibody (1:200 dilution) was incubated for 1 hr, and the pre-diluted secondary antibody was subsequently incubated with the SuperPicTure Polymer Dectection Kit (Invitrogen) for 30 min at room temperature and visualized with 3-3′ tetrachloride diaminobenzidine (DAB) chromogen (brown) for 1–2 min at room temperature. For the negative controls, BSA replaced the primary antibody step in the protocol. Low magnification of the entire erupted tooth with root structure and periodontal ligament is shown in (i) and (v). Labeled ameloblasts (ii and iv) and odontoblasts (ii and iii) show intracellular labeling and within-odontoblast cell processes (iii, arrow) and ameloblast Tomes’ processes (iv, arrow); no staining is seen in the enamel (ii and iv), dentin (i–iii), or cementum lining the root surface (vi, arrow). Active osteoblasts at the surface of the alveolar bone (v), junctional and gingival epithelium (vi) of the oral epithelium, filiform papilla of the tongue (viii), and skeletal muscle of the tongue (viii) and craniofacial complex (x) express high levels of MDA5. Proliferating chondrocytes in the cartilage (ix) are less intensively stained. The lamina propria of the tongue is negative (vii), as is the negative control (vii). Abbreviations are as follows: E, enamel; D, dentin; Od, odontoblast; P, pulp; AvC, alveolar crest; PDL, periodontal ligament; DEJ, dentin enamel junction; AvB, alveolar bone; C, cementum; JE, junctional epithelium; GE, gingival epithelium; K, keratinized layer; FP, filiform papilla; LP, lamina propria; SM, skeletal muscle; and PC, proliferating chondrocyte.
The SMS-associated amino acid substitution we identified is located in the HEL2 domain, one of the two conserved core helicase domains (Figure 4A), at an evolutionary conserved sequence motif (Figure 4B). The crystal structure of MDA5 shows that this motif is close to the bound ATP analog at the ATPase binding site (Figure 4C). It was suggested that this conserved sequence motif mediates conformational changes triggered by ATP binding to create a high-affinity nucleic acid binding site and thus leads to unwinding.8 Such a conformational change was shown later for a p.Gly821Ser altered MDA5, which resulted in a gain of function and led to spontaneously developed lupus-like autoimmune symptoms in a mouse model.9
Figure 4.
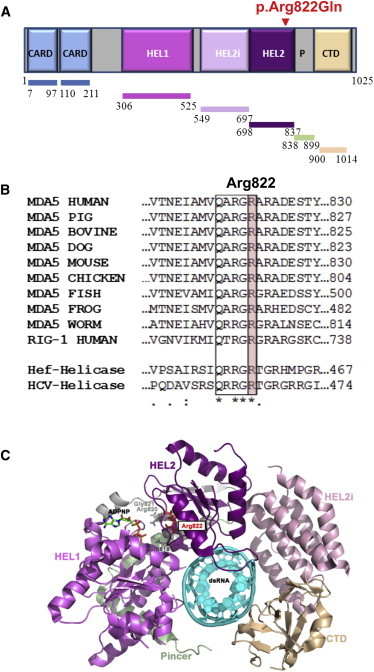
MDA5 Structure Highlighting the Location of the SMS p.Arg822Gln Substitution
(A) Color-coded schematic of the MDA5 structure shows the N-terminal region with two tandem caspase activation and recruitment domains (CARDs), central DExD/H box RNA helicase domains (HEL1 and HEL2), a helicase-insert domain (HEL2i), and a C-terminal domain (CTD), also called the regulatory or repressor domain. The latter two domains are responsible for recognizing dsRNA. The SMS p.Arg822Gln substitution is located near the HEL2 domain termination.
(B) Partial amino acid sequence alignment of human MDA5; representative orthologs show conservation of the residue altered in SMS. Multiple alignments of homologous MDA5 sequence stretches within the helicase CTD for different organisms are compared with human RIG-1, Hef-helicase from the Archaea species Pyrococcus furiosus, and the hepatitis C virus (HCV) helicase. The box indicates the region conserved among the helicase sequences, and the arginine altered in SMS is highlighted in pink. Note: this amino acid is highly conserved across evolution. Species are as follows: pig, Sus scofus; bovine, Bos taurus; dog, Canis familiaris; mouse, Mus musculus; chicken, Gallus gallus; fish, Danio rerio (zebragish); frog, Xenopus laevis (African clawed frog); and worm, Caenorhabditis elegans.
(C) 3D structure of the human MDA5 helicase region in schematic representation (PDB code 4GL2). Note: the N-terminal 2-CARD region (blue in A) is missing from the structure. The helicase domains (amino acids 306–837) are shown in shades of purple, the pincer domain (amino acids 838–899) is colored light green, and the CTD (amino acids 900–1,014) is colored tan. The ATP analog (ADPNP, in stick mode) is colored by element, the conserved amino acids Gln818, Arg820, and Gly821 of the conserved sequence motif (also in stick mode) are colored gray, the altered Arg822 (in stick mode) is shown in red, and the bound dsRNA is shown in cyan in schematic representation. (This image was prepared in PyMOL Molecular Graphics System v.1.5.0.4 [Schrödinger].)
To investigate the effect of the p.Arg822Gln substitution on MDA5 function, we cloned, sequenced, and inserted the IFIH1 open reading frames from one SMS individual and one healthy individual into expression vectors (SMS-IFIH1 and wild-type-IFIH1, respectively). Because MDA5 p.Arg822Gln is the only alteration in common between all SMS-affected individuals and non-affected individuals, we corrected c.2465G>A in the SMS-IFIH1 vector back to normal c.2465A>G (ΔSMS-IFIH1 vector) to see whether this single amino acid change can functionally and dominantly affect MDA5 function. We used HEK293T cells, with the lowest endogenous IFIH1 expression levels (Figure 5A), to overexpress MDA5. After transfection of the wild-type-IFIH1 vector, IFNB1 (interferon, beta 1, fibroblast [MIM 147640]) expression was increased in a dose-dependent manner in HEK293T cells (Figures 5B and 5C). Overexpression of the SMS-IFIH1 vector led to approximately 20× more IFNB1 expression in HEK293T cells expressing the SMS-IFIH1 vector than in cells expressing wild-type IFIH1 (Figure 5C). Overexpression of the ΔSMS-IFIH1 and wild-type-IFIH1 vectors resulted in similar amounts of IFNB1 expression, thus indicating that the p.Arg822Gln substitution is functional and dominant in controlling β-interferon production (Figure 5C). Given that Arg822 is located in one of the motifs implicated in nucleotide binding,10–12 we next tested whether the p.Arg822Gln substitution affects MDA5 function in inducing β-interferon production in cells after non-self-RNA stimulation. We established HEK293T cell lines stably overexpressing the SMS-IFIH1 or ΔSMS-IFIH1 vector by using a lentiviral delivery system and challenged them with the dsRNA analog poly(I:C). As expected, SMS-IFIH1 HEK293T cells had a higher level of IFNB1 expression than did ΔSMS-IFIH1 HEK293T cells after poly(I:C) stimulation (Figure 5D). These data indicate that the SMS-IFIH1 vector is hyperactive to non-self-dsRNA. In agreement with the elevated expression level of IFNB1 by SMS-IFIH1 signaling, there was also higher expression of interferon signature genes (ISGs; IFI27 [MIM 600009], IFI44L [MIM 613975], IFIT1 [MIM 147690], ISG15 [MIM 147571], RSG15, RSAD2 [MIM 607810], and SIGLEC1) in whole-blood samples from SMS individuals as measured by quantitative real-time PCR (Figure 6A). Similar results were obtained with isolated dental enamel organ epithelial cells from extracted SMS and control third molars (Figure 6B). Compared with that of healthy individuals, the interferon score of SMS individuals was significantly altered (p < 0.0001 by unpaired t test; Figure 6C).
Figure 5.
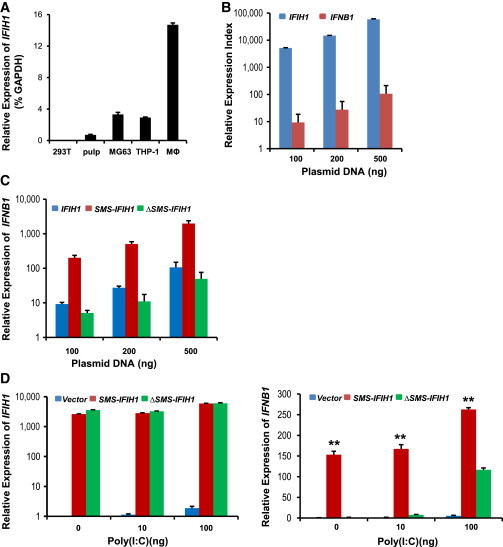
The p.Arg822Gln Substitution in MDA5 Enhances IFNB1 Induction
The coding region of human MDA5 was amplified from dental cells from one SMS individual (SMS-IFIH1) and one unaffected individual (IFIH1) and cloned into pcDNA3.1 (Life Technologies) and pCDH-CMV-MCS-EF1-copGFP (System Biosciences). SMS-IFIH1 c.2465A>G was corrected back to c.2465G>A (ΔSMS-IFIH1) with the GENEART Site-Directed Mutagenesis System according to the manual instructions. The primer sets are detailed in Table S2. For lentivirus generation, recombinant pCDH vector and package vectors were transfected into HEK293T cells. Concentrated virus with SMS-IFIH1, ΔSMS-IFIH1, or empty vector alone was used to infect HEK293T cells for establishing cell lines. Cells were then sorted by flow cytometry at the same fluorescence identity.
(A) IFIH1 was expressed at very low levels in HEK293T cells. Relative expression of endogenous IFIH1 in different types of cells was analyzed by real-time PCR in triplicates and normalized to GAPDH.
(B) IFNB1 was dose-dependently induced in HEK293T cells by transient transfection of IFIH1. HEK293T cells in 24-well plates were transfected with pcDNA3-IFIH1 at the indicated concentration for 24 hr. Relative expression of IFIH1 and IFNB1 was analyzed by real-time PCR in triplicates and normalized to cells transfected with empty vector.
(C) MDA5 with the p.Arg822Gln substitution induced a higher level of IFNB1 in HEK293T cells. HEK293T cells in 24-well plates were transfected with pcDNA3-IFIH1 (IFIH1), pcDNA3-IFIH1 (SMS-IFIH1), and pcDNA3-ΔSMS-IFIH1 (ΔSMS-IFIH1) at the indicated concentration for 24 hr. IFIH1 and IFNB1 mRNA was analyzed by real-time PCR in triplicates and normalized to HEK293T cells transfected with empty vector.
(D) Elevated mRNA level of IFNB1 in SMS-IFIH1 overexpressing HEK293T cells after stimulation with poly(I:C). HEK293T cells with SMS-IFIH1, ΔSMS-IFIH1, or empty vector (vector) were seeded in 24-well plates and transfected with the indicated amount of poly(I:C) for 24 hr. Relative expression of IFIH1 and IFNB1 mRNA was analyzed by real-time PCR in triplicates and normalized to unstimulated HEK293T cells with empty vector. ∗∗p < 0.01 by two-tailed Student’s t test, indicating a significant difference between the SMS-IFIH1 group and the ΔSMS-IFIH1 group.
Error bars represent the SD.
Figure 6.
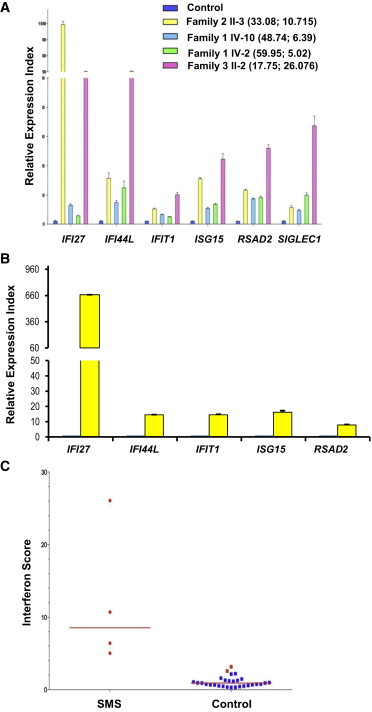
Effects of the SMS-Associated MDA5 Amino Acid Substitution on the Induction of ISGs
(A) Quantitative analysis of ISGs in whole-blood samples from four SMS family members. The expression of IFI27, IFI44L, IFIT1, ISG15, RSG15, RSAD2, and SIGLEC1 was analyzed by qPCR in three technical replicates. The relative expression of each gene in SMS blood samples was normalized to controls and is represented as the mean ± SD. Numbers in parentheses refer to the proband’s age (in years) at sampling (left) and the interferon score calculated from the median of relative change for the tested ISGs (right).
(B) Relative expression of ISGs in SMS versus control primary dental enamel organ epithelial (EOE) cells. Values represent the mean ± SD from two independent experiments, each of which had three technical replicates (n = 6). p values < 0.05 by two-tailed Student’s t test. Note: SIGLEC1 was not detected in the EOE cells.
(C) The interferon score of blood samples in affected (n = 4) and unaffected (n = 29) family members was calculated as the median of relative change for the tested ISGs. The red bar indicates the median interferon score in mutation-positive individuals. The interferon score was significantly higher in the SMS individuals than in the control individuals (unpaired t test p < 0.0001).
IFIH1 has been linked to a number of autoimmune diseases. Genome-wide association studies (GWASs) have associated IFIH1 SNPs with psoriasis risk13,14 and with systemic lupus erythematosus and dilated cardiomyopathy.15,16 RNF114 (MIM 612451), a regulator of RIG-I and MDA5 signaling, has also been identified as a psoriasis susceptibility gene.17 Genetic screens have also identified a number of IFIH1 SNPs, which are associated with resistance to type 1 diabetes due to MDA5 loss of function.18–20 Six IFIH1 point mutations, affecting the coding helicase domains, have been associated with a spectrum of neurological phenotypes, including Aicardi-Goutières syndrome (AGS [MIM 225750]), all showing an upregulated type I interferon response.21,22 On the basis of a similar gain-of-function IFIH1 mutation, SMS is allelic with AGS. However, the SMS phenotype differs substantially from the AGS phenotype. The core SMS findings of aortic calcification, dental anomalies, widened medullary cavities, osteoporosis, and acro-osteolysis have not been reported in AGS individuals, and none of our SMS cohort demonstrated microcephaly, intracranial calcification visible on plain skull X-rays, or significant neurological or developmental manifestations. Glaucoma occurs at low frequency in both entities though,2,23 and a specific search for minimal manifestations of the above signs in both entities has not been undertaken. Additional studies might reveal a continuum of features between AGS and SMS. Alternatively, the p.Arg822Gln substitution might specifically alter MDA5 function and lead to SMS, whereas other IFIH1 mutations cause related but distinct effects on protein function and lead to AGS and related neurological phenotypes. Because type I interferon production is elevated in both syndromes, the latter is more reasonable. Conformational changes by different amino acid substitutions could make MDA5 distinctly active to cytoplasmic dsRNA from self-RNA as well as to exogenous pathogens.
The p.Arg822Gln substitution identified in this study was previously reported in the ESP Exome Variant Server in a single individual with cardiac and pulmonary phenotypes. It could be that this individual demonstrates unrecognized manifestations of SMS or is asymptomatic, as occurred in two members of a recently reported IFIH1-mutation-positive family despite the presence of high sustained induction of interferon signaling.21 We also note significant phenotypic variation present in the SMS-affected family 1. This clinical variability is most likely explainable by modifying genetic factors, epigenetic factors, and exogenous influences such as exposure to different pathogens. The rapid alveolar bone loss in SMS individuals is one of the key characteristics of aggressive periodontitis. Periodontitis is typically caused by oral pathogenic bacteria. We hypothesize that the p.Arg822Gln substitution in MDA5 dysregulates host immune response as a result of hyperactivity to pathogenic viruses or bacteria and/or aberrant cross-talk with other pattern-recognition receptors. Furthermore, expression of the c.2465G>A mutant IFIH1 during development might alter the integrity of mineralized structures of the tooth.
Taken together, our data demonstrate that the MDA5 gain-of-function substitution p.Arg822Gln causes the autosomal-dominant disorder SMS through dysregulation of the human innate immune response. The altered MDA5 triggers type I interferon production, which is increased in SMS individuals and which returns to baseline with correction of the missense mutation. This heightened inflammatory state most likely leads to early-onset aggressive periodontitis and abnormal root resorption. We hypothesize a similar process in cardiac and vascular tissue. Further studies are needed to elucidate the cellular and molecular mechanisms of this SMS-associated MDA5 substitution in contribution to dysregulation of the innate immune response and subsequent disease.
Acknowledgments
We thank the participating families for their generous support, all clinicians for contributing samples, and the University of Alabama at Birmingham School of Dentistry Institute of Oral Health Research and Global Center for Craniofacial Oral and Dental Disorders for partial funding. F.R. and I.B. received funding from the Arbeitsgemeinschaft Pädiatrische Stoffwechselstörungen (Germany). We thank Ulrike Botschen for expert technical assistance and Christine Müller and Juan Dong for data collection. We also thank Johannes Roth, Helmut Wittkowski, and Katazyrna Barczik for fruitful discussions on the immunologic phenotype of the affected individuals.
Contributor Information
Frank Rutsch, Email: rutschf@ukmuenster.de.
Mary MacDougall, Email: macdougall@uab.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/
Exome Aggregation Consortium (ExAC) Browser, http://exac.broadinstitute.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://snp.gs.washington.edu/EVS
UCSC Genome Browser, http://genome.ucsc.edu/
Varbank, https://varbank.ccg.uni-koeln.de/
References
- 1.Singleton E.B., Merten D.F. An unusual syndrome of widened medullary cavities of the metacarpals and phalanges, aortic calcification and abnormal dentition. Pediatr. Radiol. 1973;1:2–7. doi: 10.1007/BF00972817. [DOI] [PubMed] [Google Scholar]
- 2.Feigenbaum A., Müller C., Yale C., Kleinheinz J., Jezewski P., Kehl H.G., MacDougall M., Rutsch F., Hennekam R.C. Singleton-Merten syndrome: an autosomal dominant disorder with variable expression. Am. J. Med. Genet. A. 2013;161A:360–370. doi: 10.1002/ajmg.a.35732. [DOI] [PubMed] [Google Scholar]
- 3.Lu C., Mamaeva O.A., Cui C., Amm H., Rutsch F., MacDougall M. Establishment of Singleton-Merten syndrome pulp cells: evidence of mineralization dysregulation. Connect. Tissue Res. 2014;55(1):57–61. doi: 10.3109/03008207.2014.923880. [DOI] [PubMed] [Google Scholar]
- 4.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeo G., Burge C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 7.Goubau D., Deddouche S., Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall M.C., Matson S.W. Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol. 1999;34:867–877. doi: 10.1046/j.1365-2958.1999.01659.x. [DOI] [PubMed] [Google Scholar]
- 9.Funabiki M., Kato H., Miyachi Y., Toki H., Motegi H., Inoue M., Minowa O., Yoshida A., Deguchi K., Sato H. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity. 2014;40:199–212. doi: 10.1016/j.immuni.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 10.Tanner N.K., Linder P. DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol. Cell. 2001;8:251–262. doi: 10.1016/s1097-2765(01)00329-x. [DOI] [PubMed] [Google Scholar]
- 11.Cordin O., Banroques J., Tanner N.K., Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 12.Wu B., Peisley A., Richards C., Yao H., Zeng X., Lin C., Chu F., Walz T., Hur S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 13.Strange A., Capon F., Spencer C.C., Knight J., Weale M.E., Allen M.H., Barton A., Band G., Bellenguez C., Bergboer J.G., Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheng Y., Jin X., Xu J., Gao J., Du X., Duan D., Li B., Zhao J., Zhan W., Tang H. Sequencing-based approach identified three new susceptibility loci for psoriasis. Nat. Commun. 2014;5:4331. doi: 10.1038/ncomms5331. [DOI] [PubMed] [Google Scholar]
- 15.Dou Q., Peng Y., Zhou B., Lin J., Li Y., Yang H., Xie Q., Li C., Zhang L., Rao L. Association of innate immune IFIH1 gene polymorphisms with dilated cardiomyopathy in a Chinese population. Immunol. Invest. 2014;43:627–637. doi: 10.3109/08820139.2014.909455. [DOI] [PubMed] [Google Scholar]
- 16.Bijlmakers M.J., Kanneganti S.K., Barker J.N., Trembath R.C., Capon F. Functional analysis of the RNF114 psoriasis susceptibility gene implicates innate immune responses to double-stranded RNA in disease pathogenesis. Hum. Mol. Genet. 2011;20:3129–3137. doi: 10.1093/hmg/ddr215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cen H., Leng R.X., Wang W., Zhou M., Feng C.C., Zhu Y., Yang X.K., Yang M., Zhai Y., Li B.Z. Association study of IFIH1 rs1990760 polymorphism with systemic lupus erythematosus in a Chinese population. Inflammation. 2013;36:444–448. doi: 10.1007/s10753-012-9564-0. [DOI] [PubMed] [Google Scholar]
- 18.Smyth D.J., Cooper J.D., Bailey R., Field S., Burren O., Smink L.J., Guja C., Ionescu-Tirgoviste C., Widmer B., Dunger D.B. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 2006;38:617–619. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- 19.Concannon P., Onengut-Gumuscu S., Todd J.A., Smyth D.J., Pociot F., Bergholdt R., Akolkar B., Erlich H.A., Hilner J.E., Julier C., Type 1 Diabetes Genetics Consortium A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes. 2008;57:2858–2861. doi: 10.2337/db08-0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nejentsev S., Walker N., Riches D., Egholm M., Todd J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rice G.I., del Toro Duany Y., Jenkinson E.M., Forte G.M., Anderson B.H., Ariaudo G., Bader-Meunier B., Baildam E.M., Battini R., Beresford M.W. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 2014;46:503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oda H., Nakagawa K., Abe J., Awaya T., Funabiki M., Hijikata A., Nishikomori R., Funatsuka M., Ohshima Y., Sugawara Y. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am. J. Hum. Genet. 2014;95:121–125. doi: 10.1016/j.ajhg.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato M., Suemori H., Hata N., Asagiri M., Ogasawara K., Nakao K., Nakaya T., Katsuki M., Noguchi S., Tanaka N., Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.