

Abstract
PARK2, a gene associated with Parkinson disease, is a tumor suppressor in human malignancies. Here, we show that c.823C>T (p.Arg275Trp), a germline mutation in PARK2, is present in a family with eight cases of lung cancer. The resulting amino acid change, p.Arg275Trp, is located in the highly conserved RING finger 1 domain of PARK2, which encodes an E3 ubiquitin ligase. Upon further analysis, the c.823C>T mutation was detected in three additional families affected by lung cancer. The effect size for PARK2 c.823C>T (odds ratio = 5.24) in white individuals was larger than those reported for variants from lung cancer genome-wide association studies. These data implicate this PARK2 germline mutation as a genetic susceptibility factor for lung cancer. Our results provide a rationale for further investigations of this specific mutation and gene for evaluation of the possibility of developing targeted therapies against lung cancer in individuals with PARK2 variants by compensating for the loss-of-function effect caused by the associated variation.
Main Text
Lung cancer (MIM 211980) is one of the most common and deadly malignancies worldwide1 and originates from complex gene-environment interactions. Prolonged exposure to carcinogens found in tobacco smoke and other environmental carcinogens that interact with various genetic susceptibility factors contribute to lung cancer development in humans. Although germline polymorphisms have been associated with lung cancer susceptibility by multiple genome-wide association studies (GWASs),2–5 little is known about the existence of variants of large effect in familial lung cancer.
PARK2 (MIM 602544; RefSeq accession number NM_004562.2) is known to encode a RING-between-RING-type E3 ubiquitin ligase. Although PARK2 alterations are a causal factor of early-onset Parkinson disease (EOPD [MIM 600116]), generally under a recessive mode of inheritance,6,7 PARK2 functions are also implicated in a wide variety of biological processes regulating cell growth and survival, such as the cell cycle, mitochondria homeostasis, metabolism, xenophagy, protein turnover, and stress response.8 Mutations in PARK2 abrogate the growth-suppressive effects of wild-type PARK2 in different human cancer cell lines, including lung cancer cell lines,9 making PARK2 a candidate gene for involvement in lung cancer risk. In this study, we performed exome sequencing in the probands of a specific lung-cancer-affected family and identified an interesting PARK2 mutation. The follow-up Sanger sequencing screen focusing on PARK2 in a large number of lung-cancer-affected families established that the recurrent germline mutation was significantly associated with familial lung cancer and had a large effect size.
Samples and clinical data used in this study were collected by the following familial lung cancer recruitment sites of the Genetic Epidemiology of Lung Cancer Consortium (GELCC): University of Cincinnati, University of Colorado, Karmanos Cancer Institute, Louisiana State University Health Sciences Center, Mayo Clinic, Johns Hopkins University, and University of Toledo. All sites accrued participants according to institutional review board (IRB)-approved protocols and obtained informed consent from each participant. The Medical College of Wisconsin Cancer Center also maintained an IRB-approved protocol for analysis of the data. We first studied a GELCC family in which eight members across three generations developed lung cancer (family A; Tables S1 and S2). Whole-exome sequencing (WES) was conducted on germline DNA for three lung cancer individuals (III-1, III-4, and IV-18) in this family. The detailed WES procedures can be seen in our previous publication.10 In brief, whole-exome capture was carried out according to the protocol for Agilent’s SureSelect Human All Exon Kit. Then the captured fragments were sequenced on an Illumina HiSeq 2000 with 100 bp paired-end reads. To achieve high-level sensitivity and accuracy for variant detection, we sequenced each sample at a mean depth of 149×. Short sequence reads were aligned to a reference genome (UCSC Genome Browser hg19) with the Burrows-Wheeler Aligner (BWA).11 The BWA assigned each alignment a mapping quality score,11 which is the Phred-scaled probability that the alignment is incorrect. We removed reads with low mapping quality scores (<5) to reduce the false-positive rate. The PCR duplicates were detected and removed by the Picard program. We then performed local realignment of the BWA-aligned reads by using the Genome Analysis Toolkit (GATK).12 GATK was also used for base quality-score recalibration and germline-variant calling. For cross-validation, VarScan 213 was also used to call germline variants according to the local-realignment results. Both GATK and VarScan 2 were utilized to identify the shared genetic variants between family members affected by lung cancer. Default parameters in VarScan 2 were used. The lists of shared single-nucleotide variants and indels were then annotated with ANNOVAR.14
To identify the most significant germline mutations associated with familial lung cancer risk in family A, we applied the following filters for the variants called by our exome sequencing pipeline: (1) the variants had to be present in all three affected family members who were subjected to exome sequencing; (2) variants present in the SNP databases with a minor allele frequency (MAF) > 0.5% were excluded from further studies because they are more likely to be functionally neutral polymorphisms (the SNP databases that we used for filtering are dbSNP build 137, 1000 Genomes, the NHLBI ESP6500 dataset, and the 69-whole-genome dataset [variant calls and allele-frequency information] from Complete Genomics); (3) the variants had to belong to the category of missense or nonsense mutations or indels, which would result in altered or truncated protein products; (4) the variants had to lie in the 6q23–q26 region, where we previously detected significant linkage signals for familial lung cancer in this family (denoted as family 102 in our previous publication) by whole-genome scanning;15,16 (5) the variants had to be predicted to be functionally “not benign” or “not tolerated” and related to cancer by bioinformatics programs such as PolyPhen-2, SIFT, and FATHMM. The workflow of the variant-prioritization steps is shown in Figure 1. Completing the first four filtering steps resulted in five candidate variants in the 6q23–q26 region (Table 1). Among them, the two UTRN (MIM 128240; RefSeq NM_007124.2) variants were not associated with cancer as predicted by FATHMM (scores > −0.75; Table 1), whereas the PACRG (MIM 608427; RefSeq NM_152410.2) variant was predicted to be benign by PolyPhen-2 (score < 0.2), and the NOX3 (MIM 607105; RefSeq NM_015718.2) variant was predicted to be tolerated according to SIFT (score = 1; Table 1). In addition, the UTRN c.7794G>C (p.Gln2598His) variant and the PACRG variant were not at the evolutionary conservation site as predicted by GERP++ (scores < 2; Table 1). Only the PARK2 c.823C>T mutation, which resulted in the substitution of arginine with tryptophan at residue 275, stood out as the most critical germline variant in family A.
Figure 1.
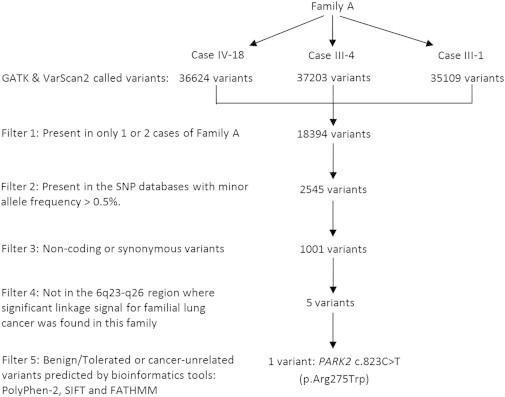
Filtering Strategy in Family A for the Three NSCLC Individuals Subjected to Exome Sequencing
The filtering criteria are written on the left side of the workflow. The number of variants that remained after each filtering step is shown in the workflow.
Table 1.
Characteristics of the Five Candidate Variants in 6q23–q26 for Familial Lung Cancer in Family A
| Gene | Genomic Positiona | Genomic Mutation | Exon | Protein Alteration |
Predicted Effect of Missense Mutation |
dbSNP137 | ESP MAFf | |||
|---|---|---|---|---|---|---|---|---|---|---|
| PolyPhen-2b | SIFTc | FATHMMd | GERP++e | |||||||
| PARK2 | chr6: 162,206,852 | c.823C>T | 7 | p.Arg275Trp | probably damaging (1.00) | deleterious (0.00) | potentially associated with cancer (−3.47) | 4.87 | rs34424986 | 0.002 |
| UTRN | chr6: 144,808,805 | c.3944A>C | 28 | p.Asn1315Thr | probably damaging (0.992) | deleterious (0.00) | not associated with cancer (1.78) | 3.95 | NA | NA |
| UTRN | chr6: 145,021,364 | c.7794G>C | 52 | p.Gln2598His | probably damaging (0.99) | deleterious (0.00) | not associated with cancer (1.58) | −2.12 | NA | NA |
| NOX3 | chr6: 155,776,042 | c.158G>T | 3 | p.Trp53Leu | probably damaging (0.996) | tolerated (1.00) | potentially associated with cancer (−3.48) | 5.91 | rs200865731 | NA |
| PACRG | chr6: 163,149,345 | c.78A>C | 2 | p.Gln26His | benign (0.19) | deleterious (0.01) | not associated with cancer (0.77) | −1.18 | rs80012280 | NA |
Abbreviations are as follows: ESP, NHLBI Exome Sequencing Project; and NA, not available.
Genomic positions are given according to the UCSC Genome Browser hg19 reference assembly.
PolyPhen-2 scores 0.85–1 are interpreted as probably damaging, scores 0.2–0.85 are possibly damaging, and scores 0–0.2 are benign.
SIFT scores range from 0 to 1. The amino acid substitution is predicted to be damaging if the score is ≤0.05 and tolerated if the score is >0.05.
Predictions with FATHMM scores less than −0.75 indicate that the mutation is potentially associated with cancer; otherwise, the mutation is not associated with cancer.
An indication of evolutionary conservation is made if a given site shows a GERP++ score > 2.
MAFs are according to the NHLBI GO Exome Sequencing Project (ESP6500SI-V2 release) Exome Variant Server v.0.0.21 (August 2013).
We next performed Sanger sequencing for PARK2 exon 7 in family A subjects with available DNA. We designed specific primers flanking exon 7 of PARK2. The primer sequences were as follows: 5′-CCAGTTCAACACAATTCCTTCA-3′ (forward) and 5′-ACAACCCTCCAGGATTACAGAA-3′ (reverse). The standard PCR was conducted, and the PCR amplicons were checked by gel electrophoresis and then sent out to Eton Bioscience for Sanger sequencing. The primers for PCR were also used for sequencing reactions. DNA sequences were examined for mutations with the software package Sequencher. An association between the c.823C>T (p.Arg275Trp) mutation and non-small-cell lung cancer (NSCLC [MIM 211980]) was revealed. The c.823C>T (p.Arg275Trp) mutation was detected in all the NSCLC subjects (III-1, III-4, III-7, III-10, and IV-18) in family A (Figures 2 and 3). One person (IV-12) who carried the c.823C>T (p.Arg275Trp) germline mutation had childhood leukemia and died at a relatively young age (44 years old) prior to entering the age range for substantial risk of lung cancer. One small cell lung cancer (SCLC [MIM 182280]) subject (V-1) had wild-type alleles at this locus, suggesting a possible lung-cancer-subtype specificity for the c.823C>T (p.Arg275Trp) allele. No biological specimens were available for the other two SCLC subjects (IV-7 and IV-9). Among the 25 family members who did not have lung cancer at the time of last contact, six had the c.823C>T (p.Arg275Trp) mutation, and 19 had wild-type alleles at this locus (family A in Figure 3).
Figure 2.
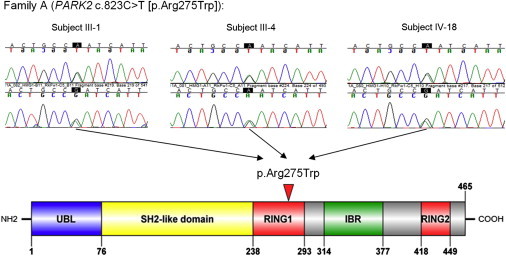
Sanger Sequencing Chromatograms of the PARK2 Mutation and the Plot of the Resulting Amino Acid Change in the PARK2 Domain
The top panel shows the sequencing chromatograms of the PARK2 germline mutation c.823C>T for the three probands in family A, and the bottom panel presents the schematic plot of PARK2 domains and the amino acid change (p.Arg275Trp) resulting from c.823C>T. Chromatograms of both forward and reverse sequences are given, and the complementary-strand alleles (G>A) are shown. The solid red arrowhead shows the location of the resulting amino acid change in PARK2.
Figure 3.
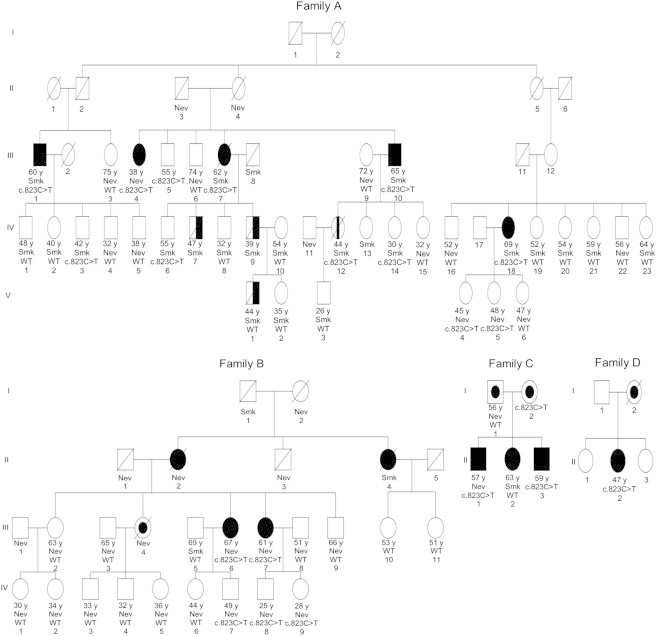
The PARK2 c.823C>T (p.Arg275Trp) Mutation in Four Lung-Cancer-Affected Families
Basic annotations are as follows: the black filled symbols indicate lung-cancer-affected individuals; an oblique line shows deceased family members; the numbers ending with “ y” under each symbol indicate the age at diagnosis (affected individuals) or last contact (control individuals); “Smk” means a current or former smoker; “Nev” indicates someone who never smoked; “c.823C>T” indicates a subject with the heterozygous c.823C>T (p.Arg275Trp) mutation; and “WT” indicates a subject with wild-type alleles at this locus. In family A, five lung-cancer-affected subjects (III-1, III-4, III-7, III-10, and IV-18 [black filled symbols]; age at onset ranging from 38 to 69 years) without SCLC had the c.823C>T (p.Arg275Trp) mutation. WES was performed for affected individuals III-1, III-4, and IV-18. Subject IV-12 (symbol with vertical black bar in the center) had leukemia. Three individuals (IV-7, IV-9, and V-1 [half-black symbols]) were affected by SCLC. Six unaffected individuals (age at exam ranging from 30 to 55 years) had the c.823C>T (p.Arg275Trp) mutation, and 19 unaffected (age at exam ranging from 26 to 75 years) had wild-type alleles at this locus. In family B, two NSCLC-affected individuals (III-6 and III-7) had the c.823C>T (p.Arg275Trp) mutation. Three unaffected individuals (age at exam ranging from 25 to 49 years) had this mutation, and 13 unaffected (age at exam ranging from 30 to 69 years) had wild-type alleles at this locus. Individual III-4 (symbol with an inner black circle) had both lung cancer and melanoma, but her DNA sample was unavailable for genetic testing. In family C, two NSCLC-affected individuals (II-1 and II-3; ages at onset of 57 and 59 years, respectively) had the c.823C>T (p.Arg275Trp) mutation. Individual I-1 (symbol with an inner black circle) had both lung cancer and prostate cancer, and I-2 (symbol with an inner black circle) had uterine cancer and potentially had lung cancer (this is unverified, and this individual has been out of contact since 2002). In family D, individual II-2 had the mutation (age at onset was 47 years) and had lung adenocarcinoma. Individual I-2 (symbol with an inner black circle) had both lung adenocarcinoma and leukemia.
Next, we conducted Sanger sequencing screening of exon 7 of PARK2 in an additional 111 unrelated familial lung cancer subjects and identified two additional families carrying the c.823C>T (p.Arg275Trp) mutation. There were no reported SCLC subjects in these families. As shown in Figure 3 and Figure S1, the mutation was detected in the two lung cancer subjects (III-6 and III-7) in family B and in two lung cancer subjects (II-1 and II-3) in family C. It should be noted that individuals I-1 and II-2 in family C had wild-type alleles at this locus. However, I-1 was affected by both lung cancer and prostate cancer, so it is likely that he and his daughter (II-2) carried a different risk allele for lung cancer than the c.823C>T (p.Arg275Trp) allele in the other two subjects in this bilineal family. We had blood DNA available for a sister and a nephew of I-1 (both unaffected by lung cancer), and sequencing showed that they also had wild-type alleles at this locus, which further strengthened our argument that the father’s side had causes of lung cancer other than the c.823C>T (p.Arg275Trp) mutation. The c.823C>T (p.Arg275Trp) variant was also found in the proband of a biospecimen-limited familial-lung-cancer-affected family (family D), for which only the proband (II-2), who had lung adenocarcinoma, was recruited (Figure 3, Figure S1). This individual was sequenced by an exome sequencing project that screened 55 independent familial lung cancer subjects. These results support PARK2 c.823C > T (p.Arg275Trp) as a lung cancer risk variant. In addition, combining the smoking-status information with the mutation data for the three multiple-case families (A–C in Table S1) showed that the frequency of c.823C>T (p.Arg275Trp) did not vary significantly according to the smoking status of the members in these families (the mutation was present in 8 of 21 smokers [frequency = 0.19] and 10 of 30 people who never smoked [frequency = 0.17]), suggesting that the mutation might act independently of smoking status in increasing lung cancer susceptibility.
The PARK2 c.823C>T (p.Arg275Trp) mutation is extremely rare in the general population; according to the ESP6500 dataset, the MAF of the mutant T allele in all ethnic groups combined is 0.002 (Table 1). We tested the association between this mutation and familial lung cancer. Because the lung-cancer-affected families we studied are all of white ethnicity, in order to exclude the potential confounding effect of ethnicity in the association analysis, we compared the frequencies between our family lung cancer cases consisting of white individuals and the control groups restricted to white ethnicity. First, ESP6500 data showed that the MAF of the c.823C>T (p.Arg275Trp) mutant A allele (the sequence read is the A allele on the complementary strand of the real PARK2 mutant T allele) in 4,300 white individuals is 0.0029 (25/8,600). In addition, an Affymetrix genotyping study in 2,134 healthy control individuals showed with the latest high-throughput Axiom Biobank Arrays that the MAF of the c.823C>T (p.Arg275Trp) mutant allele in 544 white individuals is 0 (0/1,088, see Affymetrix in the Web Resources). We also estimated the frequency by using our own exome-chip data of 2,642 unrelated subjects (1,562 white individuals and 1,080 African Americans) who were ascertained in three eye disease studies and were not known to have lung cancer. Out of the total 3,124 alleles of the 1,562 white individuals, five copies of the c.823C>T (p.Arg275Trp) mutant allele were found, yielding a MAF of 0.0016 (5/3,124). In addition, no copies of the mutant allele were observed in our other exome-chip data from 98 unrelated white probands from 49 independent pedigrees ascertained for a strong history of myopia (MIM 614292) in the Myopia Family Study, making the MAF 0 (0/196). The frequency of the c.823C>T (p.Arg275Trp) mutation in our cohort of independent familial lung cancer subjects of white ethnicity is 0.012 (4 / [112 × 2 + 55 × 2]) and is significantly higher than the combined control white populations’ frequency of 0.0023 (30/13,008, combining 25/8,600 from ESP6500, 0/1,088 from the Affymetrix study, 5/3,124 from the eye disease study, and 0/196 from the Myopia Family Study; p = 0.009; odds ratio [OR] = 5.24; 95% confidence interval = 1.33–15.00; two-tailed Fisher’s exact test). The effect size of the c.823C>T (p.Arg275Trp) mutation is larger than those reported for variants from a lung cancer GWAS.17 These results suggest that carrying this PARK2 mutation is associated with significantly higher odds of developing familial lung cancer in white individuals. We also queried the Exome Aggregation Consortium (ExAC) database, which contains allele-frequency data generated by a wide variety of large-scale projects that sequenced a total of 61,486 exomes. It was found that in the European (non-Finnish) population, the c.823C>T (p.Arg275Trp) variant had a MAF of 0.0031 in 209/66,834 chromosomes. This is a little bigger than the frequency in our control sample (MAF = 0.0023). However, the ExAC dataset contains data from thousands of exomes (contributed by TCGA [The Cancer Genome Atlas]) from cancer-affected individuals; therefore, the estimated frequency of the c.823C>T (p.Arg275Trp) mutation according to the ExAC dataset is biased and not ideal for comparison with our lung cancer subjects.
A recent study showed that PARK2 acts as a master regulator of the stability of G1/S cyclins and functions as the tumor suppressor mediating the coordination of different classes of cyclins.18 Such coordination is fundamentally important for cell-cycle regulation and tumor suppression, and loss-of-function mutations in PARK2 have been associated with different types of human cancers.18 The p.Arg275Trp substitution is located in the highly conserved and functionally important RING finger 1 domain (amino acids 238–293; Figure 2) of PARK2.9 Previous studies have shown that it causes subcellular mislocalization of PARK2 and aggresome formation in normal cell lines, suggesting that it manifests as a loss-of-function alteration.19,20 It is located at the same amino acid residue as that affected by a somatic mutation implicated in lung cancer, i.e., c.824G>A (p.Arg275Gln).9 The latter has been shown to significantly decrease PARK2’s E3 ligase activity, compromise its ability to ubiquitinate cyclin E, result in mitotic instability, and eliminate the tumor-suppressive effect of the wild-type PARK2 in multiple lung cancer cell lines.9 This was confirmed by an independent study, which further showed that the inactivating c.824G>A (p.Arg275Gln) mutation resulted in the accumulation of cyclin D and cyclin E and subsequent highly accelerated cell-cycle progression leading to tumorigenesis.18,21 On the other hand, a logical explanation for the dysfunction of the aggregation-prone PARK2 missense mutation (c.823C>T [p.Arg275Trp]) is based on the impairment of the ubiquitin-proteasome system by protein aggregation, either by exhaustion of one or more factors (such as chaperones) required for normal function or by a “clogged” proteasome.22 Aggregation induced by PARK2 c.823C>T (p.Arg275Trp), even in cells with heterozygous mutation status, could eventually result in severe proteasomal dysfunction,19,20,23,24 which could also cause the accumulation of excessive cyclin D and cyclin E, as c.824G>A (p.Arg275Gln) did, and finally lead to the deregulated G1/S-phase cyclin turnover and ultimately cancer.18,21 In addition, cBioPortal for Cancer Genomics25 shows that PARK2 somatic mutations are found in 2.7%–3.5% of lung adenocarcinoma, 5.6% of lung squamous cell carcinoma, and 4.8% of SCLC (Figure S2). Interestingly, the list of PARK2 somatic mutations identified by TCGA includes a protein-coding mutation that affects amino acid residue 275 and results in a substitution of arginine with proline (p.Arg275Pro, highlighted in red in Table S3), again indicating the involvement of this locus in lung tumorigenesis. Taken together, all the evidence suggests that residue Arg275 of PARK2 is critical to both lung cancer susceptibility and tumorigenesis.
We designed 12 pairs of PCR primers (Table S4) for the amplification and Sanger sequencing of all 12 exons of PARK2 in the 112 unrelated subjects with familial lung cancer, which revealed four additional PARK2 missense mutations in the lung-cancer-affected families in whom the c.823C>T (p.Arg275Trp) variant was not present (Table S5). These variants cause PARK2 alterations p.Arg256Cys, p.Asp280Asn, p.Ser223Leu, and p.Arg402Cys, which are also predicted in silico by bioinformatics tools to be damaging and potentially associated with cancer (Table S5). Three of the variants (c.766C>T [p.Arg256Cys], c.838G>A [p.Asp280Asn], and c.1204C>T [p.Arg402Cys]) are rare mutations already recorded in public databases, whereas the c.668C>T (p.Ser223Leu) variant is the de novo mutation we report here (Table S5). However, they are not recurring mutations, as c.823C>T (p.Arg275Trp) was in our dataset, and it is not feasible to conduct appropriate cosegregation analysis in these families because each family has only one affected proband with an available DNA sample. We present the three pedigrees with the corresponding genotype information for the c.766C>T (p.Arg256Cys), c.838G>A (p.Asp280Asn), and c.668C>T (p.Ser223Leu) mutations in Figure S3. The pedigree information for the proband bearing c.1204C>T (p.Arg402Cys) was not available, although we archived the DNA sample from this proband.
The families in our collection have not reported a history of Parkinson disease (PD [MIM 168600]), excluding the possible confounding effect of PD on our study. A literature search revealed an inverse association between PD and spontaneous lung cancer. Such association was largely from strong epidemiologic evidence suggesting that the most prominent lung cancer risk factor, smoking, is protective against PD.26–30 Yet, studies have also shown that cohorts of individuals with PD do reveal a significant increase in the risk of malignancies such as melanoma and brain and breast cancers.30,31 Because the PARK2 mutations were generally not analyzed in the previous epidemiology studies of PD, the association status of PARK2 with lung cancer risks in PD-affected individuals is unknown. However, lung cancer and EOPD have different ranges of age of onset. EOPD occurs within the age range of 21–40 years,32 whereas only about 2.3% of lung cancer occurs before age 44 (see the corresponding link in the Web Resources). In our studied lung-cancer-affected families, only one individual (III-4 in family A) was diagnosed with NSCLC before age 40 (onset age of 38 years), whereas all other subjects were in the onset age range of 47–84 (mean onset age of 62 years). So, individuals with the PARK2 variant and EOPD might not reach the onset age for developing lung cancer, which could explain the possible situation that no increased risk of lung cancer has been found for them yet.
RGS17 (MIM 607191; RefSeq NM_012419.4) is one potential candidate gene underlying the 6q linkage to familial lung cancer, as we reported before.16 However, using exome sequencing, we did not find any functional RGS17 variants that were shared among lung cancer subjects in family A. In this family, we only detected one synonymous RGS17 mutation (c.54T>G), which did not lead to an amino acid change (p.=). Our previous studies already showed that the linkage to lung cancer in family A spanned a broad genomic region of 6q23–q26, including 6q26, which harbors PARK2.15,16 Our systematic variant-prioritization steps for family A, as shown in Figure 1, guaranteed that the PARK2 c.823C>T (p.Arg275Trp) variant was the best candidate in the region 6q23–q26. Therefore, this functional variant in PARK2 is more likely than other variants to account for the lung cancer susceptibility in family A.
Through WES of individuals from a multiple-case lung-cancer-affected family and the follow-up Sanger sequencing screen of additional lung-cancer-affected families, we identified a recurrent loss-of-function germline mutation, i.e., c.823C>T (p.Arg275Trp), in PARK2 in four lung-cancer-affected families. Although common variants (MAF > 1%) associated with lung cancer have been detected by GWASs, a considerable proportion of “missing heritability” is still unexplained and might be due to undiscovered rare sequence variants. In a recent study, two rare mutations (MAF < 0.5%) causing alterations in proteins encoded by BRCA2 (MIM 600185) and CHEK2 (MIM 604373) were revealed to have a large effect on the risk of spontaneous squamous lung cancer: BRCA2 c.9976A>T (p.Lys3326∗) (rs11571833; OR = 2.47; p = 4.74 × 10−20; 95% confidence interval = 2.04–3.00) and CHEK2 c.470T>C (p.Ile157Thr) (rs17879961; OR = 0.38; p = 1.27 × 10−13; 95% confidence interval = 0.29–0.49).17 The effect size of PARK2 c.823C>T (p.Arg275Trp) (OR = 5.24) on familial lung cancer is larger than the effect sizes of the BRCA2 and CHEK2 rare variants on spontaneous lung cancer. Our finding is similar to that of an exome sequencing study that revealed the large effect of MITF (MIM 156845) c.1075G>A (p.Glu318Lys) on familial melanoma (OR = 8.37; 95% confidence interval = 2.58–23.80).33 Our data warrant further research into the deleterious PARK2 mutations or other PARK2 genetic variants for exploration of the potential feasibility of developing targeted therapies against lung cancer in individuals with PARK2 variants by compensating for the loss-of-function effect caused by the corresponding variation.
Acknowledgments
This work was supported in part by grants and contracts from the NIH (R01CA134433, R01CA134682, R01CA113793, R01CA129533, U01CA76293, P30-ES006096, RO1CA127219, N01-HG-65404, U19CA148127, P30CA023108, K07CA181480, and HHSN268201200007C), the Intramural Research Program of the NIH National Human Genome Research Institute, and the Advancing a Healthier Wisconsin fund. C.S. is supported in part by the National Lung Cancer Partnership, Free to Breathe, and the Lung Cancer Initiative of North Carolina.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Affymetrix Axiom Biobank study on p.Arg275Trp, https://www.affymetrix.com/analysis/netaffx/mappingfullrecord.affx?pk=Axiom_BioBank1:AX-82942564/
The Cancer Genome Atlas, http://cancergenome.nih.gov/
cBioPortal for Cancer Genomics, http://www.cbioportal.org/public-portal
Eton Bioscience, http://etonbio.com/
ExAC Browser, http://exac.broadinstitute.org/
FATHMM, http://fathmm.biocompute.org.uk/
NCBI Gene, http://www.ncbi.nlm.nih.gov/gene/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Information about average age for lung cancer, http://lungcancer.about.com/od/lungcancerfacts/f/What-Is-The-Average-Age-For-Lung-Cancer.htm
OMIM, http://www.omim.org/
PolyPhen-2, http://genetics.bwh.harvard.edu./pph2/
Sequencher, http://genecodes.com/
SIFT, http://sift.jcvi.org/
UCSC Human Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
VarScan 2, http://varscan.sourceforge.net/index.html
References
- 1.Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J. Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Amos C.I., Wu X., Broderick P., Gorlov I.P., Gu J., Eisen T., Dong Q., Zhang Q., Gu X., Vijayakrishnan J. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Z., Wu C., Shi Y., Guo H., Zhao X., Yin Z., Yang L., Dai J., Hu L., Tan W. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat. Genet. 2011;43:792–796. doi: 10.1038/ng.875. [DOI] [PubMed] [Google Scholar]
- 4.Thorgeirsson T.E., Geller F., Sulem P., Rafnar T., Wiste A., Magnusson K.P., Manolescu A., Thorleifsson G., Stefansson H., Ingason A. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. doi: 10.1038/nature06846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y., Broderick P., Webb E., Wu X., Vijayakrishnan J., Matakidou A., Qureshi M., Dong Q., Gu X., Chen W.V. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat. Genet. 2008;40:1407–1409. doi: 10.1038/ng.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 7.Lücking C.B., Dürr A., Bonifati V., Vaughan J., De Michele G., Gasser T., Harhangi B.S., Meco G., Denèfle P., Wood N.W., French Parkinson’s Disease Genetics Study Group. European Consortium on Genetic Susceptibility in Parkinson’s Disease Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 8.Xu L., Lin D.C., Yin D., Koeffler H.P. An emerging role of PARK2 in cancer. J. Mol. Med. 2014;92:31–42. doi: 10.1007/s00109-013-1107-0. [DOI] [PubMed] [Google Scholar]
- 9.Veeriah S., Taylor B.S., Meng S., Fang F., Yilmaz E., Vivanco I., Janakiraman M., Schultz N., Hanrahan A.J., Pao W. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 2010;42:77–82. doi: 10.1038/ng.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiong D., Li G., Li K., Xu Q., Pan Z., Ding F., Vedell P., Liu P., Cui P., Hua X. Exome sequencing identifies MXRA5 as a novel cancer gene frequently mutated in non-small cell lung carcinoma from Chinese patients. Carcinogenesis. 2012;33:1797–1805. doi: 10.1093/carcin/bgs210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amos C.I., Pinney S.M., Li Y., Kupert E., Lee J., de Andrade M.A., Yang P., Schwartz A.G., Fain P.R., Gazdar A. A susceptibility locus on chromosome 6q greatly increases lung cancer risk among light and never smokers. Cancer Res. 2010;70:2359–2367. doi: 10.1158/0008-5472.CAN-09-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.You M., Wang D., Liu P., Vikis H., James M., Lu Y., Wang Y., Wang M., Chen Q., Jia D. Fine mapping of chromosome 6q23-25 region in familial lung cancer families reveals RGS17 as a likely candidate gene. Clin. Cancer Res. 2009;15:2666–2674. doi: 10.1158/1078-0432.CCR-08-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y., McKay J.D., Rafnar T., Wang Z., Timofeeva M.N., Broderick P., Zong X., Laplana M., Wei Y., Han Y. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet. 2014;46:736–741. doi: 10.1038/ng.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gong Y., Zack T.I., Morris L.G., Lin K., Hukkelhoven E., Raheja R., Tan I.L., Turcan S., Veeriah S., Meng S. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat. Genet. 2014;46:588–594. doi: 10.1038/ng.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cookson M.R., Lockhart P.J., McLendon C., O’Farrell C., Schlossmacher M., Farrer M.J. RING finger 1 mutations in Parkin produce altered localization of the protein. Hum. Mol. Genet. 2003;12:2957–2965. doi: 10.1093/hmg/ddg328. [DOI] [PubMed] [Google Scholar]
- 20.Sriram S.R., Li X., Ko H.S., Chung K.K., Wong E., Lim K.L., Dawson V.L., Dawson T.M. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum. Mol. Genet. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- 21.Bartek J., Hodny Z. PARK2 orchestrates cyclins to avoid cancer. Nat. Genet. 2014;46:527–528. doi: 10.1038/ng.2992. [DOI] [PubMed] [Google Scholar]
- 22.Bence N.F., Sampat R.M., Kopito R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 23.McNaught K.S., Mytilineou C., Jnobaptiste R., Yabut J., Shashidharan P., Jennert P., Olanow C.W. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J. Neurochem. 2002;81:301–306. doi: 10.1046/j.1471-4159.2002.00821.x. [DOI] [PubMed] [Google Scholar]
- 24.Petrucelli L., O’Farrell C., Lockhart P.J., Baptista M., Kehoe K., Vink L., Choi P., Wolozin B., Farrer M., Hardy J., Cookson M.R. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- 25.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernán M.A., Takkouche B., Caamaño-Isorna F., Gestal-Otero J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann. Neurol. 2002;52:276–284. doi: 10.1002/ana.10277. [DOI] [PubMed] [Google Scholar]
- 27.Nefzger M.D., Quadfasel F.A., Karl V.C. A retrospective study of smoking in Parkinson’s disease. Am. J. Epidemiol. 1968;88:149–158. doi: 10.1093/oxfordjournals.aje.a120874. [DOI] [PubMed] [Google Scholar]
- 28.Wirdefeldt K., Adami H.O., Cole P., Trichopoulos D., Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur. J. Epidemiol. 2011;26(1):S1–S58. doi: 10.1007/s10654-011-9581-6. [DOI] [PubMed] [Google Scholar]
- 29.Driver J.A., Logroscino G., Buring J.E., Gaziano J.M., Kurth T. A prospective cohort study of cancer incidence following the diagnosis of Parkinson’s disease. Cancer Epidemiol. Biomarkers Prev. 2007;16:1260–1265. doi: 10.1158/1055-9965.EPI-07-0038. [DOI] [PubMed] [Google Scholar]
- 30.Olsen J.H., Friis S., Frederiksen K., McLaughlin J.K., Mellemkjaer L., Møller H. Atypical cancer pattern in patients with Parkinson’s disease. Br. J. Cancer. 2005;92:201–205. doi: 10.1038/sj.bjc.6602279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Møller H., Mellemkjaer L., McLaughlin J.K., Olsen J.H. Occurrence of different cancers in patients with Parkinson’s disease. BMJ. 1995;310:1500–1501. doi: 10.1136/bmj.310.6993.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quinn N., Critchley P., Marsden C.D. Young onset Parkinson’s disease. Mov. Disord. 1987;2:73–91. doi: 10.1002/mds.870020201. [DOI] [PubMed] [Google Scholar]
- 33.Yokoyama S., Woods S.L., Boyle G.M., Aoude L.G., MacGregor S., Zismann V., Gartside M., Cust A.E., Haq R., Harland M. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. 2011;480:99–103. doi: 10.1038/nature10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.