

Abstract
Aims
Acute coronary syndromes (ACSs) are driven by inflammation within coronary plaque. Interleukin-1 (IL-1) has an established role in atherogenesis and the vessel-response to injury. ACS patients have raised serum markers of inflammation. We hypothesized that if IL-1 is a driving influence of inflammation in non-ST elevation ACS (NSTE-ACS), IL-1 inhibition would reduce the inflammatory response at the time of ACS.
Methods and results
A phase II, double-blinded, randomized, placebo-controlled, study recruited 182 patients with NSTE-ACS, presenting <48 h from onset of chest pain. Treatment was 1:1 allocation to daily, subcutaneous IL-1receptor antagonist (IL-1ra) or placebo for 14 days. Baseline characteristics were well matched. Treatment compliance was 85% at 7 days. The primary endpoint (area-under-the-curve for C-reactive protein over the first 7 days) was: IL-1ra group, 21.98 mg day/L (95%CI 16.31–29.64); placebo group, 43.5 mg day/L (31.15–60.75) (geometric mean ratio = 0.51 mg/L; 95%CI 0.32–0.79; P = 0.0028). In the IL-1ra group, 14-day achieved high-sensitive C-reactive protein (P < 0.0001) and IL-6 levels (P = 0.02) were lower than Day 1. Sixteen days after discontinuation of treatment (Day 30) high-sensitive C-reactive protein levels had risen again in the IL-1ra group [IL-1ra; 3.50 mg/L (2.65–4.62): placebo; 2.21 mg/L (1.67–2.92), P = 0.022]. MACE at Day 30 and 3 months was similar but at 1 year there was a significant excess of events in the IL-1ra group.
Conclusion
IL-1 drives C-reactive protein elevation at the time of NSTE-ACS. Following 14 days IL-1ra treatment inflammatory markers were reduced. These results show the importance of IL-1 as a target in ACS, but also indicate the need for additional studies with anti-IL-1 therapy in ACS to assess duration and safety.
Clinical Trial Registration
EUCTR: 2006-001767-31-GB: www.clinicaltrialsregister.eu/ctr-search/trial/2006-001767-31/GB.
Keywords: Myocardial infarction, Drugs, Interleukins
See page 337 for the editorial comment on this article (doi:10.1093/eurheartj/ehu369)
Introduction
Acute coronary syndromes (ACSs) are common conditions that account for about 15% of all deaths and cause considerable morbidity among survivors. Coronary plaque destabilization, and thus presentation of ACS, is associated with inflammation in the vessel wall1–3 and a systemic inflammatory response. However, it is unknown whether specific anti-inflammatory treatments modify ACS-associated inflammation and subsequent clinical events.
Interleukin-1 (IL-1) is a cytokine at the start of the innate inflammatory response, released mainly from macrophages and monocytes, which transduces signals from bacterial products through pattern recognition receptors, complement activation, and TNFα.4 IL-1 is implicated in arterial wall inflammation in atherosclerosis and is viewed as a therapeutically tractable system. IL-1 is expressed in the vessels of humans with coronary atherosclerosis.5,6 Vessel wall IL-1 increases after balloon injury,7 and inhibition of IL-1 is associated with reduced neointima formation in a variety of experimental models.8 In Apo-E deficient mice interleukin-1 receptor antagonist (IL-1ra) inhibits the formation of intimal fatty streaks9 and genetic deletion of the IL-1 receptor is associated with a reduction in atherosclerosis in response to fat feeding.10
C-reactive protein rises following ACS and elevation of C-reactive protein is associated with future cardiovascular events.11–16 IL-1 induces IL-6 production,16 both of which stimulate hepatocyte production of C-reactive protein. Serum levels of IL-6 are elevated in patients with ACS17 and an elevation is an independent marker of increased mortality in ACS.18 Thus IL-1 may act as a driver of ACS and the associated inflammatory response. IL-1 signalling can be blocked in man by human recombinant IL-1 receptor antagonist (IL-1ra).4 The main objective of this study was to examine the effect of IL-1ra given for 14 days upon high-sensitive C-reactive protein levels in patients with non-ST elevation ACS (NSTE-ACS).
Methods
Study design
The aims and methods of the MRC ILA Heart Study have been described in detail previously and the protocol and statistical analysis plan are available in the online appendix.19 The protocol and statistical analysis plan are available in the online appendix. In brief, this was a randomized, double blind, placebo-controlled multi-centre clinical trial evaluating the effect of a 14-day regimen of IL-1ra on high-sensitive C-reactive protein level after NSTE-ACS. Additional outcomes of interest were levels of von Willebrand factor (vWF), IL-6, troponin, infarct size estimated by cardiac magnetic resonance (CMR) and burden of ischaemia by continuous ECG monitoring. Patients were recruited from five UK hospitals from the cardiac and medical admission wards. The study protocol complies with the Declaration of Helsinki. Ethical approval was obtained from the nationally approved Leeds West Research Ethics Committee and each centre obtained local specific ethical approval. All patients gave fully informed written consent to participate. The study complied with regulatory requirements of the European Union. Adverse events were reviewed by experienced staff blinded to treatment allocation to verify classification and potential association with treatment. An independent data safety monitoring board reviewed safety data throughout the study.
Participants
Eligible patients had typical cardiac chest pain <48 h from onset of symptoms, ECG changes of ischaemia, and an elevated troponin prior to randomization. The main exclusion criteria were ST elevation on the ECG, need for immediate reperfusion, or prior revascularization.
Randomization and masking
Eligible patients were randomly assigned in a 1:1 ratio using a central 24 h telephone system, stratified by study centre to receive either a 14-day supply of the recombinant IL-1ra Anakinra (Kineret™ from Amgen) or matching placebo. Qualified research personnel were masked to treatment and remained so from the time of randomization. Patients were taught to administer their own injections, but in cases where this was not possible, injections were administered by health professionals or trained family members.
Procedures
After randomization, either 100 mg of Anakinra or 100 mg of placebo was injected subcutaneously once daily in the morning into the abdomen or thigh. Other drugs were left to the treating physicians' discretion. Blood was drawn prior to the injection of study drug at the same time each day (±2h), daily for 7 days and then at Days 14 and 30. Measurements were: high-sensitive C-reactive protein at baseline, Days 1–7, 14, and 30; troponin and full blood count at baseline, 1–7, and Day 14; creatinine, electrolytes, liver function tests at baseline, Days 1–7 and 14; IL-6 and vWF at baseline, Days 1–3, Days 14 and 30. Patients were followed-up for 30 days. For measurement of high-sensitive c-reactive protein, troponin, Vwf, and IL-6, blood samples were processed and then frozen at −80°C before transportation on dry ice in batches to the core laboratories.
ST Holter Monitor Sub-study: between Days 2 and 4 post-randomization, patients at one centre (Sheffield) underwent three lead, continuous ECG monitoring for 24 h. These were analysed by an automated algorithm for ST-segment deviation, and independently reviewed by a blinded cardiac physiologist and a physician (see Supplementary material online). Cardiac Magnetic Resonance Sub-study: Patients at one centre (Leeds) were scheduled for baseline CMR imaging during their index admission with a follow-up study at 30 days, using an identical imaging protocol (see Supplementary material online).
The primary endpoint was the log-transformed area-under-the-curve (AUC) of serum high-sensitive C-reactive protein over the first 7 days. Secondary endpoints included high-sensitive C-reactive protein at 7, 14, and 30 days, log transformed AUC of Troponin-I, vWF and IL-6, total duration of ST-segment depression on Holter monitoring, myocardial scar, area at risk, and left-ventricular function determined by CMR. The cumulative incidence of major adverse cardiovascular events (MACE) including death, stroke, and new myocardial infarction were recorded at 30 days, 3 months, and 12 months.
Sample size and statistical analysis
To detect a relative reduction of one-third in the high-sensitive C-reactive protein AUC between the treatment and the placebo groups, which was the reduction seen in IL-1ra treatment of rheumatoid arthritis at this dose (Amgen data on file), with 80% power at a 5% level of significance required 80 patients in each group (n = 160) based upon the log-transformed value of the area under the curve of high-sensitive C-reactive protein and a standardized difference [difference/ standard deviation (D)] of ∼0.45. To allow for data loss of 10% in each arm and two interim analyses, a final sample size of 184 patients was projected with a P-value of <0.045 considered significant.20 Patients with at least four follow-up measurements of high-sensitive C-reactive protein were included in the primary analysis on an intention-to-treat basis and missing data was imputed using a Markov Chain Monte Carlo (MCMC) simulation method.21 A Student's t-test was used to estimate the treatment effect (in form of geometric mean ratios) for the primary endpoint and some secondary endpoints. The time-to-event endpoints were compared using the log-rank test. Other serious adverse events were analysed using a Fisher exact test. All analyses were done using Stata version 10.1 and SAS version 9.1.3.
Results
A total of 182 patients (93 IL-1ra treated, 89 placebo) were recruited at five UK centres between July 2007 and March 2010 and followed for 1 year. Patient flow through the study is summarized in Figure 1.19 Baseline characteristics were well matched between the two groups (Table 1). The mean age was 61 years, there was an excess of males, and 26% had prior MI. In-hospital revascularization rates were similar between groups (Table 4). The patients were well treated with contemporary therapy; at 14 days 95% were taking aspirin, 90% clopidogrel, 95% statins, and 80% ACE-inhibitors or angiotensin receptor blockers. Patients were randomized within 48 h from onset of chest pain and there was no difference in time from onset of chest pain to first dose between group (see Supplementary material online). In total, 85% of patients in the IL-1ra group and 84% in the placebo group received all 7 daily doses of treatment and 81 and 84%, respectively, continued treatment up to 14 days (all comparisons non-significant). Over two-thirds of patients successfully self-injected medication upon discharge home (no difference between groups), the remaining patients medication being administered by the nursing team. The reasons for non-compliance to treatment are shown in the Supplementary material online.
Figure 1.

Consort flow diagram detailing the number of participants who were randomly assigned and followed-up for the duration of the study up to 1 year.
Table 1.
Baseline characteristics of patients
| Variable | Active (n = 93) | Placebo (n = 89) | P-value |
|---|---|---|---|
| Age (years), mean (SD) | 61.4 (11.7) | 61.3 (12.3) | 0.9556 |
| Male, n (%) | 63 (67.7) | 67 (75.3) | 0.3249 |
| White British, (%) | 89 (95.7) | 82 (92.1) | 0.6845 |
| BMI (kg/m2) | 30.0 (7.1) | 28.4 (4.7) | 0.0687 |
| SBP (mmHg) | 131.4 (20.7) | 126.5 (16.7) | 0.0795 |
| DBP (mmHg) | 75.2 (12.5) | 74.3 (11.3) | 0.6063 |
| Current smoking, n (%) | |||
| Current | 34 (36.6) | 31 (34.8) | 0.4140 |
| Ex | 34 (36.6) | 27 (30.3) | |
| Never | 24 (25.8) | 31 (34.8) | |
| NA | 1 (1.1) | 0 (0) | |
| Previous MI, n (%) | 23 (24.7) | 24 (27.0) | 0.9313 |
| Prior stroke, n (%) | 1 (1.1) | 3 (3.4) | 0.3600 |
| Prior TIA, n (%) | 8 (8.6) | 2 (2.2) | 0.0769 |
| Family history of IHD, n (%) | 48 (51.6) | 47 (52.8) | 1.0000 |
| Hypertension, n (%) | 31 (33.3) | 29 (32.6) | 1.0000 |
| Hyperlipidaemia, n (%) | 27 (29.0) | 28 (31.5) | 0.9344 |
| IDDM, n (%) | |||
| Insulin dependent | 2 (2.2) | 2 (2.2) | 0.0876 |
| Non-insulin dependent | 7 (7.5) | 6 (6.7) | |
| Diet controlled | 6 (6.5) | 0 (0) | |
| No history | 77 (82.8) | 81 (91.0) | |
| NA | 1 (1.1) | 0 (0) | |
NA, not applicable; MI, myocardial infarction.
Table 4.
Summary statistics for analysis of MACE and other serious adverse events
| Parameter | Active (n = 93) | Placebo (n = 89) | P-value |
|---|---|---|---|
| Time-to-event analysisa | |||
| MACE at 30 days | 3 (46.4) | 2 (32.2) | 0.6800 |
| MACE at 3 months | 7 (38.0) | 2 (10.7) | 0.0980 |
| MACE at 1 year | 13 (18.9) | 4 (5.4) | 0.0233 |
| Death | 5 (6.8) | 2 (2.7) | 0.2440 |
| MI | 8 (11.4) | 2 (2.7) | 0.0581 |
| Stroke | 1 (1.3) | 0 (0.0) | 0.3205 |
| Event analysisb | |||
| All bleeds | 2 (2.2) | 1 (1.1) | 1.0000 |
| Neutropaenia | 0 (0.0) | 0 (0.0) | NA |
| Serious infection | 2 (2.2) | 1 (1.1) | 1.0000 |
| Revascularization | 84 (90.3) | 76 (85.4) | 0.3664 |
| Any (-cath only) | 60 (64.5) | 54 (60.7) | 0.6468 |
| CABG only | 3 (3.2) | 1 (1.1) | 0.6212 |
| Cath and CABG | 4 (4.3) | 5 (5.6) | 0.7431 |
| Cath and PCI | 53 (57.0) | 48 (53.9) | 0.7656 |
| PCI only | 1 (1.1) | 0 (0.0) | 1.0000 |
| Cath only | 28 (30.1) | 26 (29.2) | 1.0000 |
| Injection site reactions | 24 (25.8) | 4 (4.5) | <0.0001 |
| Other SAE | 30 (32.3) | 21 (23.6) | 0.2478 |
| Cardiac | 19 (20.4) | 14 (15.7) | 0.4463 |
| Non-cardiac | 15 (16.1) | 7 (7.9) | 0.1120 |
| Unable to specify | 2 (2.2) | 5 (5.6) | 0.2704 |
MACE, major adverse cardiovascular event; NA, not applicable.
aP-value was calculated from log-rank test. Number of patients with events (incidence rate/100) is presented.
bP-value was calculated from Fisher exact test. Number of patients with events (%) is presented.
As pre-specified in the statistical analysis plan, only patients with four or more readings of high-sensitive C-reactive protein were included in the primary outcome analysis. Ten patients in the IL-1ra group and 12 in the placebo group had <4 readings, these patients were excluded from the primary outcome analysis. high-sensitive C-reactive protein over the first 7 days are shown in Figure 2. high-sensitive C-reactive protein levels were similar on the first 2 days of treatment but then diverged with suppression in the IL-1ra group. The high-sensitive C-reactive protein AUC for the first 7 days in the IL-1ra group was 21.98 mg day/L (95%CI 16.31–29.64) and 43.50 mg day/L (31.15–60.75) in the placebo group, with the geometric mean ratio between IL-1ra and placebo being 0.51 (95% CI: 0.32–0.79), P = 0.0028 (Table 2 and Figure 2). This suggests that IL-1ra reduced high-sensitive C-reactive protein AUC by almost half over the first 7 days. The significant difference in absolute levels seen at Day 7 was maintained at Day 14. Table 3 shows routine blood results at baseline and Day 14. There was a statistically significant suppression of white cell count throughout treatment but no overt neutropaenia.
Figure 2.
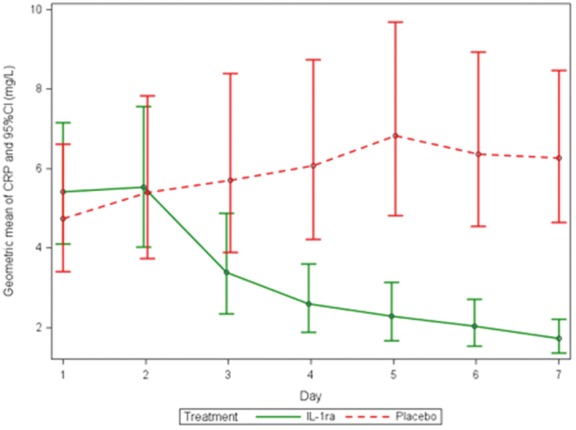
Geometric mean of high sensitivity C-reactive protein over the first 7 days of treatment with IL-1ra or placebo (95% CI) calculated for primary outcome analysis.
Table 2.
Primary and secondary outcome results
| Outcome | Active |
Placebo |
Treatment effect |
|||
|---|---|---|---|---|---|---|
| n | GM (95% CI) | n | GM (95% CI) | GM Ratio (95% CI) | P-value | |
| Imputed high-sensitive C-reactive protein AUC (Days 1–7, mg.day/L)a | 83 | 21.98 (16.31, 29.64) | 77 | 43.50 (31.15, 60.75) | 0.51 (0.32, 0.79) | 0.0028 |
| High-sensitive C-reactive protein at Day 1 (mg/L) | 89 | 5.38 (4.12, 7.04) | 85 | 5.21 (3.75, 7.22) | 1.03 (0.68, 1.57) | 0.88 |
| High-sensitive C-reactive protein at Day 7 (mg/L) | 77 | 1.53 (1.12, 2.11) | 71 | 6.77 (4.92, 9.31) | 0.25 (0.15, 0.35) | <0.0001 |
| High-sensitive C-reactive protein at Day 14 (mg/L) | 80 | 1.74 (1.28, 2.39) | 73 | 4.60 (3.21, 6.59) | 0.38 (0.24, 0.61) | 0.0001 |
| High-sensitive C-reactive protein at Day 30 (mg/L) | 78 | 3.50 (2.65, 4.62) | 74 | 2.21 (1.67, 2.92) | 1.58 (1.07, 2.34) | 0.022 |
| Imputed troponin AUC (Days 1–7, μg.day/L) | 83 | 3.23 (2.15, 4.87) | 77 | 3.83 (2.46, 5.96) | 0.84 (0.46, 1.54) | 0.58 |
| Troponin at Day 1 (μg/L) | 89 | 0.97 (0.63, 1.49) | 85 | 1.25 (0.79, 1.97) | 0.77 (0.41, 1.45) | 0.42 |
| Troponin at Day 7 (μg/L) | 77 | 0.12 (0.08, 0.18) | 71 | 0.14 (0.09, 0.23) | 0.83 (0.43, 1.56) | 0.55 |
| Troponin at Day 14 (μg/L) | 80 | 0.025 (0.017, 0.036) | 73 | 0.026 (0.016, 0.041) | 0.96 (0.54, 1.69) | 0.88 |
| Troponin at Day 30 (μg/L) | 79 | 0.014 (0.001, 0.021) | 74 | 0.012 (0.007, 0.018) | 1.23 (0.71, 2.11) | 0.46 |
| Imputed vWF AUC (Days 1–3, IU.day/mL) | 89 | 2.98 (2.75, 3.22) | 84 | 3.04 (2.83, 3.27) | 0.98 (0.88, 1.09) | 0.68 |
| vWF at Day 1 (IU/mL) | 90 | 1.45 (1.34, 1.56) | 84 | 1.43 (1.32, 1.54) | 1.01 (0.91, 3.39) | 0.80 |
| vWF at Day 14 (IU/mL) | 81 | 1.35 (1.24, 1.47) | 74 | 1.41 (1.29, 1.54) | 0.96 (0.85, 1.08) | 0.48 |
| vWF at Day 30 (IU/mL) | 80 | 1.39 (1.28, 1.51) | 72 | 1.38 (1.26, 1.53) | 1.01 (0.89, 1.14) | 0.92 |
| Imputed IL-6 AUC (Days 1–3, pg.day/mL) | 87 | 8.24 (6.71, 10.11) | 82 | 12.32 (9.66, 15.73) | 0.67 (0.49, 0.92) | 0.012 |
| IL-6 at Day 1 (pg/mL) | 86 | 6.01 (4.68, 7.72) | 81 | 5.23 (3.98, 6.88) | 1.15 (0.80, 1.66) | 0.45 |
| IL-6 at Day 14 (pg/mL) | 76 | 2.51 (2.04, 3.08) | 70 | 3.82 (2.82, 5.16) | 0.66 (0.46, 0.94) | 0.022 |
| IL-6 at Day 30 (pg/mL) | 77 | 3.38 (2.80, 4.16) | 71 | 2.88 (2.38, 3.49) | 1.18 (0.90, 1.56) | 0.22 |
aPrimary outcome excludes patients with <4 high-sensitive C-reactive protein readings.
Table 3.
Routine blood results at baseline and Day 14
| Variable | Baseline |
Day 14 |
||||
|---|---|---|---|---|---|---|
| Active (n = 93) | Placebo (n = 89) | P-value | Active (n = 85) | Placebo (n = 79) | P-value | |
| Haemogloblin (g/dL) | 14.18 ± 1.46 | 14.31 ± 1.44 | 0.54 | 13.65 ± 1.41 | 13.35 ± 1.48 | 0.19 |
| White cell count (×109/L) | 9.16 ± 2.61 | 8.87 ± 2.35 | 0.44 | 7.13 ± 1.70 | 7.78 ± 1.91 | 0.027 |
| Platelets (×109/L) | 266.92 ± 63.77 | 269.70 ± 99.44 | 0.82 | 268.81 ± 75.18 | 299.17 ± 86.91 | 0.022 |
| Urea (mmol/L) | 5.56 ± 1.76 | 5.43 ± 1.89 | 0.64 | 6.19 ± 2.25 | 5.39 ± 2.21 | 0.028 |
| Creatinine (µmol/L) | 91.14 ± 17.71 | 92.05 ± 24.91 | 0.78 | 96.59 ± 20.59 | 93.42 ± 25.18 | 0.40 |
| Sodium (mmol/L) | 138.07 ± 2.98 | 138.00 ± 2.96 | 0.88 | 138.89 ± 3.05 | 138.24 ± 3.80 | 0.25 |
| Potassium mmol/L) | 4.16 ± 0.32 | 4.22 ± 0.37 | 0.29 | 4.39 ± 0.28 | 4.45 ± 0.40 | 0.32 |
| eGFR (mL/min) | 73.36 ± 15.86 | 75.06 ± 21.20 | 0.55 | 69.96 ± 15.48 | 74.25 ± 19.71 | 0.14 |
| Aspartate transaminase (IU/L) | 35.30 (30.74, 40.53) | 36.56 (30.97, 43.15) | 0.74 | 25.82 (23.73, 28.10) | 22.51 (20.86, 24.29) | 0.02 |
| Alkaline transaminase (IU/L)a | 26.34 (23.66, 29.32) | 25.92 (23.39, 28.73) | 0.83 | 29.09 (25.91, 32.67) | 23.64 (21.48, 26.02) | 0.007 |
| Alkaline phosphatise (IU/L)a | 96.77 (86.02, 108.88) | 88.27 (78.80, 98.88) | 0.27 | 88.93 (78.27, 101.04) | 88.80 (77.75, 101.42) | 0.99 |
| Total bilirubin (µmol/L) | 11.04 (10.05, 12.13) | 12.28 (11.07, 13.62) | 0.13 | 11.11 ± 4.99 | 10.71 ± 5.34 | 0.65 |
| CK (IU/L) | 147.28 (110.46, 196.36) | 199.04 (156.50, 253.14) | 0.11 | – | – | – |
| CKMB (µg/L)a | 5.44 (3.57, 8.28) | 4.83 (3.01, 7.78) | 0.71 | – | – | – |
| Troponin I (µg/L)a | 1.40 (0.94, 2.10) | 2.42 (1.58, 3.71) | 0.07 | – | – | – |
| Troponin T (µg/L)a,b | 0.15 (0.05, 0.52) | 0.27 (0.11, 0.67) | 0.39 | – | – | – |
aGeometric mean (95% CI).
bn = 17; 8 active, 9 placebo.
At Day 30, an increase in the absolute high-sensitive C-reactive protein from Day 14 in the IL-1ra treated group to 3.50 mg/L (2.65–4.62) was seen. This is in contrast to placebo treatment where high-sensitive C-reactive protein continued to decline to 2.21 mg/L (1.67–2.92). This resulted in a significant difference between groups at Day 30.
There were no differences in troponin AUC (Days 1–7) or any absolute measures of troponin (see Supplementary material online, Figure). The AUC of IL-6 (Days 1–3) in the IL-1ra group was 8.24 pg day/mL (6.71–0.11) and in the placebo group was 12.32 pg/mL (9.66–15.73) P = 0.012. IL-6 levels remained suppressed by IL-1ra at Day 14 but by Day 30 there was no significant difference between groups. There were no significant differences in any vWF measurements.
Clinical outcomes, MACE, and adverse events are summarized in Table 4 and Figure 3. As expected, there was a significant increase in injection site reactions in the IL-1ra treated group (26 compared with 4% with placebo, P < 0.0001). There was no significant difference in MACE at 30 days or 3 months. Although this study was not powered for the analysis of clinical outcomes, at 1 year there was a significant increase in MACE in the IL-1ra treated group, driven by a non-significant increase in recurrent myocardial infarction.
Figure 3.

Kaplan–Meier event curves for major adverse cardiovascular events, death, myocardial infarction, and stroke by treatment.
There were no significant differences between groups in either of the ST segment Holter or CMR sub-studies (see Supplementary material online).
Discussion
This is the first randomized trial to investigate the effects of a specific inhibitor of interleukin-1 (IL-1ra, Anakinra) in the NSTE-ACS and demonstrates powerful suppression of markers of inflammation in this condition. Widespread coronary inflammatory activation has become the established mechanism of ACS.22,23 The link between this paradigm and blood-based markers of inflammation has been suggested by a number of studies, often based on secondary analysis of large trials of a variety of therapeutic compounds,12 and a 30- patient pilot study investigating the effect of IL-1RA on cardiac remodelling post-STEMI showed a significant reduction in high-sensitive C-reactive protein at 72 h.24 These studies have suggested that relative elevations of high-sensitive C-reactive protein, IL-6, and vWF, among others, may predict a number of adverse outcomes plausibly linked to continued coronary inflammation.12,17,18,25 Prospective studies are based mainly in the stable population where small elevations of high-sensitive C-reactive protein appear to predict continued risk of AMI and other CV events although this effect may be small.15,26 Others, looking specifically at the time of ACS, have not seen a predictive property of high-sensitive C-reactive protein.27
This study indicates that IL-1 drives a significant part of the elevation of high-sensitive C-reactive protein that occurs at the time of NSTE-ACS. The study has examined the treatment effect on high-sensitive C-reactive protein in two ways: the AUC of high-sensitive C-reactive protein (up to Day 7) reflecting total high-sensitive C-reactive protein production in this time, and achieved high-sensitive C-reactive protein at Day 14 (patients were prepared to self-inject for 14 days, but had daily samples drawn for only 7 days). high-sensitive C-reactive protein was rapidly suppressed into the normal range in treated patients and was persistently lower than in the control group for the duration of therapy with IL-1ra. Since, IL-1ra has no known functions other than blockade of the IL-1 signalling receptor and is highly effective therapy in IL-1 driven auto-inflammatory syndromes, these data strongly suggest the importance of IL-1 as a driving influence on inflammation in these patients.
At the time of discontinuation of IL-1ra at 14 days high-sensitive C-reactive protein was suppressed, suggesting inhibition of inflammation, but by Day 30, high-sensitive C-reactive protein levels rose in the IL-1ra treated group, whereas those in the placebo group continued to fall resulting in a significant difference between the groups at 30 days. This indicates a loss of the IL-1ra mediated inhibition of inflammation. These results suggest that either the inflammatory events around the time of NSTE-ACS may continue beyond Day 14 and/or that IL-1-stimulated inflammation may normally drive its own later resolution. Both explanations are plausible. IL-1 receptor occupation leads to a number of events that are associated with either latent IL-1 expression that is not translated or IL-1 production that is not released. It is also possible that withdrawal of IL-1 receptor inhibition leads to increased sensitivity to IL-1 via the loss of endogenous IL1-ra normally driven by IL-1 receptor stimulation.
IL-1ra therapy was in general very well tolerated. There was the expected increase in injection site reactions with the treatment and a mild but significant lowering of the white cell count, which is presumably the withdrawal of IL-1 mediated white cell survival effects.28 This study was not powered for clinical endpoints, however, the MACE composite endpoint at 1 year showed an increase in events in the IL-1ra-treated group. This may be for a number of reasons including pharmacological issues (dose and duration of therapy), or biological consequences of IL-1 inhibition that impairs natural healing driving the late MACE, but this will need to be explored in other appropriately designed studies. Considerable caution, however, should be given to the validity of these MACE observations because of power limitations.
IL-1ra therapy was without effect upon infarct size, as measured by troponin and CMR (see Supplementary material online). There is debate from studies using experimental models of the effect of IL-1 inhibition on infarct size. Our results do not support a reduction in infarct size by IL-1ra in the NSTE-ACS patients we studied, but do not exclude either an effect in more extensive or full-thickness myocardial infarction as may occur in ST elevation ACS. The findings suggest that the effects of IL-1ra are not due to a reduction in infarct size but there is a reduction of the associated inflammation. Whether the inflammatory stimulus derives from the plaque or the myocardium is unknown and unexplored in this study.
This study has some important limitations, which have become apparent given the results. The most obvious is that treatment was given for only one duration—14 days—and our data suggest that a longer duration would have been be desirable. The treatment was given as early as possible but as this was a trial requiring consent, we cannot exclude a difference in results if the IL-1ra had been given immediately by the study team. The size of infarcts is certainly not large, as shown in the CMR substudy. A bigger infarct may have given more opportunity for an effect on infarct size modulation. The study was not powered for late events and as a result we have very few details on the nature of these late events that give rise to the apparent excess MACE at 1 year.
These results, therefore, give both encouragement and potential warning for the use of IL-1 inhibition in patients with coronary artery disease that has presented as ACS. We show that IL-1 is certainly an important mediator of the inflammation at the time of NSTE-ACS and this trial met its primary endpoint. The duration of treatment and perhaps dose and timing of treatment with this inhibitor in ACS may, however, be critical. These results indicate that treatment for 14 days is insufficient to achieve durable suppression of inflammation following discontinuation of the drug. Our results have also indicated the possibility of an increased rate of late re-infarction following a 2-week period of IL-1 inhibition. This demands further study as IL-1 is so clearly an important mediator in ACS as shown by our results. The large prospective study of long-term IL-1 inhibition in patients with CAD in the CANTOS29 study will throw more light on this important area, but additional studies around the time of ACS are now urgently needed.
Statement on analysis error of this manuscript
In July 2010, the preliminary results for the MRC-ILA-HEART Study were prepared and disseminated to authors for review. The Chief Investigator queried the results on the basis of the equivalence of injection site reaction rate with the Clinical Trials and Evaluation Unit (CTEU) in September 2010, which was re-checked and verified by the study statistician. A manuscript was subsequently prepared but was never published. In July 2012, the Chief Investigator requested an audit of the study results, which was undertaken in August 2012. This identified a simple yet critical error in the linkage of the randomization list with the patient ID list which produced an incorrect result. When the analysis was repeated with the correct data, the primary endpoint of the trial had been met as reported here. An abstract of the incorrect data was published (http://heart.bmj.com/content/97/Suppl_1/A13.1) but the error has been notified by Heart.
A subsequent investigation led by the Chair of the trial Data Monitoring Committee in August 2012 confirmed the simple but critical error in the linkage with the randomization list and recommended the re-analysis of the data by the study statistician and also an independent re-analysis by a second statistician. The Trial Steering Committee in a meeting in November 2012 endorsed this course of action. Therefore, D.W. was appointed to conduct an independent analysis of the data. The findings of D.W. were consistent with the revised analysis by the study statistician and it is this verified data that is reported in this manuscript.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This work was supported by UK Medical Research Council Experimental Medicine Grant number G0502131 and a UK Medical Research Council Clinical Research Training Fellowship MR/K002406/1 (A.M.K.R.). IL-1ra (Anakinra) and matching placebo were donated by Amgen Corporation. Funding to pay the Open Access publication charges for this article was provided by the University of Sheffield.
Conflict of interest: D.C.C. has received grant funding from Novartis.
Acknowledgements
We are grateful to all the patients who agreed to participate in this study and all the health professionals who took part.
References
- 1.Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med. 2013;368:2004–2013. doi: 10.1056/NEJMra1216063. [DOI] [PubMed] [Google Scholar]
- 2.Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 3.Glass CK, Witztum JL. Atherosclerosis: the road ahead review. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 4.Dinarello CA, Simon A, van der Meer JWM. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16:1000–1006. doi: 10.1161/01.atv.16.8.1000. [DOI] [PubMed] [Google Scholar]
- 6.Satterthwaite G, Francis SE, Suvarna K, Blakemore S, Ward C, Wallace D, Braddock M, Crossman D. Differential gene expression in coronary arteries from patients presenting with ischemic heart disease: further evidence for the inflammatory basis of atherosclerosis. Am Heart J. 2005;150:488–499. doi: 10.1016/j.ahj.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 7.Chamberlain J, Gunn J, Francis S, Holt C, Crossman D. Temporal and spatial distribution of interleukin-1 beta in balloon injured porcine coronary arteries. Cardiovasc Res. 1999;44:156–165. doi: 10.1016/s0008-6363(99)00175-3. [DOI] [PubMed] [Google Scholar]
- 8.Chamberlain J, Evans D, King A, Dewberry R, Dower S, Crossman D, Francis S. Interleukin-1beta and signaling of interleukin-1 in vascular wall and circulating cells modulates the extent of neointima formation in mice. Am J Pathol. 2006;168:1396–1403. doi: 10.2353/ajpath.2006.051054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elhage R, Maret A, Pieraggi MT, Thiers JC, Arnal JF, Bayard F. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation. 1998;97:242–244. doi: 10.1161/01.cir.97.3.242. [DOI] [PubMed] [Google Scholar]
- 10.Chamberlain J, Francis S, Brookes Z, Shaw G, Graham D, Alp NJ, Dower S, Crossman DC. Interleukin-1 regulates multiple atherogenic mechanisms in response to fat feeding. PLoS ONE. 2009;4:e5073. doi: 10.1371/journal.pone.0005073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG, Pepys MB, Maseri A. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N Engl J Med. 1994;331:417–424. doi: 10.1056/NEJM199408183310701. [DOI] [PubMed] [Google Scholar]
- 12.Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, Pfeffer MA, Braunwald E. C-reactive protein levels and outcomes after statin therapy. N Eng J Med. 2005;352:20–28. doi: 10.1056/NEJMoa042378. [DOI] [PubMed] [Google Scholar]
- 13.Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation. 1998;97:2007–2011. doi: 10.1161/01.cir.97.20.2007. [DOI] [PubMed] [Google Scholar]
- 14.Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–1565. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- 15.Emerging Risk Factors Collaboration. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367:1310–1320. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schindler R, Mancilla J, Endres S, Ghorbani R, Clark SC, Dinarello CA. Correlations and interactions in the production of interleukin-6 (IL-6), IL-1, and tumor necrosis factor (TNF) in human blood mononuclear cells: IL-6 suppresses IL-1 and TNF. Blood. 1990;75:40–47. [PubMed] [Google Scholar]
- 17.Nijm J, Wikby A, Tompa A, Olsson AG, Jonasson L. Circulating levels of proinflammatory cytokines and neutrophil-platelet aggregates in patients with coronary artery disease. Am J Cardiol. 2005;95:452–456. doi: 10.1016/j.amjcard.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Biasucci LM, Liuzzo G, Fantuzzi G, Caligiuri G. Increasing levels of interleukin (IL)-1Ra and IL-6 during the first 2 days of hospitalization in unstable angina are associated with increased risk of in-hospital coronary events. Circulation. 1999;99:2079–2084. doi: 10.1161/01.cir.99.16.2079. [DOI] [PubMed] [Google Scholar]
- 19.Crossman DC, Morton AC, Gunn JP, Greenwood JP, Hall AS, Fox KA, Lucking AJ, Flather MD, Lees B, Foley CE. Investigation of the effect of Interleukin-1 receptor antagonist (IL-1ra) on markers of inflammation in non-ST elevation acute coronary syndromes (The MRC-ILA-HEART Study) Trials. 2008;9:8. doi: 10.1186/1745-6215-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979;35:549–556. [PubMed] [Google Scholar]
- 21.Schafer J. Analysis of Incomplete Multivariate Data. New York: Chapman and Hall; 1997. [Google Scholar]
- 22.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buffon A, Biasucci LM, Liuzzo G, D'Onofrio G, Crea F, Maseri A. Widespread coronary inflammation in unstable angina. N Engl J Med. 2002;347:5–12. doi: 10.1056/NEJMoa012295. [DOI] [PubMed] [Google Scholar]
- 24.Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, Oddi C, Roberts CS, Melchior RD, Mueller GH, Abouzaki NA, Rengel LR, Varma A, Gambill ML, Falcao RA, Voelkel NF, Dinarello CA, Vetrovec GW. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) Pilot Study] Am J Cardiol. 2013;111:1394–1400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montalescot G, Collet JP, Lison L, Choussat R, Ankri A, Vicaut E, Perlemuter K, Philippe F, Drobinski G, Thomas D. Effects of various anticoagulant treatments on von Willebrand factor release in unstable angina. J Am Coll Cardiol. 2000;36:110–114. doi: 10.1016/s0735-1097(00)00695-1. [DOI] [PubMed] [Google Scholar]
- 26.Sever PS, Poulter NR, Chang CL, Hingorani A, Thom SA, Hughes AD, Welsh P, Sattar N on behalf of the ASCOT Investigators. Evaluation of C-reactive protein prior to and on-treatment as a predictor of benefit from atorvastatin: observations from the Anglo-Scandinavian Cardiac Outcomes Trial. Eur Heart J. 2012;33:486–494. doi: 10.1093/eurheartj/ehr262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaski JC, Fernández-Bergés DJ, Consuegra-Sánchez L, Fernández JMC, Garcia-Moll X, Mostaza JM, Cebada RT, Juanatey JRG, Martinez GG, Marrugat J. A comparative study of biomarkers for risk prediction in acute coronary syndrome—results of the SIESTA (Systemic Inflammation Evaluation in non-ST-elevation Acute coronary syndrome) study. Atherosclerosis. 2010;212:636–643. doi: 10.1016/j.atherosclerosis.2010.06.026. [DOI] [PubMed] [Google Scholar]
- 28.Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, Giri JG, Dower SK, Sims JE, Mantovani A. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science. 1993;261:472–475. doi: 10.1126/science.8332913. [DOI] [PubMed] [Google Scholar]
- 29.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]