

Abstract
AIM: To determine how glucocorticoids (GCs) may affect the growth and chemosensitivity of common carcinoma cells.
METHODS: The effect of dexamethasone (DEX) on growth and chemosensitivity was assessed in 14 carcinoma cell lines. The function of GC receptors (GR) was assessed by MMTV reporter assay. Overexpression of GR was done by in vitro transfection and expression of a GR-expressing vector. Immunohistochemical stain of tissues and cells were done by PA1-511A, an anti-GR monoclonal antibody.
RESULTS: DEX inhibited cell growth of four (MCF-7, MCF-7/MXR1, MCF-7/TPT300, and HeLa), increased cisplatin cytotoxicity of one (SiHa), and decreased cisplatin cytotoxicity of two (H460 and Hep3B) cell lines. The GR content of the seven cell lines affected by DEX was significantly higher than those of the seven cell lines unaffected by DEX (5.2±2.5×104 sites/cell vs 1.3±1.4×104 sites/cell, P = 0.005). Only two DEX-unresponsive cell lines (NPC-TW01 and NPC-TW04) contained high GR amounts in the range (1.9-8.1×104 sites/cell) of the seven DEX-responsive cell lines. The GR function of NPC-TW01 and NPC-TW04, however, was found to be impaired. The importance of high cellular amount of GR in mediating DEX susceptibility of the cells was further exemplified by GR dose-dependent drug resistance to cisplatin of AGS, a cell line with low GR content and was unaffected by DEX before transfection of GR-expressing vector. Immunohistochemical studies of human cancer tissues showed that 5 of the 45 (11.1%) breast cancer and 43 of the 85 (50.6%) non-small cell lung cancer had high GR contents at the ranges of the GC-responsive carcinoma cell lines.
CONCLUSION: The growth and chemosensitivity of human carcinomas with high GR contents may be affected by GC. However, in light of the heterogeneous and even contradictive effects of GC on these cells, routine examination of GR contents of human carcinoma tissues may not be clinically useful until other markers that help predict the ultimate effect of GC on individual patients are identified.
Keywords: Glucocorticoids, Glucocorticoid receptor, Carcinoma, Cell growth, Chemosensitivity, Drug resistance
INTRODUCTION
Although glucocorticoids (GCs) are effective in inducing apoptosis via uncharacterized pathways in many hematological malignancies[1], they are generally thought to be ineffective in the treatment of non-hematological solid tumors. However, in the treatment of patients with solid tumors, co-administration of GC with anti-cancer drugs is a common clinical practice to prevent drug-induced allergic reaction or nausea/vomiting. Although GC receptors (GRs) are ubiquitous in cancer cells and have been linked to signal transduction pathways pertinent to their growth, defense, and apoptosis[2-4], little is known regarding the effects of GC on the growth and chemosensitivity of common human carcinomas. Several studies have shown diverse effects of GC on chemosensitivity in non-hematological neoplastic cells. GC has been reported to inhibit cell growth[5-7] and affect the chemosensitivity[8-12] in some sarcoma and carcinoma cell lines. Further, the pretreatment with dexamethasone (DEX) may increase antitumor activity of carboplatin and gemcitabine in tumor xenografts due to increased intra cancer cell drug accumulation was also reported[13]. More comprehensive study is needed to clarify the role of GC on chemosensitivity of non-hematological neoplastic cells.
In the present study, we examined the effects of DEX on the chemosensitivity of 14 carcinoma cell lines. We found that GC exerted a GR-related differential effect on the growth or chemosensitivity of half of carcinoma cells. Further, we have demonstrated that a substantial portion of human carcinomas do contain high GR contents and, therefore, the tumor response to chemotherapy may be affected by the co-administered GC.
MATERIALS AND METHODS
Cell culture and chemicals
SiHa, HeLa, Caski (human cervical carcinoma), H460 (human lung carcinoma), Hep3B, Huh 7 (human hepatocellular carcinoma), and MCF-7 (human breast cancer) were obtained from the American Type Culture Collection (Rockville, MD, USA). MCF-7/MXR1 and MCF-7/TPT300 cells were derived from MCF-7 cells by selection for growth in increasing the concentrations of mitoxantrone or topotecan, respectively. MCF-7/MXR1 cells were gifts from Dr. Kenneth Cowan (National Cancer Institute, USA). MCF-7/TPT300 cells were selected as previously described[14]. NPC-TW01 and NPC-TW04 (nasopharyngeal cancer) were obtained as previously described[15,16]. They were maintained in Dulbecco’s modified Eagle’s medium supplemented with 2 mmol/L glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Sigma Chemical Co., St. Louis, MO, USA), and 10% heat-inactivated fetal bovine serum (Life Technologies, Inc., Gaithersburg, MD, USA). AGS, N87, and SNU1 cells (human gastric cancer) were obtained from the American Type Culture Collection (Rockville, MD, USA) and maintained in RPMI 1640 (Sigma Chemical Co.) supplemented with 2 mmol/L glutamine, 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cisplatin was obtained from Pharmacia-Upjohn (Kalamazoo, MI, USA). DEX was purchased from Sigma Chemical Co., and [3H] DEX (specific activity 35-50 Ci/mmol) was from Blossom Biotechnologies Inc. (Blossom, TX, USA).
Cytotoxicity assay
The in vitro growth inhibitory effects of the drugs in 14 carcinoma cell lines were determined by the MTT assay as previously described with slight modification[17]. Briefly, cells were plated in 96-well plates at 5×103 cells/well. After overnight incubation, various concentrations of drugs were added in triplicate samples to each culture. Cells were exposed to drugs continuously. After 3-4 d of culture, when cells in drug-free wells reached 90% confluency, 50 μL of 2.5 mg/mL MTT (Sigma Chemical Co.) in PBS was added to each well, followed by incubation for 4 h at 37 °C. The formazan crystals were dissolved in DMSO. The absorbance was determined with an ELISA reader at 540 nm. Absorbance values were normalized to the values obtained for the vehicle-treated cells to determine the percentage of surviving cells. Each assay was performed in triplicate.
Measurement of GR content of cancer cell lines by [3H]-labeled ligand binding assay
The GR content was measured by a whole-cell binding assay as previously described with minor modification[18]. Briefly, cells with 90% confluency were subcultured and allowed to grow overnight, and then trypsinized and suspended in a culture medium containing 10% fetal bovine serum (pH 7.2) to a density of 1-10×106 cells/mL. Cells were exposed to various concentrations of [3H] DEX from 1 to 100 nmol/L in the presence or absence of 10 μmol/L unlabeled DEX, followed by incubation for 1 h at 37 °C, and harvested by centrifugation at 1 200 r/min for 1 min. Cells were then washed thrice in 3.0 mL of Hank’s balanced salt solution and finally suspended in 1.6 mL of the same solution. A 0.2-mL aliquot of this suspension was used for the determination of cell number, and 1.0 mL was assayed for radioactivity by a liquid scintillation counter. The presence of at least 200-fold excess of unlabeled DEX effectively competed out all of the binding of [3H] DEX to specific GR. The binding of [3H] DEX to specific GR was represented as the difference in disintegrations per minute per cell between those samples incubated with [3H] DEX alone and those with at least 200-fold excess of unlabeled DEX. Using the specific activity of [3H] DEX, the number of receptors per cell was calculated, assuming that each receptor binds to one DEX molecule.
Examination of endogenous GR function by MMTV reporter assay
The human GR-expressing plasmid, pS-hGR, and the luciferase reporter plasmid, MMTV reporter plasmid were gifts from Prof. Chawnshang Chang (George H Whipple Laboratory for Cancer Research, University of Rochester, Rochester, NY, USA). The MMTV reporter plasmid contains the 1.4-kb MMTV LTR which encompasses the natural GRE sequences[19]. MCF-7, TW01, and TW02 cells were either transiently transfected with MMTV reporter plasmid cells, or co-transfected with MMTV reporter plasmid and pS-hGR (in a ratio of 5:1) by Lipofectamine 2000 (Life Technologies, Inc. [GIBCO BRL], Gaithersburg, MD, USA) according to the manufacturer’s protocol. Forty-eight hours after transfection, 1×105 transfected cells were stimulated with 1 μmol/L DEX and incubated for an additional 6 h. Reporter gene activity was determined with the Luciferase Reporter Assay System (Packard, the Netherlands).
Transfection of GR-expressing vector into cancer cell lines with low GR content
AGS cells were transfected with the pS-hGR by Lipofectamine 2000 (Life Technologies, Inc. [GIBCO BRL], Gaithersburg, MD, USA) according to the manufacturer’s protocol. The stable clones were selected by 400 μg/mL hygromycin for 20 d. Single cell clones were obtained by limiting dilution of the hygromycin-resistant cells. The success of transfection was verified by Western blot analysis of GR and β-actin using rabbit polyclonal antibodies purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) with signals visualized by an enhanced chemiluminescence kit followed by exposure to X-ray films. The GR number in different clones of transfected cells was measured by [3H]-labeled ligand binding assay.
GR immunohistochemistry study in cell lines and human cancer tissue samples
The GR immunocytochemistry study was performed in the 14 carcinoma cell lines. Cells with 90% confluency were subcultured and allowed to grow overnight, then were harvested and centrifuged at 400 r/min for 1 min. The cell pellets were fixed in 4% buffered formalin and embedded in paraffin.
Eighty-five human non-small cell lung cancer and 45 breast cancer tissue specimens which had been fixed in 4% buffered formalin and embedded in paraffin were used for immunohistochemistry study. Use of these tissue materials had followed the regulation of the research ethical committee of the National Taiwan University Hospital.
Five micrometers of paraffin-embedded cell block sections prepared on coated slides (DAKO Ltd.) were dewaxed and rehydrated, and endogenous peroxidase activity was blocked by incubation with 3% H2O2-methanol solution for 10 min. Heat retrieval was used for antigen retrieval. Endogenous biotin was blocked by normal goat serum (DAKO Ltd). Slides were then incubated with anti-GR antibody (PA1-510A, 511A and 512, Affinity BioReagents, Golden, CO, USA). The DAKO Envision system (Copenhagen, Denmark) was used to further avoid endogenous biotin contamination. Sites of bound peroxidase were visualized using liquid 3’,5’-diaminobenzidine (DAKO, Glostrup, Denmark) and counte-rstained with hematoxylin. Under this staining condition, only the carcinoma cells with high (>10 000 sites/cell) GR content were positively stained. Tumor tissue samples were considered to be having high GR expression, when more than 20% of tumor cells were positively stained.
Statistical analysis
Independent t test was used to assess the correlation of GR content with the effect of DEX.
RESULTS
DEX affects either growth or chemosensitivity in 7 of the 14 carcinoma cell lines
DEX (0.01-1.0 μmol/L) inhibited cell growth in MCF-7, MCF-7/MXR1, MCF-7/TPT300, and HeLa cells (Figure 1A). However, DEX alone, up to 20 μmol/L, had no effect on the growth in the other 10 cell lines, including AGS, N87, SNU1, SiHa, Caski, Hep3B, Huh 7, TW01, TW04, and H460 (representative data shown in Figure 1A). The latter 10 carcinoma cell lines were further tested for the effect of GC on the chemosensitivity of carcinoma cells toward cisplatin. Pretreatment of SiHa cells with 1 μmol/L DEX for 3 h decreased the IC50 of cisplatin from 18.6±1.9 to 9.7±2.0 mmol/L (Figure 1B). This cytotoxicity-enhancing effect could be observed even when the concentration of DEX was as low as 1 nmol/L (data not shown). In contrast, DEX slightly increased cisplatin resistance in H460 and Hep3B cells (Figures 1C and D). DEX had no effect on the chemosensitivity of AGS, N87, SNU1, Huh-7, Caski, NPC-TW01, and NPC-TW04 cells (representative data shown in Figure 1E).
Figure 1.
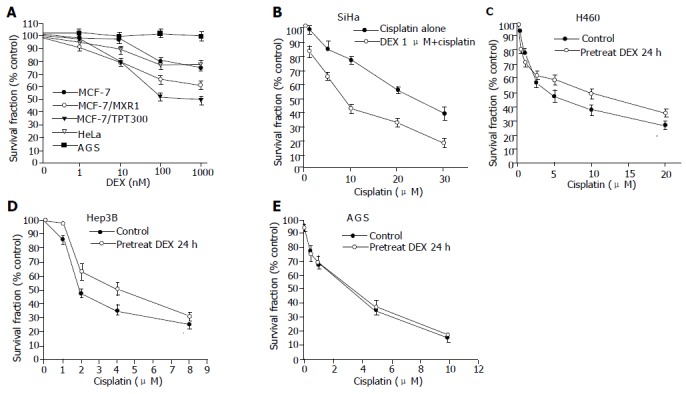
Effect of DEX on the growth and chemosensitivity in carcinoma cell lines. Cell numbers were measured by MTT assay and were plotted as a percentage of the control (cells not exposed to drugs); A: Growth of MCF-7, MCF-7/MXR1, MCF-7/TPT300, and HeLa cells were suppressed by DEX. Data of AGS represent the other 10 cell lines, growth of which was not affected by DEX; B: SiHa cells pretreated with DEX for 3 h were more sensitive to cisplatin; C and D: Pretreatment with DEX 1 mmol/L for 24 h diminished cisplatin cytotoxicity in H460 cells and Hep3B cells; E: Data of AGS represent the seven cell lines (AGS, N87, Caski, Hut 7, SNU1, NPC-TW01, and NPC-TW04), of which cytotoxicity of cisplatin was not affected by DEX. All values represent mean±SD of six separate wells.
Effects of GC correlate well with GR content of the cells
The GR contents of these 14 cell lines are listed in Table 1. The GR contents of the seven cell lines affected by DEX were significantly higher than those of the other seven cell lines unaffected by DEX (5.22.5104 sites/cell vs 1.31.4104 sites/cell, P = 0.005) suggesting GR is one of the pivotal mediators of the effect of DEX on carcinoma cell. The GR content of human lymphocytes, the internal control for these experiments, was tested parallel and was within the reported range (2 500-5 400 sites/cell)[20].
Table 1.
Correlation of GR content with the effect of DEX in carcinoma cells
| Cells | Origin | Effect of DEX | GR (sites/cell) |
| MCF7 | Breast | Growth inhibition | 6.37×104 |
| MCF7/TPT300 | Breast | Growth inhibition | 7.43×104 |
| MCF7/MXR1 | Breast | Growth inhibition | 5.9×104 |
| HeLa | Uterine cervix | Growth inhibition | 1.94×104 |
| SiHa | Uterine cervix | Increased sensitivity toward cisplatin | 8.1×104 |
| H460 | Lung | Increased resistance toward cisplatin | 2.1×104 |
| Hep3B | Liver | Increased resistance toward cisplatin | 4.3×104 |
| NPC-TW01 | Nasopharyngeal | No effect | 4.2×104 |
| NPC-TW04 | Nasopharyngeal | No effect | 2.0×104 |
| Caski | Uterine cervix | No effect | 7.2×103 |
| Huh 7 | Liver | No effect | 7.9×103 |
| AGS | Stomach | No effect | 5.7×103 |
| N87 | Stomach | No effect | 5.0×103 |
| SNU1 | Stomach | No effect | 1.97×103 |
DEX: dexamethasone; GR: glucocorticoid receptor.
GC-unresponsive GR-rich carcinoma cells have dysfunctional GR
As shown in Table 1, DEX had no effect on NPC-TW01 and NPC-TW04, two cell lines with GR content as high as that of the seven GC-responsive cell lines. The function of the GR in these two cell lines was further examined. As shown in Figure 2, MCF-7 cells contained endogenous DEX-responsive GR, while NPC-TW01 and NPC-TW04 cells did not. Further, when NPC-TW01 and NPC-TW04 cells were co-transfected with MMTV reporter plasmid and pS-GR, which contains functional human GR gene, the response to DEX were restored (Figure 2). These data strongly suggested that the function of endogenous GR of NPC-TW01 and NPC-TW04 cells was probably impaired.
Figure 2.
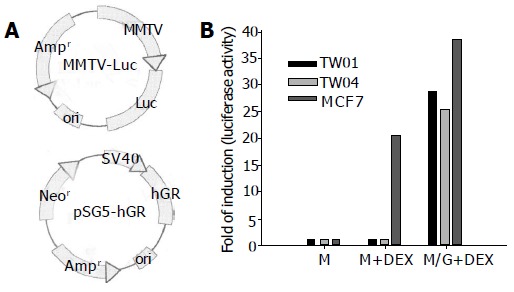
Functional assay of the GR in NPC-TW01 and NPC-TW04 cells. NPC-TW01, NPC-TW04, and MCF-7 cells were transiently transfected with MMTV reporter plasmid (lanes M and M+DEX) or co-transfected with MMTV reporter plasmid and pS-hGR (lane M/G+DEX). The cells were then treated with 1 μmol/L DEX for 6 h (lanes M+DEX and M/G+DEX). Then the luciferase activity was assayed and represented in terms of folds of the induction activity of the control (lane M). All values represent mean±SD of three experiments(A,B).
Expression of GR in GR low-expressing cells increases their responsiveness to DEX
To further examine whether the GR content is pivotal in mediating the susceptibility to DEX in carcinoma cell, pS-hGR was transfected into AGS cells, a GR low-expressing cell line. The GR content in empty vector-transfected AGS cells, and pooled stably pS-hGR-transfected AGS cells were 5.2103 and 1.42104/cell, respectively. The GR contents of clone 1, 2, and 3 were 1.54104, 1.32104, and 5.8103/cell, respectively. Treatment of DEX alone had no effect on cell growth in these cells (data not shown). However, as shown in Figure 3, pS-hGR transfected AGS cells that expressed high GR content became susceptible to the effect of DEX with increasing drug resistance toward cisplatin. The cells that express higher GR content (pooled cells, clones 1 and 2) were more resistant toward cisplatin, while cells with lower GR content (empty vector and clone 3) remained non-susceptible to DEX treatment.
Figure 3.
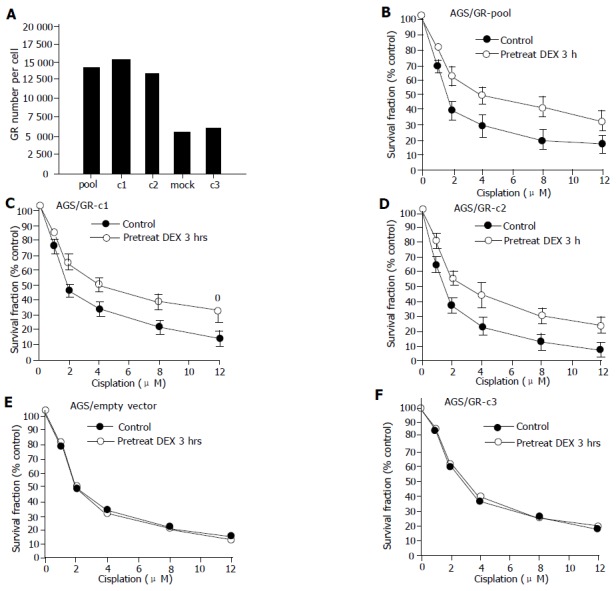
Increased drug resistance to cisplatin in pS-hGR-transfected AGS. AGS cells were transfected with pS-hGR and MTT assay were performed. A: GR number measured by [3H]-labeled ligand binding assay. Pool: AGS/GR-pool; AGS cells transfected with pS-hGR, pooled cells. c1: AGS/GR-c1; AGS cells transfected with pS-hGR, single cell cloned, clone 1. c2: AGS/GR-c2; AGS cells transfected with pS-hGR, single cell cloned, clone 2. Mock: AGS/empty vector; AGS cells transfected with empty vector. c3: AGS/GR-c3; AGS cells transfected with pS-hGR, single cell cloned, clone 3; B-D: Pretreatment with DEX 1 μmol/L for 3 h diminished cisplatin cytotoxicity in AGS/GR-pool, AGS/GR-c1, and AGS/GR-c2 cells; E and F: Pretreatment with DEX 1 μmol/L for 3 h had no effect on the cisplatin cytotoxicity in AGS/empty vector cells and AGS/GR-c3. All values represent mean±SD of six separate wells.
Tumor tissues of common human carcinomas may express high level of GR
GR expression was examined by immunohistochemical stain in the carcinoma cell lines (Figure 4A), and tumor tissue samples of human breast cancer and human non-small cell lung cancer (Figure 4B). Positive GR immunoreactivity (representing high GR content) was observed in 5 of the 45 tumor samples (11.1%) of breast cancer and 43 of the 85 tumor samples (51%) of non-small cell lung cancer patients.
Figure 4.
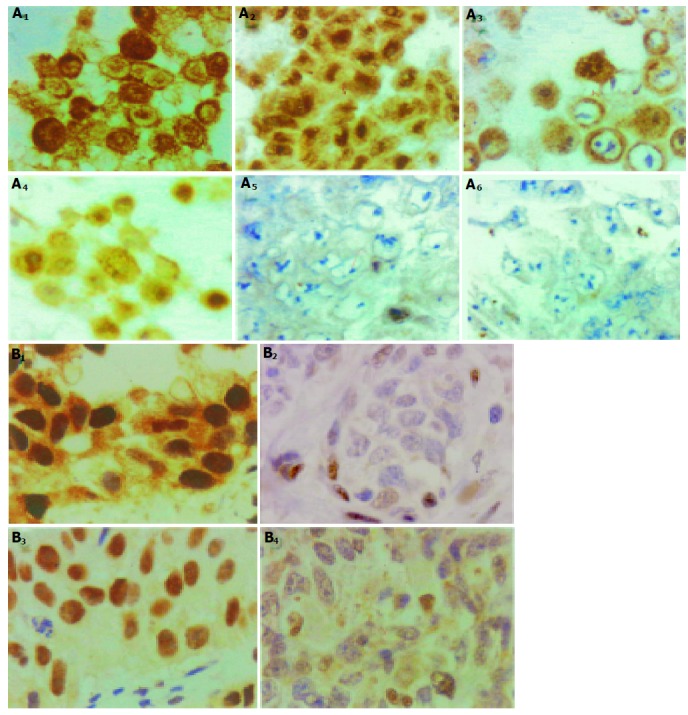
A: Immunocytochemical stain for GR expression in representing carcinoma cell lines. (A1) and A2): SiHa cells, which had GR content about 8.1×104/cell according to ligand binding assay; (A3) and (A4): HeLa cells, with GR content about 1.97×104/cell; (A5) and (A6): N87 cells, with GR content about 5.0×103/cell. (A2), (A4), and (A5): cells treated with DEX for 3 h before harvest. In (A1) and (A3), the GR immunoreactivity localized in cytoplasm. After DEX treatment, the immunoreactive GR translocalized to nuclei (A2) and (A4). The low GR content cancer cells N87 showed negligible immunoreactivity. B: Immunohistochemical stain for GR expression in human carcinoma tumor tissue samples. Non-small cell lung cancer tumor samples, with high GR expression (Ba), and low GR expression (B2). Breast cancer tumor samples, with high GR expression (B3), and low GR expression (B4).
DISCUSSION
This study has demonstrated that GC affects either growth or chemosensitivity in a substantial portion of carcinoma cells. Since GC is commonly co-administered with anticancer drugs such as taxanes and platinums, how GC alters the effect of chemotherapy may have to be taken into consideration in clinical practice. As shown in this study, it may not be difficult to identify those carcinoma patients of whom tumor response is going to be affected by GC, since only cells with high GR content are affected. However, how GC will actually affect the growth of tumors of these cancer patients remains uncertain since the effects of GC, as disclosed in this study, are extremely diverse and even contradictory.
A direct correlation between GR content of the cells and the magnitude of physiologic response to GC has been reported in hematologic malignancies[21]. The sensitivity of many lymphoid cell lines to GC-induced growth arrest and apoptosis is directly proportional to intracellular receptor content[22-24]. Several studies also identified a correlation between reduced GR expression and a poor treatment response as well as poor prognosis in patients with acute lymphocytic leukemia, suggesting that reduced GR expression could lead to clinical GC resistance[22,25-27]. Our study demonstrated that the susceptibility to the effect of DEX on cell growth or chemosensitivity in carcinoma cells also correlate well with the level of GR content. However, the GR contents of the GC-responsive carcinoma cells are almost 10 times higher than that of lymphoid cells[27-29], suggesting that the cellular contexts or the signal transduction pathways for the interaction of GC and GR are probably different between epithelial and lymphoid malignant cells.
In this study, the only two cell lines, NPC-TW01 and NPC-TW04, which have relatively high GR content but not susceptible to the growth regulatory effect of DEX, were found to have non-functional endogenous GR. Previous studies on both human and mouse cell lines have shown that somatic mutation in the GR gene is the principal mechanism for in vitro acquisition of GC resistance[28-30]. However, we failed to detect GR gene mutations in NPC-TW01 and NPC-TW04 (data not shown). Beside the GR number and GR gene mutations, the phosphorylation status of GR has also been reported to be correlated with GR resistance[31]. The expression level of GR co-regulators was also important to normal GR function[32]. Therefore, there should be other mechanisms associated with the malfunction of GR in these two cell lines.
Activated GR may activate or suppress gene expression through interaction with respective positive or negative cis-acting regulatory elements in the promoters regions[33,34]. Activated GR can also regulate the expression of GC responsive genes indirectly through protein-protein interactions with other transcription factors such as NF-κB and AP-1[35-37]. Inactivation of NF-κB or AP-1 has been shown to alter the vulnerability of cancer cells to several cytotoxic agents[38,39]. Our preliminary data have indicated that suppression of NF-κB by GC is one of the major mechanisms of increasing cisplatin sensitivity in SiHa cell[40]. Some studies have demonstrated variable potential other mechanisms of GC related growth arrest in non-hematologic cancer cells that included upregulation of p21Cip1[7], p57 Kip2[8], and inhibition of ERK/MAPK kinase pathway[9], and mechanisms of GC related chemosensitivity alteration include induction of GC-inducible protein kinase-1 (SGK-1) and mitogen-activated protein kinase phosphatase-1 (MKP-1)[14], and modulation of bcl-x expression[11]. However, it remains difficult to explain the diverse and even contradictive effect of GC on GR-rich carcinoma cells. The possibilities that the specific presence of certain co-regulators of GR in different carcinoma cells may dictate the ultimate effect of GC needs to be clarified.
The [3H] labeled DEX ligand binding assay has been the standard method to measure the GR content in cell lines and in vivo human leukemia samples. The GR amount measured by [3H] labeled DEX ligand binding assay is expressed as GR number per cell. However, this method is hard to apply to solid tumor tissue samples, since it is difficult to quantify the cell number in tissue samples. There were other methods to detect the expression of GR in the in vivo solid tumor samples, including cytosol DCC-competitive (dextran-coated charcoal) protein binding assay[41,42], and RT-PCR[43]. These methods all use tissue homogenizer and the results were normalized by the total protein or RNA amount. Therefore, the result was the average of the GR content of the whole tissue sample, which includes the cancer cells as well as the stroma cells and adjacent normal tissues component. Besides, the cytosol DCC-competitive protein binding assay required fresh frozen tissue which limited its applicability. In this study, we have shown an ideal staining condition by which only cancer cells with high GR content were stained positively (Figure 4). We have also demonstrated that a substantial portion of common human carcinoma do express high level of GR, and therefore are potentially susceptible to the growth and chemosensitivity-regulatory effect of GC.
Clinically, the serum concentration of DEX was found to be around 0.12 μmol/L, lasting for 1-3 h, after a single oral dose of 7.5 mg of DEX[44]. However, the serum concentration of DEX may reach 2 μmol/L after a single intravenous infusion of 80-100 mg of DEX[45]. Since the administration of relatively high-dose DEX, at the range of 10-50 mg/d, or its equivalents, has been widely used for the prevention of cisplatin-induced nausea/vomiting and taxanes-induced allergic reactions, the possible effect of GC on the chemosensitivity of some cancer patients needs to be seriously considered.
In summary, the results of this study suggest that GC exerts a GR-dependent effect on the growth or chemosensitivity in a substantial portion of carcinoma cells. The clinical relevance and the cellular mechanisms that dictate the disparate effects of GC need to be further clarified.
Footnotes
Supported by grants from the National Science Council No.NSC 93-2314-B-002-006, Taiwan, and grants from National Taiwan University Hospital 91-N006
Science Editor Guo SY Language Editor Elsevier HK
References
- 1.Frankfurt O, Rosen ST. Mechanisms of glucocorticoid-induced apoptosis in hematologic malignancies: updates. Curr Opin Oncol. 2004;16:553–563. doi: 10.1097/01.cco.0000142072.22226.09. [DOI] [PubMed] [Google Scholar]
- 2.Wright AP, Zilliacus J, McEwan IJ, Dahlman-Wright K, Almlöf T, Carlstedt-Duke J, Gustafsson JA. Structure and function of the glucocorticoid receptor. J Steroid Biochem Mol Biol. 1993;47:11–19. doi: 10.1016/0960-0760(93)90052-x. [DOI] [PubMed] [Google Scholar]
- 3.Schwartzman RA, Cidlowski JA. Glucocorticoid-induced apoptosis of lymphoid cells. Int Arch Allergy Immunol. 1994;105:347–354. doi: 10.1159/000236781. [DOI] [PubMed] [Google Scholar]
- 4.McEwan IJ, Wright AP, Gustafsson JA. Mechanism of gene expression by the glucocorticoid receptor: role of protein-protein interactions. Bioessays. 1997;19:153–160. doi: 10.1002/bies.950190210. [DOI] [PubMed] [Google Scholar]
- 5.Rogatsky I, Trowbridge JM, Garabedian MJ. Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Mol Cell Biol. 1997;17:3181–3193. doi: 10.1128/mcb.17.6.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samuelsson MK, Pazirandeh A, Davani B, Okret S. p57Kip2, a glucocorticoid-induced inhibitor of cell cycle progression in HeLa cells. Mol Endocrinol. 1999;13:1811–1822. doi: 10.1210/mend.13.11.0379. [DOI] [PubMed] [Google Scholar]
- 7.Greenberg AK, Hu J, Basu S, Hay J, Reibman J, Yie TA, Tchou-Wong KM, Rom WN, Lee TC. Glucocorticoids inhibit lung cancer cell growth through both the extracellular signal-related kinase pathway and cell cycle regulators. Am J Respir Cell Mol Biol. 2002;27:320–328. doi: 10.1165/rcmb.4710. [DOI] [PubMed] [Google Scholar]
- 8.Wolff JE, Denecke J, Jürgens H. Dexamethasone induces partial resistance to cisplatinum in C6 glioma cells. Anticancer Res. 1996;16:805–809. [PubMed] [Google Scholar]
- 9.Chang TC, Hung MW, Jiang SY, Chu JT, Chu LL, Tsai LC. Dexamethasone suppresses apoptosis in a human gastric cancer cell line through modulation of bcl-x gene expression. FEBS Lett. 1997;415:11–15. doi: 10.1016/s0014-5793(97)01083-1. [DOI] [PubMed] [Google Scholar]
- 10.Naumann U, Durka S, Weller M. Dexamethasone-mediated protection from drug cytotoxicity: association with p21WAF1/CIP1 protein accumulation? Oncogene. 1998;17:1567–1575. doi: 10.1038/sj.onc.1202071. [DOI] [PubMed] [Google Scholar]
- 11.Benckhuijsen C, Osman AM, Hillebrand MJ, Smets LA. Glucocorticoid effect on melphalan cytotoxicity, cell-cycle position, cell size, and [3H]uridine incorporation in one of three human melanoma cell lines. Cancer Res. 1987;47:4814–4820. [PubMed] [Google Scholar]
- 12.Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64:1757–1764. doi: 10.1158/0008-5472.can-03-2546. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Li M, Rinehart JJ, Zhang R. Pretreatment with dexamethasone increases antitumor activity of carboplatin and gemcitabine in mice bearing human cancer xenografts: in vivo activity, pharmacokinetics, and clinical implications for cancer chemotherapy. Clin Cancer Res. 2004;10:1633–1644. doi: 10.1158/1078-0432.ccr-0829-3. [DOI] [PubMed] [Google Scholar]
- 14.Yang CH, Schneider E, Kuo ML, Volk EL, Rocchi E, Chen YC. BCRP/MXR/ABCP expression in topotecan-resistant human breast carcinoma cells. Biochem Pharmacol. 2000;60:831–837. doi: 10.1016/s0006-2952(00)00396-8. [DOI] [PubMed] [Google Scholar]
- 15.Lin CT, Wong CI, Chan WY, Tzung KW, Ho JK, Hsu MM, Chuang SM. Establishment and characterization of two nasopharyngeal carcinoma cell lines. Lab Invest. 1990;62:713–724. [PubMed] [Google Scholar]
- 16.Lin CT, Chan WY, Chen W, Huang HM, Wu HC, Hsu MM, Chuang SM, Wang CC. Characterization of seven newly established nasopharyngeal carcinoma cell lines. Lab Invest. 1993;68:716–727. [PubMed] [Google Scholar]
- 17.Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936–942. [PubMed] [Google Scholar]
- 18.Harmon JM, Thompson EB. Isolation and characterization of dexamethasone-resistant mutants from human lymphoid cell line CEM-C7. Mol Cell Biol. 1981;1:512–521. doi: 10.1128/mcb.1.6.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheidereit C, Geisse S, Westphal HM, Beato M. The glucocorticoid receptor binds to defined nucleotide sequences near the promoter of mouse mammary tumour virus. Nature. 1983;304:749–752. doi: 10.1038/304749a0. [DOI] [PubMed] [Google Scholar]
- 20.Lippman ME, Yarbro GK, Leventhal BG. Clinical implications of glucocorticoid receptors in human leukemia. Cancer Res. 1978;38:4251–4256. [PubMed] [Google Scholar]
- 21.Vanderbilt JN, Miesfeld R, Maler BA, Yamamoto KR. Intracellular receptor concentration limits glucocorticoid-dependent enhancer activity. Mol Endocrinol. 1987;1:68–74. doi: 10.1210/mend-1-1-68. [DOI] [PubMed] [Google Scholar]
- 22.Gehring U, Mugele K, Ulrich J. Cellular receptor levels and glucocorticoid responsiveness of lymphoma cells. Mol Cell Endocrinol. 1984;36:107–113. doi: 10.1016/0303-7207(84)90089-3. [DOI] [PubMed] [Google Scholar]
- 23.Bourgeois S, Newby RF. Diploid and haploid states of the glucocorticoid receptor gene of mouse lymphoid cell lines. Cell. 1977;11:423–430. doi: 10.1016/0092-8674(77)90060-5. [DOI] [PubMed] [Google Scholar]
- 24.Chapman MS, Askew DJ, Kuscuoglu U, Miesfeld RL. Transcriptional control of steroid-regulated apoptosis in murine thymoma cells. Mol Endocrinol. 1996;10:967–978. doi: 10.1210/mend.10.8.8843413. [DOI] [PubMed] [Google Scholar]
- 25.Bloomfield CD, Smith KA, Peterson BA, Munck A. Glucocorticoid receptors in adult acute lymphoblastic leukemia. Cancer Res. 1981;41:4857–4860. [PubMed] [Google Scholar]
- 26.Pui CH, Dahl GV, Rivera G, Murphy SB, Costlow ME. The relationship of blast cell glucocorticoid receptor levels to response to single-agent steroid trial and remission response in children with acute lymphoblastic leukemia. Leuk Res. 1984;8:579–585. doi: 10.1016/0145-2126(84)90006-7. [DOI] [PubMed] [Google Scholar]
- 27.Pui CH, Costlow ME. Sequential studies of lymphoblast glucocorticoid receptor levels at diagnosis and relapse in childhood leukemia: an update. Leuk Res. 1986;10:227–229. doi: 10.1016/0145-2126(86)90046-9. [DOI] [PubMed] [Google Scholar]
- 28.Wells RJ, Mascaro K, Young PC, Cleary RE, Bachner RL. Glucocorticoid receptors in the lymphoblasts of patients with glucocorticoid-resistant childhood acute lymphocytic leukemia. Am J Pediatr Hematol Oncol. 1981;3:259–264. [PubMed] [Google Scholar]
- 29.Powers JH, Hillmann AG, Tang DC, Harmon JM. Cloning and expression of mutant glucocorticoid receptors from glucocorticoid-sensitive and -resistant human leukemic cells. Cancer Res. 1993;53:4059–4065. [PubMed] [Google Scholar]
- 30.Ashraf J, Thompson EB. Identification of the activation-labile gene: a single point mutation in the human glucocorticoid receptor presents as two distinct receptor phenotypes. Mol Endocrinol. 1993;7:631–642. doi: 10.1210/mend.7.5.8316249. [DOI] [PubMed] [Google Scholar]
- 31.Buttgereit F, Saag KG, Cutolo M, da Silva JA, Bijlsma JW. The molecular basis for the effectiveness, toxicity, and resistance to glucocorticoids: focus on the treatment of rheumatoid arthritis. Scand J Rheumatol. 2005;34:14–21. doi: 10.1080/03009740510017706. [DOI] [PubMed] [Google Scholar]
- 32.McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344. doi: 10.1210/edrv.20.3.0366. [DOI] [PubMed] [Google Scholar]
- 33.Beato M, Chávez S, Truss M. Transcriptional regulation by steroid hormones. Steroids. 1996;61:240–251. doi: 10.1016/0039-128x(96)00030-x. [DOI] [PubMed] [Google Scholar]
- 34.Reichardt HM, Schutz G. Glucocorticoid signaling–multiple variations of a common theme. Mol Cell Endocrinol. 1998;146:1–6. doi: 10.1016/s0303-7207(98)00208-1. [DOI] [PubMed] [Google Scholar]
- 35.McEwan IJ, Wright AP, Gustafsson JA. Mechanism of gene expression by the glucocorticoid receptor: role of protein-protein interactions. Bioessays. 1997;19:153–160. doi: 10.1002/bies.950190210. [DOI] [PubMed] [Google Scholar]
- 36.Dumont A, Hehner SP, Schmitz ML, Gustafsson JA, Lidén J, Okret S, van der Saag PT, Wissink S, van der Burg B, Herrlich P, et al. Cross-talk between steroids and NF-kappa B: what language? Trends Biochem Sci. 1998;23:233–235. doi: 10.1016/s0968-0004(98)01212-2. [DOI] [PubMed] [Google Scholar]
- 37.Göttlicher M, Heck S, Herrlich P. Transcriptional cross-talk, the second mode of steroid hormone receptor action. J Mol Med (Berl) 1998;76:480–489. doi: 10.1007/s001090050242. [DOI] [PubMed] [Google Scholar]
- 38.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 39.Wang CY, Mayo MW, Baldwin AS. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 40.Chuang SE, Yeh PY, Lu YS, Lai GM, Liao CM, Gao M, Cheng AL. Basal levels and patterns of anticancer drug-induced activation of nuclear factor-kappaB (NF-kappaB), and its attenuation by tamoxifen, dexamethasone, and curcumin in carcinoma cells. Biochem Pharmacol. 2002;63:1709–1716. doi: 10.1016/s0006-2952(02)00931-0. [DOI] [PubMed] [Google Scholar]
- 41.Allegra JC, Lippman ME, Thompson EB, Simon R, Barlock A, Green L, Huff KK, Do HM, Aitken SC. Distribution, frequency, and quantitative analysis of estrogen, progesterone, androgen, and glucocorticoid receptors in human breast cancer. Cancer Res. 1979;39:1447–1454. [PubMed] [Google Scholar]
- 42.Liu SH, Otal-Brun M, Webb TE. Glucocorticoid receptors in human tumors. Cancer Lett. 1980;10:269–275. doi: 10.1016/0304-3835(80)90080-4. [DOI] [PubMed] [Google Scholar]
- 43.Smith RA, Lea RA, Curran JE, Weinstein SR, Griffiths LR. Expression of glucocorticoid and progesterone nuclear receptor genes in archival breast cancer tissue. Breast Cancer Res. 2003;5:R9–12. doi: 10.1186/bcr556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weijtens O, Schoemaker RC, Cohen AF, Romijn FP, Lentjes EG, van Rooij J, van Meurs JC. Dexamethasone concentration in vitreous and serum after oral administration. Am J Ophthalmol. 1998;125:673–679. doi: 10.1016/s0002-9394(98)00003-8. [DOI] [PubMed] [Google Scholar]
- 45.Brady ME, Sartiano GP, Rosenblum SL, Zaglama NE, Bauguess CT. The pharmacokinetics of single high doses of dexamethasone in cancer patients. Eur J Clin Pharmacol. 1987;32:593–596. doi: 10.1007/BF02455994. [DOI] [PubMed] [Google Scholar]