

Abstract
AIM: To evaluate the hepatic dysfunction in leptospirosis is usually mild and resolved eventually. However, sequential follow-up of liver biochemical data remained lacking..
METHODS: The biochemistry data and clinical symptoms of 11 sporadic patients were collected and analyzed, focusing on the impacts of leptospirosis upon liver biochemistry tests.
RESULTS: The results disclosed that of the 11 cases, 5 or 45% died. The liver biochemistry data in the beginning of the disease course were only mildly elevated. Nevertheless, late exaggerated aspartate transaminase (AST) elevations were noted in three cases who finally died when compared with the typical course. Besides, significant higher AST/alanine transaminase (ALT) ratios (AARs) of the peak levels for transaminase were also noted in the cases who eventually succumbed. The mean±SD of AARs for the survival group and dead group were 5.652.27 (n = 5) and 1.860.64 (n = 6) respectively (P = 0.006). The ratios of the cases who finally died were all more than 3.0. Conversely, the survival group’s ratios were less than 3.0.
CONCLUSION: Serial follow-up of transaminase might provide evidence to predict some rare evolutions in leptospirosis. If AST elevated progressively without a concomitant change of ALT, it might indicate an acute disease course with ensuing death. Additionally, AAR is another prognostic parameter for leptospirosis. Once the value was higher than 3.0, a grave prognosis is inevitable.
Keywords: Leptospirosis, Liver, Aspartate transaminase, Alanine transaminase, AST/ALT ratios
INTRODUCTION
Leptospirosis is caused by a variety of pathogenic spirochetes of the genus Leptospira[1], the human beings are generally the dead-end host after transmission from animal contact[2]. This disease is characterized by a broad spectrum of manifestations, which are usually subclinical[1,2]. Its diagnosis relies upon strong suspicions of this disease from some nonspecific clinical presentations[3]. Confirmation of this infection is usually established by serological studies[4] or PCR[5,6].
In terms of pathologic findings and biochemical manifestations, liver is not the main target of spirochete infection[2,3,7,8]. Hepatic dysfunction in leptospirosis was usually mild and resolved eventually. However, if jaundice occurs, serum bilirubin levels might be markedly elevated as compared with aspartate transaminase (AST) and alanine transaminase (ALT)[9]. Weil’s disease, defined as severe icteric leptospirosis with renal failure, accounts for mortality associated with leptospirosis[1]. In some cases, aside from severe jaundice, the elevation of AST was so remarkable as acute viral hepatitis could be, which attracted our attention in the endemic area of the HBV. However, the data regarding sequential follow-up of liver biochemical data of leptospirosis remained lacking. Herein, our experiences of the sporadic leptospirosis are presented and the possible implication of exaggerated AST change in leptospirosis is discussed.
MATERIALS AND METHODS
During the period from May 1998 to January 2000, 11 patients with sporadic leptospirosis in intensive care unit, Chang Gung Memorial Hospital, Taiwan, were reviewed to evaluate the impact of severe leptospirosis on liver biochemistry tests. There were seven males and four females, and their age ranged from 30 to 78 years (mean: 57 years). None of the patients were alcoholics, nor suggestive of acute viral infection or chronic virus carrier by serological tests excluding the following: hepatitis A, B, and C, Epstein-Barr virus, cytomegalovirus and Hanta virus. Bacterial and fungal cultures were all sterile. There was no history of previous liver disease in any of the patients. Leptospirosis was confirmed by either urine PCR tests or serum microscopic agglutinin test (single serum titer ≥100-fold or paired serum titers rise ≥4-fold).
The hepatic biochemistry parameters including AST (normal 0-34 U/L), ALT (normal 0-36 U/L), alkaline phosphatase (Alk-p, normal 28-94 U/L), γ-glutamyl transpeptidase (r-GT, normal <26 U/L), direct bilirubin (bili-D, normal 0-0.4 mg/dL), total bilirubin (bili-T, normal 0-1.3 mg/dL), creatine phosphokinase (CPK, normal 15-130 U/L) were chronologically collected and then analyzed. Student’s t-test was used to compare the means. A probability value of less than 0.05 was considered statistically significant. Furthermore, the influence of leptospirosis on liver function tests was discussed.
RESULTS
The disease onset was severe and abrupt in all patients with fever and chills. The clinical manifestations included conjunctival suffusion in 9, jaundice in 9, abdominal pain in 7, myalgia in 7, consciousness disturbance in 4, hepatomegaly in 4, upper gastrointestinal bleeding 2, rhabdomyolysis in 2, and hemophagocytosis, hemolytic uremic syndrome each for one case. This disease progressed to acute renal failure in all and acute respiratory failure in six. All patients received parenteral penicillin and hemodialysis. Finally, five icteric cases died exclusively of multiple organ failure.
Table 1 illustrates the characteristics and liver biochemical data of the patients. Of the 11 patients, 5 (cases 1-5) or 45% contributes to the mortality rate. One of four (25%) younger than 50 years, and four out of seven (57%) older than 50 years expired. One hundred percent of the cases got abnormal liver biochemistry parameters in spite of various degrees. Alk-p and r-GT were only elevated mildly during the entire course and never exceeded 4-folds of the normal limit. Upon admission, eight had hyperbilirubinemia; however, in the 3rd wk of the disease, one of the patients who originally had normal bilirubin levels became icteric. Therefore, there were nine cases of Weil’s disease by the definition mentioned before. The highest bilirubin level attained 35.1 mg%. CPK ranged from 178 to 2 889 U/L.
Table 1.
Biochemistry parameters of the patients with leptospirosis, presented with data upon entry (initial admission)/peak levels
|
Biochemical parameters (entry/peak) |
||||||||||||
| Case . No. | Age (yr) | Sex | AST (U/L) | ALT (U/L) | AAR | Alk-P (U/L) | r-GT (U/L) | Bili (D) (mg%) | CPK (U/L) | DA | Remark | |
| 1 | 60 | f | 190/901 | 78/144 | 6.26 | 232/232 | 100/100 | 2.7/4.6 | 628 | 12 | A | B1 |
| 2 | 70 | m | 103/132 | 9/22 | 6 | 198/212 | 125/174 | 10.6/13.8 | 99 | 47 | A | |
| 3 | 42 | f | 86/1216 | 50/308 | 3.94 | 40/446 | 6/285 | 14.6/16.4 | N.A. | 65 | A | B3 |
| 4 | 58 | f | 63/63 | 1/8 | 7.88 | 74/241 | 30/98 | 0.2/3 | 985 | 78 | A | |
| 5 | 75 | m | 226/580 | 139/139 | 4.17 | 195/195 | 80/80 | 6/15.7 | 99 | 19 | A | B5 |
| Mean | 61 | 133.6/578 | 55.4/214.6 | 5.65 | 147.8/265.2 | 68.2/286.8 | 6.82/10.7 | 452.8 | 44.2 | |||
| (SD) | 12.7 | 70.5/493.8 | 21/166 | 2.27 | 85.0/102.6 | 49.3/84.9 | 5.83/6.39 | 433.7 | 28.5 | |||
| 6 | 78 | m | 74/127 | 23/45 | 2.82 | 104/114 | 32/24 | 2.5/2.7 | 178 | 35 | ||
| 7 | 41 | m | 165/208 | 149/149 | 1.39 | 68/78 | 32/64 | 6/14.3 | 2889 | 33 | ||
| 8 | 30 | m | 203/253 | 185/102 | 2.48 | 40/45 | 6/7 | 0.1/0.1 | 704 | 10 | ||
| 9 | 65 | m | 29/34 | 25/25 | 1.36 | 198/198 | 50/70 | 0.3/0.3 | N.A. | 16 | ||
| 10 | 65 | m | 21/29 | 16/17 | 1.7 | 104/104 | 20/20 | 1.4/1.4 | 61 | 38 | ||
| 11 | 43 | f | 130/156 | 102/112 | 1.39 | 141/141 | 80/80 | 3.3/3.3 | 69 | 21 | ||
| Mean | 53.661 | 103.6/134.5 | 83.3/175 | 1.85 | 109.2/136 | 36.7/144.2 | 2.26/13.68 | 780.2 | 25.5 | |||
| (SD) | 0.35 | 74.3/90.8 | 72.9/53.6 | 0.64 | 55.6/52.8 | 25.8/30.7 | 2.2/5.35 | 1028.2 | 11.43 | |||
N.A.: not assessed; D.A.: d of admission; AAR: AST/ALT ratio of peak levels; m: male; f: female. A: Complicated with multiple organ failure and fatal; B: exaggerated AST peak levels; B1: hemophagocytosis. B2: shock, hemorrhage. B3: DIC.
The most evident difference of liver biochemistry parameters between the dead and survival group was bilirubin levels (Table 1). The mean±SD of Bili (D) of the peak level for the former are significantly higher than the latter (P = 0.03). All those who died had jaundice; however, four cases with jaundice recovered.
Upon admission, both AST and ALT levels of all the patients elevated modestly with an average of 117.27 and 70.63 U/L respectively. However, three patients (cases 1, 3, and 5) with Weil’s disease had unusually exaggerated AST elevation more than 2-3 wk after the onset of disease (Table 1, the peak AST level for case 1, 3, and 5 were 901, 1 216 and 580 U/L respectively). A representative case (case 1) showed disproportional progressive elevations of AST levels since the first admission day, which was the 12th d since the disease onset (Figure 1). Although ALT peaked at 144 U/L, it did not increase in proportion with AST, which peaked at 901 U/L. Meanwhile, CPK was 628 U/L. Due to pancytopenia, a bone marrow biopsy was performed for this patient and revealed hemophagocytosis. Case 3 displayed peak AST and ALT levels (1 216 and 308 U/L) (Table 1) after an episode of septic shock. However, they returned to normal 4 d later. Case 5 obtained peak levels of AST (580 U/L) and ALT (139 U/L) 18 d after the onset of disease. CPK was only 99 U/L then. A peripheral blood smear for pancytopenia demonstrated burr cells and toxic granules in WBCs. A disseminated intravascular coagulopathy (DIC) profile revealed positive finding, while both direct and indirect Coomb’s tests yielded negative findings. All three patients eventually died of multiple organ failure.
Figure 1.
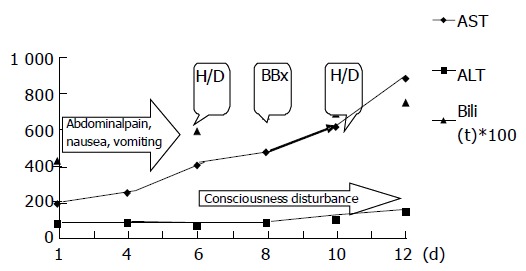
Clinical manifestations and biochemical changes in the whole course of case 1. Unit: AST (U/L), ALT (U/L), Bili(T) (mg/dL), H/D: hemodialysis, BBx: bone marrow biopsy.
In addition to exaggerated transaminase elevations of the late stage in the three cases mentioned above, significant higher AST/ALT ratios (AARs) of the peak levels for transaminase were also noted in the cases who eventually succumbed (Table 1 and Figure 2). The mean±SD of the AAR for the survival group and dead group were 5.65±2.27 (n = 5) and 1.86±0.64 (n = 6) respectively (P = 0.006). The ratios of the cases who finally died were all higher than 3.0. Conversely, the survival group’s ratios were lower than 3.0 (Figure 2).
Figure 2.
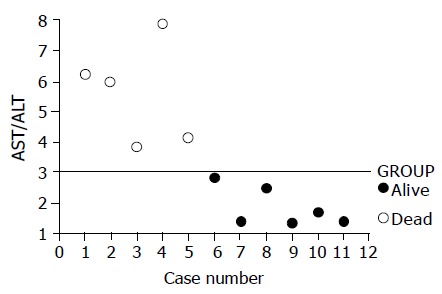
Scattered figure of AST/ALT (AAR) values. All the dead cases are shown as AAR >3.0. On the contrary, the ratios of all the survival cases were <3.0.
DISCUSSION
Age is reported as the most significant host factor related to mortality of leptospirosis. The mortality rate rose from 10% (in patients less than 50 years) to 56% (in those over 51 years)[1]. As the average age (57 years) of our cases was much higher than those of teenagers and young adults, who were considered as the major victims of leptospirosis, they were rarely admitted to hospital on account of their subclinical symptoms[1,3,6]. Furthermore, all the cases were enrolled from the intensive care unit, it is reasonable why the overall mortality (45%) of this hospitalized series was much higher than other published reports (5-16%)[1,2].
On hospitalization, the liver biochemistry tests disclosed only modest elevations in AST, ALT, Alk-p, and r-GT. However, total bilirubin ranged from normal limits to as high as 28.2 mg/dL. Such a pattern was compatible with the fact that the liver dysfunction in leptospirosis is typically mild, resolved finally and serum bilirubin can be markedly elevated as compared with other liver enzymes[1,2,3,9]. Hyperbilirubinemia is not only related to hepatocellular dysfunction but also magnified by impairing bilirubin excretion from renal failure, and bilirubin overproduction from tissue hemorrhage and intravascular hemolysis[8]. Five Weil’s disease accounted for the mortality (Table 1, cases 1-5), it was also consistent with that leptospirosis is never fatal without jaundice[1,8]. It is because the pathogenesis in jaundice resulted from leptospirosis is as complex as mentioned above[8].
Nevertheless, among the five cases dying of Weil’s disease, three (cases 1, 3, and 5) had exaggerated AST elevation 2-3 wk after the onset of disease. In contrast, among the four Weil’s disease of the survival group (cases 6, 7, 9, and 10), none had been found with exaggerated AST elevation through the whole disease course. All the cases 1, 3, and 5 developed progressive AST elevation without concomitant ALT change. Therefore, the extrahepatic origin of AST should be considered. Myositis is a common presentation in leptospirosis and usually abated by the 2nd wk[10,11]. If the extrahepatic AST had come from muscle, AST would return to normal within 2 wk rather than being progressively elevated as seen in the two cases. Moreover, it has been reported that for severe myositis in leptospirosis, CPK elevation up to 8 880 U/L only correlated with AST elevation as high as 440 U/L[12]. Myositis alone could not contribute to AST elevation in the two cases, as their AST elevations (901 and 580 U/L) were not in proportion to CPK (628 and 99 U/L) elevations in terms of myositis. Therefore, the muscle origin of AST could not simply explain this phenomenon, and the exaggerated AST elevation must have had another source.
The pathogenesis of the systemic lesions in leptospirosis was ascribed to directly damaging the parenchymal cellular membranes. It may initially cause functional disorder of these membranes, and only later lead to necrosis[13]. Nevertheless, complete cell necrosis is not prominent in leptospirosis, which is in accordance with the low level of serum transaminase in this disease[7]. Notably, hemophagocytosis and DIC were proved in the two cases (cases 1 and 5) respectively. Both are rare presentations in human leptospirosis[13,14], and they indicated a severe form of infections and disease progression. Besides, clinical presentations (respiratory failure, renal failure, consciousness disturbance, etc.,), they indicated grave multiple organ failure. It was suggested that, in rare and deteriorated situations, the systemic parenchymal cellular dysfunction in leptospirosis could evolve into extensive cellular necrosis. Namely, severe and extensive destruction of cells in various organs -not only muscle and liver -might lead to the unusual AST elevations. It had been documented that, in acute and/or fatal leptospirosis, the pathogenesis of the pathologic features are related to the presence of the organisms in the tissues, and was usually multi-system involvement[15,16].
All the five fatal cases displayed multiple organ failure (Table 1). The major difference between those who with (cases 1, 3, and 5) and without (cases 2 and 4) exaggerated AST elevation was the natural course of disease. The duration from disease onset to death appeared much shorter in the former (12-19 d) as compared to the latter (47-78 d). The clinical course of leptospirosis can be divided into two stages, a 3-7-d septic stage followed by an immune stage lasting for 4-30 d[2]. All the exaggerated AST occurred in the immune stage of leptospirosis. The balance between the host and parasite determines, whether the infection runs an acute or a protracted course[17]. In acute leptospirosis, the main pathogenesis was probably due to the direct toxic damage of spirochetes[13,18,19]. However, an immune complex-mediated mechanism was suggested when the course was protracted[18]. Because the infections ran acute courses in cases 1, 3, and 5, it might indicate that although the exaggerated AST elevations occurred in the immune stages (after 1 wk of disease onset), the hosts could not clear the spirochetes then from the blood by immune reactions efficiently. The spirochetes were consistently virulent to the systemic parenchymal cells. Finally unavoidable deaths with fulminant courses of the hosts ensued.
Aside from AST, AAR of peak levels for transaminase might predict the outcome of leptospirosis. AAR was significantly higher in the fatal cases. The ratios were higher than 3.0 (Table 1 and Figure 2) in all of the deaths. In contrast, all the cases who finally recovered had the ratios of less than 3.0. The ratios seemed to stop at 3. Both multiple major organ failure and systemic parenchymal cells necrosis might contribute to the higher ratios in the fatal cases.
In endemic areas of hepatitis B infection such as Taiwan, viral hepatitis was the first consideration for icteric patients with abrupt disease onset associated with transaminase elevation. Such impression might delay the appropriate management of the true culprit-leptospirosis. Typically acute viral hepatitis presented as exaggerated transaminase elevations initially and recovered thereafter regardless of the existence of jaundice. Besides, the ALT elevation usually sustains longer than that of AST. On the contrary, leptospirosis showed as initial modest transaminase elevations accompanied with extremely high CPK levels[1,2,8]. The initial amplitude of transaminase could be a clue to differentiate viral hepatitis and leptospirosis. However, in a chronic HBV carrier, the delayed transaminase elevation would confuse the physician, if the elevation is due to acute exacerbation of chronic HBV infection. We were convinced that delayed and disproportional AST elevation is in favor of nonhepatic origin. In leptospirosis, it might hint a fulminant disease course with high mortality.
Conclusively, in addition to clinical symptoms, serial follow-up of transaminase could provide some evidence to predict the rare evolution in leptospirosis. Late disproportional AST elevation indicates an ominous sign, mortality that typically ensues from a fulminant disease course. AAR is a parameter responsive to the prognosis of leptospirosis, the ratio >3.0 means a grave prognosis. Furthermore, in endemic areas of viral hepatitis, Weil’s disease still should be considered in icteric cases of unknown origin regardless of AST level, since exaggerated AST might herald the unusual fulminant mortality.
Footnotes
Supported by the Chang Gung Medical Research Project fund, No. CMRPG 33014
Science Editor Guo SY Language Editor Elsevier HK
References
- 1.Isselbacher KJ, Braunwald E, Wilson JD. Harrison's principles of internal medicine. 14th ed. St. Louis: McGraw-Hill; 1993. pp. 1036–1038. [Google Scholar]
- 2.Taber LH, Feigin RD. Spirochetal infections. Pediatr Clin North Am. 1979;26:377–413. doi: 10.1016/s0031-3955(16)33713-0. [DOI] [PubMed] [Google Scholar]
- 3.Farr RW. Leptospirosis. Clin Infect Dis. 1995;21:1–6; quiz 7-8. doi: 10.1093/clinids/21.1.1. [DOI] [PubMed] [Google Scholar]
- 4.Lupidi R, Cinco M, Balanzin D, Delprete E, Varaldo PE. Serological follow-up of patients involved in a localized outbreak of leptospirosis. J Clin Microbiol. 1991;29:805–809. doi: 10.1128/jcm.29.4.805-809.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merien F, Baranton G, Perolat P. Comparison of polymerase chain reaction with microagglutination test and culture for diagnosis of leptospirosis. J Infect Dis. 1995;172:281–285. doi: 10.1093/infdis/172.1.281. [DOI] [PubMed] [Google Scholar]
- 6.Ralph D, McClelland M, Welsh J, Baranton G, Perolat P. Leptospira species categorized by arbitrarily primed polymerase chain reaction (PCR) and by mapped restriction polymorphisms in PCR-amplified rRNA genes. J Bacteriol. 1993;175:973–981. doi: 10.1128/jb.175.4.973-981.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brito de T, Penna DO, Hoshino S, Pereira VG, Caldas AC, Rothstein W. Cholestasis in human leptospirosis: a clinical, histochemical, biochemical and electron microscopy study based on liver biopsies. Beitr Pathol Anat. 1970;140:345–361. [PubMed] [Google Scholar]
- 8.Sherlock S, Dooley J. Disease of the liver and biliary system. 10th ed. Malden: Blackwell Sci Pub; 1997. pp. 507–510. [Google Scholar]
- 9.den Haan PJ, van Vliet AC, Hazenberg BP. Weil's disease as a cause of jaundice. Neth J Med. 1993;42:171–174. [PubMed] [Google Scholar]
- 10.Johnson WD, Silva IC, Rocha H. Serum creatine phosphokinase in leptospirosis. JAMA. 1975;233:981–982. [PubMed] [Google Scholar]
- 11.Heath CW, Alexander AD, Galton MM. Leptospirosis in the United States. N Engl J Med. 1965;273:857–864 contd. doi: 10.1056/NEJM196510142731606. [DOI] [PubMed] [Google Scholar]
- 12.Farkas PS, Knapp AB, Lieberman H, Guttman I, Mayan S, Bloom AA. Markedly elevated creatinine phosphokinase, cotton wool spots, and pericarditis in a patient with leptospirosis. Gastroenterology. 1981;80:587–589. [PubMed] [Google Scholar]
- 13.Nicodemo AC, Duarte MI, Alves VA, Takakura CF, Santos RT, Nicodemo EL. Lung lesions in human leptospirosis: microscopic, immunohistochemical, and ultrastructural features related to thrombocytopenia. Am J Trop Med Hyg. 1997;56:181–187. doi: 10.4269/ajtmh.1997.56.181. [DOI] [PubMed] [Google Scholar]
- 14.Yang CW, Pan MJ, Wu MS, Chen YM, Tsen YT, Lin CL, Wu CH, Huang CC. Leptospirosis: an ignored cause of acute renal failure in Taiwan. Am J Kidney Dis. 1997;30:840–845. doi: 10.1016/s0272-6386(97)90091-3. [DOI] [PubMed] [Google Scholar]
- 15.Nally JE, Chantranuwat C, Wu XY, Fishbein MC, Pereira MM, Da Silva JJ, Blanco DR, Lovett MA. Alveolar septal deposition of immunoglobulin and complement parallels pulmonary hemorrhage in a guinea pig model of severe pulmonary leptospirosis. Am J Pathol. 2004;164:1115–11127. doi: 10.1016/S0002-9440(10)63198-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown PD, Carrington DG, Gravekamp C, van de Kemp H, Edwards CN, Jones SR, Prussia PR, Garriques S, Terpstra WJ, Levett PN. Direct detection of leptospiral material in human postmortem samples. Res Microbiol. 2003;154:581–586. doi: 10.1016/S0923-2508(03)00166-9. [DOI] [PubMed] [Google Scholar]
- 17.Yasuda PH, Hoshino-Shimizu S, Yamashiro EH, De Brito T. Experimental leptospirosis (L. interrogans serovar icterohae-morrhagiae) of the guinea pig: leptospiral antigen, gamma globulin and complement C3 detection in the kidney. Exp Pathol. 1986;29:35–43. doi: 10.1016/s0232-1513(86)80004-4. [DOI] [PubMed] [Google Scholar]
- 18.Alves VA, Vianna MR, Yasuda PH, De Brito T. Detection of leptospiral antigen in the human liver and kidney using an immunoperoxidase staining procedure. J Pathol. 1987;151:125–131. doi: 10.1002/path.1711510205. [DOI] [PubMed] [Google Scholar]
- 19.Alves VA, Gayotto LC, De Brito T, Santos RT, Wakamatsu A, Vianna MR, Sakata EE. Leptospiral antigens in the liver of experimentally infected guinea pig and their relation to the morphogenesis of liver damage. Exp Toxicol Pathol. 1992;44:425–434. doi: 10.1016/S0940-2993(11)80185-5. [DOI] [PubMed] [Google Scholar]