

Abstract
AIM: To study Helicobacter pylori (H pylori) infection in relation to E-cadherin (E-cad) promoter polymorphism and hypermethylation in GCs.
METHODS: Specimens were taken from representative cancerous lesions and adjacent non-cancerous epithelia of 67 resected GCs. H pylori was detected by real-time PCR of the cagA gene from non-neoplastic epithelium. E-cad promoter polymorphism and hypermethylation were determined by restriction fragment length polymorphism analysis and methylation-specific PCR, respectively. Expression of E-cad protein was determined by immunohistochemistry.
RESULTS: H pylori was found in 57% of patients with GC. H pylori infection was more frequently found in tumors with the -160C/C genotype than those with the -160C/A and -160A/A genotypes (74% vs 47%, P = 0.02). H pylori infection was associated with E-cad methylation in non-neoplastic epithelium; however, no significant difference in H pylori was observed between methylated and unmethylated cancerous lesions.
CONCLUSION: Patients with the -160C/C genotype might require H pylori infection to promote the inactivation of CDH1, suggesting that H pylori infection might affect GC in an initial stage because polymorphism is germ line. Mechanism of hypermethylation of CDH1 promoter in GC is complex, and H pylori infection might affect it in an initial stage.
Keywords: H pylori, E-cadherin, -160 C→A polymorphism, Hypermethylation, Gastric carcinoma
INTRODUCTION
E-cadherin (E-cad, also known as CDH1) is a member of a family of transmembrane glycoproteins expressed on epithelial cells and is responsible for calcium-dependent cell-to-cell adhesion[1]. E-cad forms complexes and connects actin filaments with α-, β-, and γ-catenins[2,3], which is crucially involved in neoplastic transformation and metastasis[4,5]. Loss of cell adhesion may contribute to loss of contact inhibition of growth, which is an early step in the neoplastic process. Furthermore, loss of cadherin activity may result in cancer cell detachment and metastasis[6,7].
Gastric carcinogenesis is a multistep process with morphological progression involving multiple genetic and epigenetic events, whereas CDH1 is an important putative tumor suppressor gene. In gastric carcinoma (GC), the percentage reduction in E-cad expression varies from 17% to 92%, and is more frequent in diffuse type carcinomas than in intestinal types[8-13]. Germ-line mutation of the CDH1 gene is found in familial GCs[14,15]. Somatic mutations of E-cad are found in more than 50% of gastric carcinomas of the diffuse type[16-18]. In addition to the classic two-hit inactivation mechanism, CDH1 can be silenced by cytosine-guanosine (CpG) sequence methylation in GC[17,18]. Moreover, Li et al, reported that -160C/A polymorphism has a direct effect on the transcriptional regulation of CDH1[19].
Helicobacter pylori (H pylori) is the single most important etiological factor for gastric cancer development. The strongest evidence comes from three independent, nested case-control studies in which pre-existing infection markedly increased the risk of GC[20-22]. It is estimated that approximately 60% of all GC cases can be attributed to H pylori infection[20]. However, there are few data in the molecular profiles of E-cad of H pylori-positive and H pylori-negative GCs[23].
El-Omar et al[24], reported that interleukin-1 polymorphisms that cause the up-regulation of interleukin-1β with H pylori infection are associated with an increased risk of GC. Moreover, Hmadcha et al[25], found that interleukin-1β might induce gene methylation through the production of nitric oxide and the subsequent activation of DNA methyltransferase. It is possible that H pylori infection induces methylation through the production of interleukin-1β. In this study, we investigated the relationship between H pylori infection and CDH1 methylation. Since -160C/A polymorphism is germ line, we also investigated the relationship between H pylori infection and polymorphism, to find if H pylori infection affects tumorigenesis in the initial stage.
MATERIALS AND METHODS
Patients and samples
The specimens were surgically obtained from 67 patients with GC between 2000 and 2002, at the Division of General Surgery, Department of Surgery, Tri-Service General Hospital, Taipei, Taiwan. None of the subjects received preoperative anti-cancer therapy. Clinical information was obtained from medical records. Samples were taken from representative cancerous lesions and the corresponding non-cancerous epithelia. All tumor DNA samples were obtained by microdissection from 5-µm-thick hematoxylin and eosin-stained paraffin-embedded tissue sections[26]. Non-cancerous DNA was extracted from tissues which were flash-frozen in liquid nitrogen and stored in -80 °C. All 67 samples were classified according to the Lauren criteria[27]: 26 as intestinal, 41 as diffuse types. The tumors were staged at the time of surgery using the standard criteria for TNM staging, with the unified international gastric cancer staging classification[28].
H pylori detection with real-time PCR
Real time PCR amplification of the cagA gene was conducted by adding 62.5 ng of DNA from stomach normal tissues isolates to 20 μL reactions containing 10 μL of 2X QuantiTect SYBR Green PCR Master Mix (QIAGEN, Hilden, Germany), 0.5 μmol/L S1227 Sense Primer (nt 154-175:5’-GATAACAGGCAAGCTTTTGAGG-3’), 0.5 μmol/L AS1576 Antisense Primer (nt 503-583:5’-CTGCAAA-AGATTGTTTGGCAG-3’) and 0.4 U of Uracil-N-glycosylase (New England Biolabs, Boston, USA).
Real-time PCR amplification was carried out in a LightCyclerTM (Roche, Basel, Switzerland). After a preincubation step at 50 °C for 2 min, 95 °C for 15 min in order to activate the Uracil-N-glycosylase and HotStar Taq DNA polymerase, amplification was performed during 65 cycles including denaturation (94 °C, 15 s with a temperature transition rate of 20 °C/s), annealing (60 °C, 25 s, with a temperature transition rate of 20 °C/s) and extension (72 °C, 18 s with a temperature transition rate of 20 °C/s). A single fluorescent signal was obtained once per cycle at the end of the extension step with detection channel F1. After amplification, melting curve analysis was performed on the products by heating to 95 °C for 0 s, cooling to 65 °C for 15 s, followed by a temperature increase to 95 °C with a temperature transition rate of 0.2 °C/s while continuously collecting the fluorescent signal.
To analyze the data of the real-time PCR assay, the derivative melting curves were obtained with the LightCycler data analysis software, version 3.5 (Roche). The melting temperature (Tm) of the melting curve in each sample was used to detect the H pyloric cagA gene.
Restriction-fragment length polymorphism analysis to identify nucleotide changes at -160 of the CDH1 promoter
The -160 polymorphic site contained either a C or a A residue. The tumor type was determined by BstEII digestion of the PCR products amplified using the primer set 5’-TGATCCCAGGTCTTAGTGAG-3’ (upstream) and 5’-AGTCTGAACTGACTT CCGCA-3’ (downstream). The 318-bp PCR product was cut into two fragments (208 and 110 bp) it contained the A residue. To ensure that the observed polymorphism was specific and not an experimental artifact, the results were confirmed by direct DNA sequencing.
Methylation-specific PCR and bisulfite-modified genomic sequencing to detect promoter hypermethylation of CDH1
The bisulfite modification procedure was carried out by the Zymo’s EZ DNA Methylation Kit (Zymo Research, USA). One microliter of bisulfite-modified DNA was amplified in a total volume of 25 μL containing 1×PCR Buffer II, 2 mmol/L MgCl2, 0.25 mmol/L deoxynucleotide triphosphate (dNTPs), 0.2 μmol/L each primer, and 0.04 U of Ampli Taq GoldTM DNA polymerase (Applied Biosystems, USA) at 95 °C for 30 s, the specific annealing temperature for 30 s, and 72 °C for 45 s. Water was used as a negative control. Seven millimole per liter/L of PCR product was directly loaded onto 3% agarose gel stained with ethidium bromide and directly visualized under UV illumination. Samples were scored as methylation, when there was a clearly visible band on the gel with the methylation primers[29].
Immunohistochemical staining and evaluation of E-cad expression
In immunohistochemical studies, sections (5 µm thick) from fixed, paraffin-embedded tumors were reacted with monoclonal anti-E-cad antibody (Cappel, Aurora, OH, USA), then with secondary antibody, and the signal was detected using an avidin-biotin complex and diaminobenzidine (DAB) kit (Vector Laboratories, Burlingame, CA, USA). DAB produced a yellowish brown stain, if the sample was positive. If more than 90% of the tumor cells exhibited intense membranous staining similar to that of normal cells, the result was considered positive (++). If the staining intensity was demonstrably reduced in comparison with that of normal cells and/or the staining pattern was heterogeneous (10-90% positive), the result was deemed to be weakly positive (+). If E-cad expression was completely lost or positive in less than 10% of cells, the result was defined as negative (-).
Statistical analysis
Analyses were performed using S-Plus®2000 for Windows Statistical Software (CANdiensten, Amsterdam, The Netherlands). The level of significance was set at P<0.05 for all tests. Continuous variables are expressed as mean±SD, and were tested using Student’s t test; categorical variables were tested using Fisher’s exact test.
RESULTS
Of these 67 patients, 45 were men, and 22 were women. Their median age was 68.56 years (range, 31-88 years). All patients underwent surgical resection of the primary tumor. Twenty-six and 41 tumors were of the intestinal and diffuse histotypes, respectively. Reduced E-cad expression was observed in 88% (36/41) of diffuse type tumors and in 50% (13/26) of intestinal type tumors (P = 0.0015). Representative examples of immunohistochemical staining for E-cad expression in non-cancerous epithelium (++) and diffuse type tumor (-) are shown in Figures 1A and B.
Figure 1.
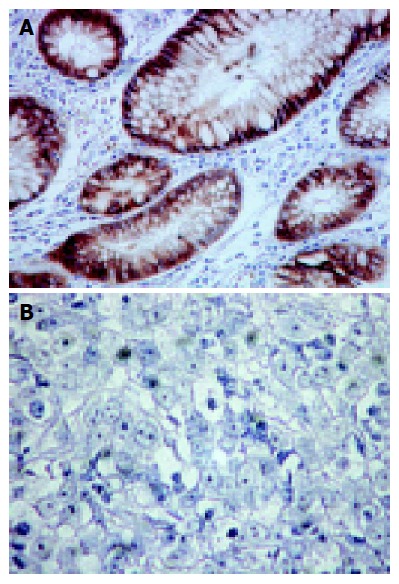
Immunohistochemical staining. A: Positive (++) immunohistochemical staining for E-cad expression in non-cancerous epithelium; B: Negative (-) immunohistochemical staining for E-cad expression in cancerous lesion in diffuse type tumor.
H pylori infection was detected by real-time PCR (iCycler iQ Real-Time Quantitative System) for H pylori cagA gene in non-cancerous gastric epithelial samples adjacent to tumors. H pylori was found in 57% of GC patients. No significant difference in H pylori infection was observed between intestinal and diffuse type tumors (23/41, 56% vs 15/26 58%). There was no significant difference in reduced E-cad expression between H pylori-positive tumors and H pylori-negative tumors (27/38, 71.1% vs 22/29, 76%).
H pylori and -160C/A polymorphism
Among the 67 patients, 27 were genotype C/C (40%), 26 were genotype A/C (39%), and 14 were genotype A/A (21%) (Figure 2). There was no significant difference in the frequency of the C/A+A/A genotypes between tissues with normal and reduced E-cad expression (13/18, 72% vs 27/49, 55%). Interestingly, analyzing 67 available cases of H pylori infection and polymorphism status, H pylori infection was more frequently observed in tumors with C/C genotype of CDH1 promoter than in C/A+A/A genotype tumors (20/27, 74% vs 18/40, 45%; P = 0.02, Table 1).
Figure 2.
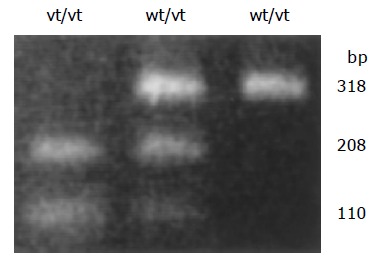
RFLP analysis of genetic polymorphism of the 160 site of the E-cad promoter. The C/A polymorphism was differentiated by BstEII digestion of PCR products homozygous for the wild-type (high-activity) allele (wt/wt, CC genotype), heterozygous for the variant (low-activity) allele (wt/vt, CA genotype), and homozygous for the low-activity allele (vt/vt, AA genotype).
Table 1.
H pylori infection in association with histotypes and E-cad expression profiles in GC
| H pylori (+) (%) | H pylori (–) (%) | P | |
| Lauren’s | |||
| Intestinal type | 23 (56.1) | 18 (43.9) | |
| Diffuse type | 15 (57.7) | 11 (42.3) | NS |
| Promoter polymorphism | |||
| C /C | 20 (74.1) | 7 (25.9) | |
| C /A+A /A | 18 (45) | 22 (55) | 0.02 |
| Methylation (noncancerous) | |||
| Methylated | 36 (94.7) | 22 (75.9) | |
| Unmethylated | 2 (5.3) | 7 (24.1) | 0.025 |
| Methylation (cancerous) | |||
| Methylated | 26 (68.4) | 19 (65.5) | |
| Unmethylated | 12 (31.6) | 10 (34.5) | NS |
| IHC | |||
| Normal (++) | 11 (28.9) | 7 (24.1) | |
| Reduced (+, –) | 27 (71.1) | 22 (75.9) | NS |
NS: not significant.
H pylori infection and hypermethylation in non-cancerous gastric epithelia
Promoter hypermethylation was found in 87% non-cancerous gastric epithelia of 67 GC patients (Figure 3A). Hypermet-hylation was frequently associated with H pylori positive than H pylori negative (36/38, 95% vs 22/29, 76%, P = 0.025, Table 1).
Figure 3.
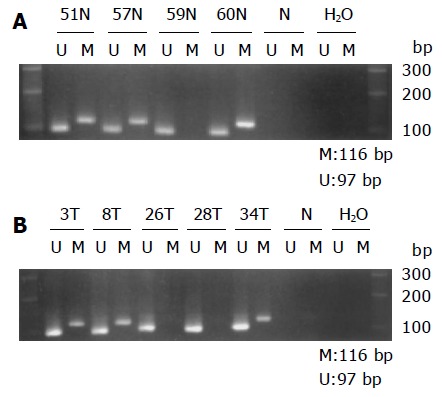
Gel electrophoresis picture demonstrating aberrant methylation in E-cad. Primer sets used for simple amplification are designed as unmethylated (U), methylated (M), unmodified DNA (N). Water is used as a negative control (H2O). Molecular weight marker is 100-bp DNA ladder. A: Methylation in non-cancerous epithelium. Samples 51, 57, and 60 N are methylated. Sample 59 N is unmethylated; B: Methylation in cancerous lesion. Samples 3, 8, and 34 T are methylated. Samples 26 and 28 T are unmethylated.
H pylori infection and hypermethylation in cancerous lesions
Results from MSP indicated that the CDH1 promoter was hypermethylated in 45 (67%) of 67 GCs (Figure 3B). Hypermethylation was more frequent in diffuse type tumors (76%, 31/41) than in intestinal type tumors (50%, 13/26; P = 0.03). Furthermore, hypermethylation was more frequent in GCs with reduced E-cad expression than normal expression (37/45, 82% vs 12/22, 55%, P = 0.02). No significant difference in H pylori infection was observed between methylated and unmethylated tumors (26/45, 58% vs 12/22, 55%, Table 1).
DISCUSSION
H pylori is responsible for the pathogenesis of atrophic gastritis and intestinal metaplasia[30,31]. Epidemiological studies have indicated that infection with H pylori is a risk factor for GC[20-22]. Moreover, Mongolian gerbils develop chronic gastritis, intestinal metaplasia, and adenocarcinoma after inoculation with H pylori into the stomach. However, the link between H pylori infection and the molecular mechanism of human gastric carcinoma remains to be investigated. Lim et al, reported that the increased expression of cell adhesion molecules (galectin 1, aldolase A, integrin α5, LMO7) and the decrease in E-cad expression induced by H pylori might contribute to cell adhesion, invasion, and possibly cell proliferation in gastric epithelial cells[32]. Kitadai et al, reported that coculture with H pylori increased the expression of interleukin-8, vascular endothelial growth factor (VEGF), angiogenin, uPA, and MMP-9 and increased angiogenic and collagenase activities in gastric carcinoma cells[33]. Sharma et al, reported that the activation of interleukin-8 gene expression by H pylori is regulated by the transcription factor, nuclear factor-kappa B, in gastric epithelial cells[34]. Akhtar et al, reported that promoter methylation regulates H pylori-stimulated cyclooxygenase-2 expression in gastric epithelial cells[35]. However, in vivo data on the differences in the molecular profile (ras, MDM2, c-erbB-2, cyclin D1, p53, CDH1) of H pylori-positive and H pylori-negative gastric carcinomas are almost non-existent[23].
In this study, H pylori was found in 57% of patients with GC. No significant difference in H pylori infection was observed between intestinal and diffuse type tumors (56% vs 58%). This is consistent with the current view that 60% of all GC cases are related to H pylori infection, irrespective of the histological tumor type[20,23].
H pylori and promoter polymorphism
Li et al, reported that the A allele of the -160C/A promoter polymorphism altered transcriptional binding, resulting in a reduction in transcriptional efficiency of 68% relative to that of the C allele[19]. In this study, H pylori was significantly more frequent in the C/C genotype of CDH1 promoter than in the C/A+A/A genotypes. The C allele shows better transcriptional activity than the A allele, so patients with the C/C type may require H pylori infection to promote the inactivation of CDH1. Since -160C/A polymorphism is germ line, H pylori infection might affect CDH1 inactivation in an early, possibly preneoplastic stage.
H pylori and promoter hypermethylation
In this study, hypermethylation of the CDH1 promoter was present in 67% GCs, and was more frequent in diffuse type tumors (76%) than in intestinal type tumors (50%, P = 0.03). CDH1 promoter hypermethylation was associated with decreased immunohistochemical expression of E-cad (82% vs 55%, P = 0.02). These data are similar to the results reported by Tamura et al, and Graziano et al[37], which indicated that CDH1 promoter methylation may play a major role, together with mutations or deletions, in causing inactivation of the CDH1 gene in GC, especially in diffuse type tumors.
Chan et al[18], investigated CDH1 hypermethylation in the gastric mucosa of 35 patients with dyspepsia and found that hypermethylation was age-related and associated with H pylori infection. In our study, hypermethylation was found in 87% non-cancerous gastric epithelia of 67 GC patients and frequently associated with H pylori positive than H pylori negative epithelia. Therefore, our results of promoter hypermethylation of E-cad in non-cancerous gastric epithelia are compatible with hypothesis proposed by El-Omar et al[24], and Hmadcha et al[25]. However, we observed no association between H pylori infection and CDH1 methylation in the gastric cancerous epithelia. It seems that H pylori is unrelated to CDH1 methylation in GC. However, on the basis of current knowledge, H pylori should be considered as a factor facilitating the multifactorially determined process of gastric carcinogenesis, but by itself does not initiate or trigger the carcinogenic process (the “hypothesis of no return”)[23,38]. Moreover, few H pylori are detected in cancerous epithelia in the human stomach. Under in vivo conditions, H pylori infects the non-cancerous epithelia surrounding the tumor and up-regulates methylation, and may indirectly stimulate hypermethylation in GCs at the tumor-normal tissue interface[33]. In addition, DNA methylation is catalyzed by DNA methyltransferase, and DNA CpG islands can be demethylated by MBD2[39]. H pylori infection might induce activation of DNA methyltransferase. However, Oue et al, reported that the mechanism of CpG island hypermethylation is complex in the GC microenvironment and may not result from a simple transcriptional up-regulation of methyltransferase or down-regulation of MBD2[40]. Thus from the results of this study and the above statement, we conclude that the mechanism of hypermethylation of CDH1 in GC is complex, and H pylori might affect hypermethylation in an initial stage.
In conclusion, patients with the -160C/C genotype might require H pylori infection to promote the inactivation of CDH1. H pylori infection might affect E-cad promoter hypermethylation of GC in an initial stage. Therefore, H pylori infection should be considered as a factor that facilitates a multifactorially determined process of gastric carcinogenesis in an initial stage.
ACKNOWLEDGMENTS
We are grateful to Ms. Pei-Ei Wu, Yi-Chien Mao and Chien-Shi Wang for their technical assistance.
Footnotes
Supported by Clinical Research Fund of the Tri-Service General Hospital and CY Foundation for Advancement of Education, Science and Medicine, Taipei, Taiwan, China
Science Editor Guo SY Language Editor Elsevier HK
References
- 1.Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 2.Takeichi M. Cadherins: a molecular family important in selective cell-cell adhesion. Annu Rev Biochem. 1990;59:237–252. doi: 10.1146/annurev.bi.59.070190.001321. [DOI] [PubMed] [Google Scholar]
- 3.Grunwald GB. The structural and functional analysis of cadherin calcium-dependent cell adhesion molecules. Curr Opin Cell Biol. 1993;5:797–805. doi: 10.1016/0955-0674(93)90028-o. [DOI] [PubMed] [Google Scholar]
- 4.Hirohashi S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol. 1998;153:333–339. doi: 10.1016/S0002-9440(10)65575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pignatelli M, Vessey CJ. Adhesion molecules: novel molecular tools in tumor pathology. Hum Pathol. 1994;25:849–856. doi: 10.1016/0046-8177(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 6.Hashimoto M, Niwa O, Nitta Y, Takeichi M, Yokoro K. Unstable expression of E-cadherin adhesion molecules in metastatic ovarian tumor cells. Jpn J Cancer Res. 1989;80:459–463. doi: 10.1111/j.1349-7006.1989.tb02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bussemakers MJ, van Moorselaar RJ, Giroldi LA, Ichikawa T, Isaacs JT, Takeichi M, Debruyne FM, Schalken JA. Decreased expression of E-cadherin in the progression of rat prostatic cancer. Cancer Res. 1992;52:2916–2922. [PubMed] [Google Scholar]
- 8.Shimoyama Y, Hirohashi S. Expression of E- and P-cadherin in gastric carcinomas. Cancer Res. 1991;51:2185–2192. [PubMed] [Google Scholar]
- 9.Oka H, Shiozaki H, Kobayashi K, Tahara H, Tamura S, Miyata M, Doki Y, Iihara K, Matsuyoshi N, Hirano S. Immunohistochemical evaluation of E-cadherin adhesion molecule expression in human gastric cancer. Virchows Arch A Pathol Anat Histopathol. 1992;421:149–156. doi: 10.1007/BF01607048. [DOI] [PubMed] [Google Scholar]
- 10.Matsuura K, Kawanishi J, Fujii S, Imamura M, Hirano S, Takeichi M, Niitsu Y. Altered expression of E-cadherin in gastric cancer tissues and carcinomatous fluid. Br J Cancer. 1992;66:1122–1130. doi: 10.1038/bjc.1992.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mayer B, Johnson JP, Leitl F, Jauch KW, Heiss MM, Schildberg FW, Birchmeier W, Funke I. E-cadherin expression in primary and metastatic gastric cancer: down-regulation correlates with cellular dedifferentiation and glandular disintegration. Cancer Res. 1993;53:1690–1695. [PubMed] [Google Scholar]
- 12.Gabbert H. Mechanisms of tumor invasion: evidence from in vivo observations. Cancer Metastasis Rev. 1985;4:293–309. doi: 10.1007/BF00048094. [DOI] [PubMed] [Google Scholar]
- 13.Shun CT, Wu MS, Lin JT, Wang HP, Houng RL, Lee WJ, Wang TH, Chuang SM. An immunohistochemical study of E-cadherin expression with correlations to clinicopathological features in gastric cancer. Hepatogastroenterology. 1998;45:944–949. [PubMed] [Google Scholar]
- 14.Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–405. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- 15.Gayther SA, Gorringe KL, Ramus SJ, Huntsman D, Roviello F, Grehan N, Machado JC, Pinto E, Seruca R, Halling K, et al. Identification of germ-line E-cadherin mutations in gastric cancer families of European origin. Cancer Res. 1998;58:4086–4089. [PubMed] [Google Scholar]
- 16.Becker KF, Atkinson MJ, Reich U, Becker I, Nekarda H, Siewert JR, Höfler H. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res. 1994;54:3845–3852. [PubMed] [Google Scholar]
- 17.Machado JC, Oliveira C, Carvalho R, Soares P, Berx G, Caldas C, Seruca R, Carneiro F, Sobrinho-Simöes M. E-cadherin gene (CDH1) promoter methylation as the second hit in sporadic diffuse gastric carcinoma. Oncogene. 2001;20:1525–1528. doi: 10.1038/sj.onc.1204234. [DOI] [PubMed] [Google Scholar]
- 18.Chan AO, Lam SK, Wong BC, Wong WM, Yuen MF, Yeung YH, Hui WM, Rashid A, Kwong YL. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut. 2003;52:502–506. doi: 10.1136/gut.52.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li LC, Chui RM, Sasaki M, Nakajima K, Perinchery G, Au HC, Nojima D, Carroll P, Dahiya R. A single nucleotide polymorphism in the E-cadherin gene promoter alters transcriptional activities. Cancer Res. 2000;60:873–876. [PubMed] [Google Scholar]
- 20.Forman D, Newell DG, Fullerton F, Yarnell JW, Stacey AR, Wald N, Sitas F. Association between infection with Helicobacter pylori and risk of gastric cancer: evidence from a prospective investigation. BMJ. 1991;302:1302–1305. doi: 10.1136/bmj.302.6788.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med. 1991;325:1132–1136. doi: 10.1056/NEJM199110173251604. [DOI] [PubMed] [Google Scholar]
- 22.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med. 1991;325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 23.Blok P, Craanen ME, Offerhaus GJ, Dekker W, Kuipers EJ, Meuwissen SG, Tytgat GN. Molecular alterations in early gastric carcinomas. No apparent correlation with Helicobacter pylori status. Am J Clin Pathol. 1999;111:241–247. doi: 10.1093/ajcp/111.2.241. [DOI] [PubMed] [Google Scholar]
- 24.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 25.Hmadcha A, Bedoya FJ, Sobrino F, Pintado E. Methylation-dependent gene silencing induced by interleukin 1beta via nitric oxide production. J Exp Med. 1999;190:1595–1604. doi: 10.1084/jem.190.11.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moskaluk CA, Kern SE. Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol. 1997;150:1547–1552. [PMC free article] [PubMed] [Google Scholar]
- 27.Lauren P. The two main histological types of gastric carcinoma diffuse and so-called intestinal type of carcinoma. Acta Pathol Microbiol Scand. 1965;64:31–49. doi: 10.1111/apm.1965.64.1.31. [DOI] [PubMed] [Google Scholar]
- 28.Sobin LH, Wittekind CH (editors) International union against cancers (UICC): TNM classification of malignant tumors. 5th ed. PMCid. New York: John Wiley; 1997. p. PMC1716062. [Google Scholar]
- 29.Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graff JR, Herman JG, Myöhänen S, Baylin SB, Vertino PM. Mapping patterns of CpG island methylation in normal and neoplastic cells implicates both upstream and downstream regions in de novo methylation. J Biol Chem. 1997;272:22322–22329. doi: 10.1074/jbc.272.35.22322. [DOI] [PubMed] [Google Scholar]
- 31.Kawaguchi H, Haruma K, Komoto K, Yoshihara M, Sumii K, Kajiyama G. Helicobacter pylori infection is the major risk factor for atrophic gastritis. Am J Gastroenterol. 1996;91:959–962. [PubMed] [Google Scholar]
- 32.Lim JW, Kim H, Kim KH. Cell adhesion-related gene expression by Helicobacter pylori in gastric epithelial AGS cells. Int J Biochem Cell Biol. 2003;35:1284–1296. doi: 10.1016/s1357-2725(03)00051-7. [DOI] [PubMed] [Google Scholar]
- 33.Kitadai Y, Sasaki A, Ito M, Tanaka S, Oue N, Yasui W, Aihara M, Imagawa K, Haruma K, Chayama K. Helicobacter pylori infection influences expression of genes related to angiogenesis and invasion in human gastric carcinoma cells. Biochem Biophys Res Commun. 2003;311:809–814. doi: 10.1016/j.bbrc.2003.10.077. [DOI] [PubMed] [Google Scholar]
- 34.Sharma SA, Tummuru MK, Blaser MJ, Kerr LD. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J Immunol. 1998;160:2401–2407. [PubMed] [Google Scholar]
- 35.Akhtar M, Cheng Y, Magno RM, Ashktorab H, Smoot DT, Meltzer SJ, Wilson KT. Promoter methylation regulates Helicobacter pylori-stimulated cyclooxygenase-2 expression in gastric epithelial cells. Cancer Res. 2001;61:2399–2403. [PubMed] [Google Scholar]
- 36.Tamura G, Yin J, Wang S, Fleisher AS, Zou T, Abraham JM, Kong D, Smolinski KN, Wilson KT, James SP, et al. E-Cadherin gene promoter hypermethylation in primary human gastric carcinomas. J Natl Cancer Inst. 2000;92:569–573. doi: 10.1093/jnci/92.7.569. [DOI] [PubMed] [Google Scholar]
- 37.Graziano F, Arduini F, Ruzzo A, Mandolesi A, Bearzi I, Silva R, Muretto P, Testa E, Mari D, Magnani M, et al. Combined analysis of E-cadherin gene (CDH1) promoter hypermethylation and E-cadherin protein expression in patients with gastric cancer: implication for treatment with demethylating drugs. Ann Oncol. 2004;15:489–492. doi: 10.1093/annonc/mdh108. [DOI] [PubMed] [Google Scholar]
- 38.Wu MS, Shun CT, Wang HP, Sheu JC, Lee WJ, Wang TH, Lin JT. Genetic alterations in gastric cancer: relation to histological subtypes, tumor stage and Helicobacter pylori infection. Gastroenterology. 1997;112:1457–1465. doi: 10.1016/s0016-5085(97)70071-4. [DOI] [PubMed] [Google Scholar]
- 39.Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nature. 1999;397:579–583. doi: 10.1038/17533. [DOI] [PubMed] [Google Scholar]
- 40.Oue N, Kuraoka K, Kuniyasu H, Yokozaki H, Wakikawa A, Matsusaki K, Yasui W. DNA methylation status of hMLH1, p16(INK4a), and CDH1 is not associated with mRNA expression levels of DNA methyltransferase and DNA demethylase in gastric carcinomas. Oncol Rep. 2001;8:1085–1089. doi: 10.3892/or.8.5.1085. [DOI] [PubMed] [Google Scholar]