

Abstract
AIM: We designed two synthetic-core-specific peptides core 1 (C1) and core 2 (C2), and an E1-specific peptide (E1). We produced specific polyclonal antibodies against these peptides and used the antibodies for detection of HCV antigens on surface and within infected peripheral blood leukocytes.
METHODS: Peripheral blood from a healthy individual who tested negative for HCV RNA was incubated with HCV type 4 infected serum for 1 h and 24 h at 37 °C. Cells were stained by direct and indirect immunofluorescence and measured by flow cytometry.
RESULTS: After 1 h of incubation, antibodies against C1, C2, and E1 detected HCV antigens on the surface of 27%, 26% and 73% of monocytes respectively, while 10%, 5% and 9% of lymphocytes were positive with anti-C1, anti-C2 and anti-E1 respectively. Only 1-3% of granulocytes showed positive staining with anti-C1, anti-C2 and anti E1 antibodies. After 24 h of incubation, we found no surface staining with anti-C1, anti-C2 or anti-E1. Direct immunostaining using anti-C2 could not detect intracellular HCV antigens, after 1 h of incubation with the virus, while after 24 h of incubation, 28% of infected cells showed positive staining. Only plus strand RNA was detectable intracellularly as early as 1 h after incubation, and remained detectable throughout 48 h post-infection. Interestingly, minus RNA strand could not be detected after 1 h, but became strongly detectable intracellularly after 24 h post-infection.
CONCLUSION: Monocytes and lymphocytes are the preferred target cells for HCV infection in peripheral blood leukocytes. Our specific anti-core and anti-E1 antibodies are valuable reagents for demonstration of HCV cell cycle. Also, HCV is capable of infecting and replicating in peripheral blood mononuclear cells as confirmed by detection of minus strand HCV RNA as well as intracellular staining of core HCV antigen.
Keywords: Flow cytometry, Hepatitis C virus, Envelope, Core, Antibodies, Indirect immunofluorescence, Minus and plus RNA strand, Peripheral blood mononuclear cells
INTRODUCTION
Hepatitis C virus (HCV) is the major etiology of non-A, non-B hepatitis that infects 170 million people worldwide. Approximately 70-80% of HCV patients develop chronic hepatitis, 20-30% of which progress to liver cirrhosis[1]. At present, there is no vaccine available to prevent HCV infection, and current therapies are not optimal. The initial steps of HCV infection (binding and entry) that are critical for tissue tropism, and hence pathogenesis, are poorly understood. Studies to elucidate this process have been hampered by the lack of robust cell culture systems or convenient small animal models that can support HCV infection. HCV is an enveloped, positive-strand RNA virus that belongs to the Flaviviridae family. Based on the sequence heterogeneity of the genome, HCV is classified into six major genotypes and 100 subtypes[1]. The viral genome (-9.6 kb) is translated into a single polyprotein of -3 000 amino acids (aa). A combination of host and viral proteases are involved in polyprotein processing to give at least nine different proteins[2]. The structural proteins of HCV are believed to comprise the core protein (~21 kDa) and two envelope glycoproteins: E1 (~31 kDa) and E2 (~70 kDa). Like other enveloped viruses, E1 and E2 proteins are most likely play a pivotal role in HCV life cycle: in the assembly of infectious particles and in the initiation of viral infection by binding to its cellular receptor(s). Since hepatocytes represent the primary site of HCV replication in vivo, the HCV genome has also been found in lymphoid cells. Infection of the lymphoid cells has been implicated in extrahepatic manifestations of HCV infection such as mixed cryoglobulinemia and B-lymphocyte proliferative disorders[3-5]. Virus-cell interaction is a multistep process and frequently involves more than a single receptor. There are, at least, three ways employed by virus to bind its target cells. First, virus can harbor two receptor-binding sites that allow binding to alternative receptors expressed on different cell types (e.g., adenovirus type 37)[6]. Second, virus can bind to a “common” surface molecule that captures and concentrates virus at the cell surface, and this event is followed by binding to a high-affinity primary receptor (e.g., herpes simplex virus)[7]. Third, virus binds to a high-affinity receptor, and this event induces conformational changes leading to the exposure of binding sites for a co-receptor (e.g., HIV type 1)[8,9]. So far, little is known about which mechanism is adopted by HCV to bind and enter target cells. The association of CD81 and the LDL-R with E2 protein or HCV virion, respectively, have led to the assumption that either one may represent the cellular receptor for HCV[10-12]. Despite several reports demonstrating the E2 binding to CD81[13,14], the interaction between HCV virion with this molecule is less clear. CD81 molecule only inhibited the binding of truncated E2 protein, but not HCV virion, to Molt-4 cells[15], suggesting that the HCV virion may use other receptor(s) for entry into cells. Also, Triyanti et al[16] demonstrated that HCV-LPs can be used as a valuable tool to study the mechanism of binding and entry of HCV infection. Further characterization of HCV-LP and its interaction with cells will help us understand the early steps of HCV infection and will facilitate studies to identify other candidate receptor(s) for HCV. In the present study, we aimed to use in-house polyclonal antibodies against HCV peptides from core and envelope regions for the detection of surface and intracellular HCV antigens in PBMCs infected with HCV type 4 positive serum, based on direct and indirect immunofluorescence labeling followed by flow cytometric analysis.
MATERIALS AND METHODS
Infected serum samples
For infection experiments, we utilized serum samples positive for HCV RNA as determined by nested RT-PCR and genotyped as genotype 4 using the method described by Ohno et al[17].
Infection of peripheral blood with HCV type 4
Whole blood cells were obtained from a healthy volunteer (only a single subject was used for the sake of fixing the host factor throughout the experiment). Serum from this blood sample was tested negative for HCV RNA by using a standard nested RT-PCR. Whole blood was incubated at 37 °C for 1, 24 and 48 h with a serum sample that tested positive for HCV RNA (viral loads: 9 million copies/mL, G4 HCV RNA) at a ratio of 20 µL serum to 110 µL whole blood). After incubation, blood cells were thoroughly washed four times with PBS. Detection of HCV RNA strands in peripheral blood cells will be described later.
Design of HCV synthetic peptides
Sequence analysis of HCV quasispecies in local patients (results not shown) revealed several conserved regions within the core and the E1 proteins. We designed four synthetic-core-specific peptides and a E1-specific peptide, and results showed that all peptides detect HCV antibodies in infected sera to varying degrees. The relative sensitivities and specificities of these synthetic peptides in detecting circulating HCV antibodies in infected human sera were described[18]. In the present study, we raised HCV-specific polyclonal antibodies against two core peptides anti-C1 (DVKFPGGGQIVGGVYLLPRR), anti-C2 (IPKA-RRPEGRTWAQPGY) as well as a E1 peptide (anti-E1, GHRMAWDMM).
Production of polyclonal antibodies against core and envelope regions of HCV
New Zealand rabbits (2 rabbits per each peptide and 2 per control, injected only with KLH) were immunized independently with purified synthetic peptides coupled with KLH protein. Equal volume of diluted core 1 (C1), core 2 (C2) and E1 synthetic peptides and Freund’s complete adjuvant were emulsified and injected subcutaneously into the rabbits in three different sites. On d 15 and 28, the rabbits were immunized again with the same protein emulsified with incomplete Freund’s adjuvant. On d 32, the rabbits were killed and sera were separated and stored at -20 °C. For direct immunofluorescence, immunized polyclonal antibodies were digested with pepsin A (porcine 1:60 000 grade (sigma P-7012), St. Louis, MO, USA) at acidic pH and the F(ab)2 portion was labeled with fluorescence isothiocyanate (FITC) according to Hudson and Hay[19].
Surface and intracellular staining of infected peripheral blood cells and flow cytometric analysis
After incubation of normal blood cells with HCV-infected serum positive for blood cells, they were washed four times with phosphate-buffered saline (PBS). Surface labeling was performed by direct and indirect immunofluorescence. Primary polyclonal antibodies (C1, C2 and E1) were incubated with cells for 30 min at 4 °C. RBCs were ruptured by lysis buffer (83% ammonium chloride). Cells were centrifuged and supernatants were removed. Cell pellets were washed twice with PBS containing 1% normal goat serum, then incubated with anti-rabbit IgG labeled with FITC (1:100 dilution, Sigma, St. Louis, MO, USA) for 30 min. Cells were washed once more, resuspended in PBS and kept at 4 °C until flow cytometric analysis. For intracellular staining, cells were incubated with 4% paraformaldehyde for 10 min and 0.1% Triton X-100 in Tris buffer (pH 7.4) for 6 min. After washing with PBS, cells were incubated with FITC-labeled F(ab)2 portion of HCV core antibody (at 1:2 000 dilution) for 30 min at 4 °C. Cells were washed thrice with PBS containing 1% normal goat serum and cells were suspended in 500 µL and analyzed.
Isolation of PBMC and extraction of RNA
Infected peripheral blood cells were isolated, as reported by Lohr et al[20] Briefly, peripheral blood samples were diluted with 5 volumes of a freshly prepared RBC lysis buffer (38.8 mmol/L NH4Cl, 2.5 mmol/L K2HCO3, 1 mmol/L EDTA, pH 8.0), incubated at room temperature for 10 min and the nucleated cells were precipitated and washed in the same buffer to remove adherent viral particles before lysis in 4 mol/L guanidinium isothiocyanate containing 25 mmol/L sodium citrate, 0.5% sarcosyl and 0.1 mol/L β-mercaptoethanol. Cellular RNA was extracted using the single-step method described originally by Chomczynski and Sacchi[21].
PCR of genomic and anti-genomic RNA strands of HCV
Reverse transcription-nested PCR was carried out according to Lohr et al[20] with few modifications. Retrotranscription was performed in 25 µL reaction mixture containing 20 U of AMV reverse transcriptase (Clontech, USA) with either 400 ng of total PBMCs RNA or 3 µL of purified RNA from serum samples as template, 40 U of RNAsin (Clontech, USA), a final concentration of 0.2 mmol/L from each dNTP (Promega, Madison, WI, USA) and 50 pmoL of the reverse primer 1CH (for plus strand) or 50 pmol of the forward primer 2CH (for minus strand). The reaction was incubated at 42 °C for 60 min and denatured at 98 °C for 10 min. Amplification of the highly conserved 5-UTR sequences was done using two rounds of PCR with two pairs of nested primers. First round amplification was done in 50 µL reaction mixture, containing 50 pmol from each of 2CH forward primer and P2 reverse primer and P2 reverse primer, 0.2 mmol/L from each dNTP, 10 µL from RT reaction mixture as template and 2 U of Taq DNA polymerase (Promega, USA) in a 1 buffer supplied with the enzyme. The thermal cycling protocol was as follows: 1 min at 94 °C, 1 min at 55 °C and 1 min at 72 °C for 30 cycles. The second round amplification was done similar to the first round, except for use of the nested reverse primer D2 and forward primer F2 at 50 pmol each. A fragment of 179 bp was identified in positive samples. Primer sequences were as follows:
1CH: 5’-ggtgcacggtctacgagacctc-3’
2CH: 5’-aactactgtcttcacgcagaa-3’
P2: 5’-tgctcatggtgcacggtcta-3’
D2: 5’-actcggctagcagtctcgcg-3’
F2: 5’-gtgcagcctccaggaccc-3’
To control false detection of negative-strand HCV RNA and known variations in PCR efficiency[22,23], specific control assays and rigorous standardization of the reaction were employed. Specific control assays were included: (1) cDNA synthesis without RNA templates to exclude product contamination; (2) cDNA synthesis without RTase to exclude Taq polymerase RTase activity; (3) cDNA synthesis and PCR step done with only the reverse or forward primer to confirm no contamination from mixed primers. These controls were consistently negative. In addition, cDNA synthesis was carried out using only one primer present followed by heat inactivation of RTase activity at 95 °C for 1 h, in an attempt to diminish false detection of negative-strand prior to the addition of the second primer.
RESULTS
Surface detection of HCV antigens by flow cytometry on infected blood leukocytes
The borders of the respective cell regions of lymphocytes, monocytes and granulocytes were clearly defined on the cytograms of flow cytometry based on the size and the granularity of cells (Figure 1). Using control healthy blood, no immunostained positive cells were found using any of the polyclonal antibodies (C1 or C 2 and anti-E1) compared with normal rabbit serum. After 1 h of incubation at 37 °C with HCV positive serum, indirect immunofluorescence-labeled anti-E1, antibody demonstrated positive staining in 9%, 73% and 1-2% of lymphocytes, monocytes and granulocyte respectively. Anti-C1 demonstrated positive staining in 10%, 27%, and 1% of lymphocytes, monocytes and granulocyte respectively. Staining with anti-C2 showed positive cells in 5%, 26% and 1% of lymphocytes, monocytes, and granulocyte respectively (Table 1 and Figure 1). Interestingly, after 24 h of incubation with HCV positive serum, no detectable viral antigens were demonstrated on the surface of any cell type.
Figure 1.
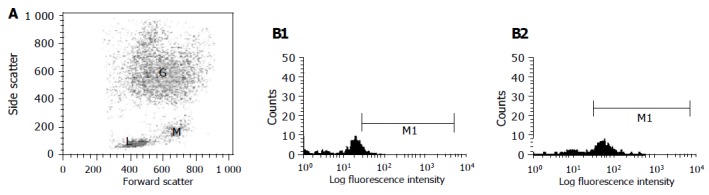
The borders of the respective cell regions of lymphocytes (L), monocytes (M) and granulocytes (G) were clearly defined on the cytograms of flow cytometry based on the size and the granularity (x-axis, forward scattered cells and y-axis, side scattered cells of cells after surface immunostaining) (A); Histograms from gated monocytes obtained from fluorescent activated cell sorter analysis of HCV-infected peripheral blood mononuclear leukocytes. Cells were stained with anti-E1 antibody against HCV E1 region. Histograms represent gated monocytes from (B1) infected cells stained with normal rabbit serum as control or (B2) infected cells stained with antibody against E1 after incubation of blood with serum sample after 1 h at 37 °C in which x-axis represents fluorescence intensity. M1 is marker for positive cell population.
Table 1.
Percentage of positive cells after 1-h incubation of peripheral blood with serum positive for HCV RNA
| Lymphocytes | Monocytes | Granulocytes | |
| Antibody | percentage of | percentage of | percentage of |
| positive cells | positive cells | positive cells | |
| E1 | 5 | 73 | 1 |
| C1 | 10 | 27 | 2 |
| C2 | 9 | 26 | 3 |
Positive cells were determined by indirect immunofluorescence and flow cytometric analysis.
Intracellular staining with direct immunofluorescence
Direct immunostaining of total peripheral mononuclear cells using FITC-F(ab)2 portion of anti C2 antibody could not detect intracellular viral antigens after 1 h of incubation with HCV-positive serum. However, after 24 h of incubation, 28% of infected cells stained positive for viral core antigen (Figure 2).
Figure 2.
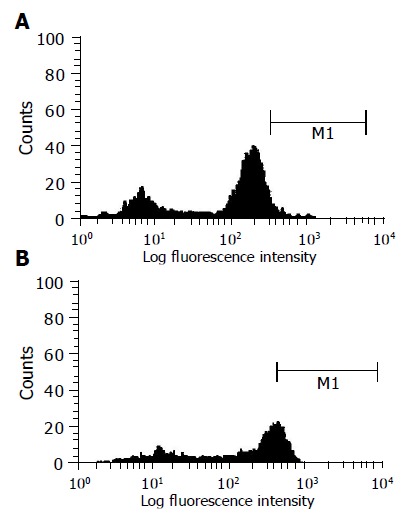
Histogram from gated leukocytes obtained from fluorescent activated cell sorter analysis of HCV-infected peripheral blood mononuclear leukocytes. Cells were stained intracellularly with anti-C2 antibody conjugated with FITC. Histogram represents gated leukocytes from (A) healthy uninfected cells or (B) infected cells stained with anti-C2 antibody conjugated with FITC after incubation of blood with serum sample after 24 h at 37 °C in which x-axis represents fluorescence intensity. M1 is marker for positive cell population.
RT-PCR for minus and plus RNA strand of HCV RNA in PBMCs
The genomic plus (+) strand was weakly detectable in PBMCs after 1 h of incubation, whereas antigenomic (-) strand was undetectable. After 24 and 48 h of incubation, both strands were detectable with stronger signal of the anti-genomic (-) strand (Figure 3).
Figure 3.
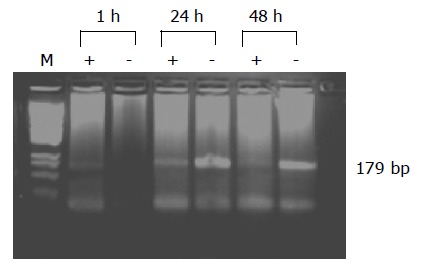
Analysis of HCV RNA plus strand and minus strand from infected PBMC. PCR amplification products of the genomic strand (+) and the replicative strand (-) of HCV from total cellular RNA of PBMC infected with HCV for 1, 24 and 48 h. Lane M represents the M.W. marker PGem.
DISCUSSION
Extrahepatic HCV replication has long been a controversial topic, since the finding of the high rate of re-infection of grafts after orthotopic liver transplantation in patients with the end-stage HCV-induced liver diseases. However, whether PBMCs are supportive for HCV replication is still uncertain. The detection of the minus strand HCV RNA is thought to be a reasonable marker for HCV replication, because the minus RNA strand is the replicative intermediate of HCV. In past decade, several reports on the detection of HCV RNA in PBMCs have been published[24-26]. Gribier et al[27] incubated PBMCs from healthy donors with HCV positive sera, and detected HCV RNA plus and minus strands using RT-PCR and in situ hybridization. Results of Gong et al[28] showed that HCV is capable of infecting and replicating in PBMCs, and HCV NS5 protein was clearly expressed in these cells. On a previous study by El-Awady et al[29] we utilized PBMCs as a cellular component to assess viral replication in chronic HCV patients. This study demonstrated higher sensitivity of concomitant PCR amplification of plus and minus strands in PBMCs together with plus strand in serum samples over the traditional amplification of viral RNA in serum. Later, we employed this apparent intracellular amplification in PBMCs as a predictor for treatment relapse after interferon therapy[30]. After 1 h of incubation, in the present study, only plus RNA strand of the virus was weakly detectable. When incubation with positive serum was extended for 24 and 48 h, both plus and minus RNA strands have become strongly detectable (Figure 3). Flow cytometry is advantageous over RT-PCR in its ability to count directly the rate of infection into the cells. Furthermore, it is more specific than traditional immunocytochemical analyses via its ability to quantify the intensity of fluorescence per cell, thus facilitating clear distinction between specific and non-specific immunofluorescence. A greater number of cells are allowed to be counted, which is very important while considering that in vitro viral infections are variable biological phenomena. This gives us the possibility of testing several replicates simultaneously and elevating the accuracy in statistical analysis. In case of cells that are relatively less susceptible, such as PBML, it is desired to count large number of cells to obtain a significant number of positives, turning manual counting very laborious. Therefore, in our case, to gain a better understanding of the cell-specific tropism of HCV, we designed the experiments to detect HCV antigen on blood leukocyte subsets, lymphocytes, monocytes and granulocytes. Simultaneously, we planned to evaluate the usefulness of specific polyclonal anti-sera raised against synthetic core and envelope HCV peptides made from conserved sequences among several HCV isolates[18]. In a parallel investigation, the antibodies currently used demonstrated 100% sensitivity and 100% specificity in detecting circulating viral antigens in HCV RNA-positive and HCV RNA-negative subjects, respectively (data not shown). Results presented here revealed that about 73% of infected monocytes were immunostained positive for envelope antigen. A well-defined HCV positive population in infected peripheral blood leukocytes showed that the monocytes are the preferred target for HCV. However, the results of previous studies have been conflicting; several investigators have reported inconsistent presence of HCV in B and T lymphocytes[26,31]. However, flow cytometry using specific monoclonal antibody could detect the presence of HCV core antigen that was demonstrated in monocytes but not in lymphocytes[32]. These reports demonstrated the presence of HCV RNA which was detected in peripheral blood mononuclear cells (PBMC) by PCR in 17 of 24 HCV-infected patients with chronic hepatitis with or without cirrhosis. In 29% of patients whose PBMC contained HCV RNA, flow cytometry with a murine monoclonal antibody to HCV core epitopes revealed cytoplasmic staining of peripheral blood monocytes, whereas cell surface of monocytes and lymphocytes did not stain for HCV core epitopes. The discrepancy between the current results and those of Bouffard et al[32] may be related to either the difference between in vivo and in vitro infection of PBMCs or to the degree of the antibody specificity. In the present study, HCV tropism in peripheral leukocytes is not only limited to polymorphonuclear leukocytes, since granulocytes were also shown to stain positive with HCV antibodies after short incubation with positive sera. Since minus RNA strand of the HCV can hardly be detected in infected serum, its detection in PBMCs can be taken as a molecular indicator of intracellular genomic replication of HCV and ruling out false positive, due to contamination of cellular preparations with infected sera. In acute HCV infection, HCV RNA minus strand is rare in PBMCs, but in the chronic group, the minus strand HCV RNA is common in the PBMCs (14 of 35, 40%), which is similar to what Chang et al[33] reported. The ratio of HCV RNA minus strand detected in chronic hepatitis C is much higher than that in acute hepatitis C, suggesting that the replication of HCV in PBMCs may play an important role in the processes of chronicity, and the mechanism could be that HCV in PBMCs escapes from host immunity, and makes the infection of HCV persistent. On one hand, the dysfunction of the HCV-infected PBMCs leads to immune function decline and this becomes more difficult for the host to clear intrahepatic HCV[34]. On the other hand, some authors are still arguing that the minus strand HCV RNA in the blood cells including PBMC may be artifacts from contamination or passive absorption by circulating virus[11] or self-priming/mis-priming during PCR reaction[35,36]. For this reason, the expression of HCV-related proteins in extrahepatic cells has become the key point for demonstrating intracellular HCV replication. HCV was repeatedly reported to replicate and express structural and non-structural proteins, primarily in PBMC[28,37,38]. In the present study, we utilized the combined results of nested RT-PCR of plus and minus RNA strands coupled with core and E1 protein expression to follow steps of viral binding, entry, replication and protein expression in human PBMCs. The detection of E1 and core antigens on cell surface as early as 1 h of incubation with virus, followed by disappearance of viral antigens after 24 h, suggests that viral entrance into cells occurs within 24 h. The concomitant intracellular appearance of E1 and core antigens suggests that binding, entry and expression of structural proteins occurs within 24 h. At the level of genomic replication, failure of minus strand detection after 1 h, followed by strong detection after 24 h supports the hypothesis that processing of HCV polyprotein and release of viral antigens with subsequent acceleration of genomic replication of viral RNA occurs within the first 24 h of infection. In conclusion, we suggest that the present method for detection of E1 and core antigens is useful in monitoring of HCV life cycle and can contribute to analysis of HCV infection more precisely. Therefore, the study of virus-host cell interaction may lead to interesting and probably non-expected concepts of HCV pathology.
Footnotes
Co-first-authors: Mostafa K El-Awady and Ashraf A Tabll
Co-correspondents: Ashraf A Tabll
Science Editor Guo SY Language Editor Elsevier HK
References
- 1.World Health Organization. Global surveillance and control of hepatitis C. Report of a WHO Consultation organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp, Belgium. J Viral Hepat. 1999;6:35–47. [PubMed] [Google Scholar]
- 2.Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol. 2000;81:1631–1648. doi: 10.1099/0022-1317-81-7-1631. [DOI] [PubMed] [Google Scholar]
- 3.Agnello V, Chung RT, Kaplan LM. A role for hepatitis C virus infection in type II cryoglobulinemia. N Engl J Med. 1992;327:1490–1495. doi: 10.1056/NEJM199211193272104. [DOI] [PubMed] [Google Scholar]
- 4.Quinn ER, Chan CH, Hadlock KG, Foung SH, Flint M. Levy S. The B cell-receptor of a hepatitis C virus (HCV)-associated non-Hodgkin lymphoma binds the viral E2 envelope protein, implicating HCV in lymphomagenesis. Blood. 2001;98:3745–3749. doi: 10.1182/blood.v98.13.3745. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt WN, Stapleton JT, LaBrecque DR, Mitros FA, Kirby PA, Phillips MJ, Brashear DL. Hepatitis C virus (HCV) infection and cryoglobulinemia: analysis of whole blood and plasma HCV-RNA concentrations and correlation with liver histology. Hepatology. 2000;31:737–744. doi: 10.1002/hep.510310326. [DOI] [PubMed] [Google Scholar]
- 6.Wu E, Fernandez J, Fleck SK, Von Seggern DJ S, Nemerow GR. A 50-kDa membrane protein mediates sialic acid-independent binding and infection of conjunctival cells by adenovirus type 37. Virology. 2001;279:78–89. doi: 10.1006/viro.2000.0703. [DOI] [PubMed] [Google Scholar]
- 7.Campadelli-Fiume G, Cocchi F, Menotti L, Lopez M. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev Med Virol. 2000;10:305–319. doi: 10.1002/1099-1654(200009/10)10:5<305::aid-rmv286>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 8.Finnegan CM, Berg W, Lewis GK, DeVico AL. Antigenic properties of the human immunodeficiency virus envelope during cell-cell fusion. J Virol. 2001;75:11096–11105. doi: 10.1128/JVI.75.22.11096-11105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klasse PJ, Bron R, Marsh M. Mechanisms of enveloped virus entry into animal cells. Adv Drug Deliv Rev. 1998;34:65–91. doi: 10.1016/S0169-409X(98)00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 11.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci USA. 1999;96:12766–12771. doi: 10.1073/pnas.96.22.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monazahian M, Böhme I, Bonk S, Koch A, Scholz C, Grethe S, Thomssen R. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J Med Virol. 1999;57:223–229. doi: 10.1002/(sici)1096-9071(199903)57:3<223::aid-jmv2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 13.Flint M, Maidens C, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, Higginbottom A, Levy S, McKeating JA. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J Virol. 1999;73:6235–6244. doi: 10.1128/jvi.73.8.6235-6244.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higginbottom A, Quinn ER, Kuo CC, Flint M, Wilson LH, Bianchi E, Nicosia A, Monk PN, McKeating JA, Levy S. Identification of amino acid residues in CD81 critical for interaction with hepatitis C virus envelope glycoprotein E2. J Virol. 2000;74:3642–3649. doi: 10.1128/jvi.74.8.3642-3649.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wünschmann S, Medh JD, Klinzmann D, Schmidt WN, Stapleton JT. Characterization of hepatitis C virus (HCV) and HCV E2 interactions with CD81 and the low-density lipoprotein receptor. J Virol. 2000;74:10055–10062. doi: 10.1128/jvi.74.21.10055-10062.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Triyatni M, Saunier B, Maruvada P, Davis AR, Ulianich L, Heller T, Patel A, Kohn LD, Liang TJ. Interaction of hepatitis C virus-like particles and cells: a model system for studying viral binding and entry. J Virol. 2002;76:9335–9344. doi: 10.1128/JVI.76.18.9335-9344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohno O, Mizokami M, Wu RR, Saleh MG, Ohba K, Orito E, Mukaide M, Williams R, Lau JY. New hepatitis C virus (HCV) genotyping system that allows for identification of HCV genotypes 1a, 1b, 2a, 2b, 3a, 3b, 4, 5a, and 6a. J Clin Microbiol. 1997;35:201–207. doi: 10.1128/jcm.35.1.201-207.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Awady MK, El-Demellawy MA, Khalil SB, Galal D, Goueli SA. Synthetic peptide-based immunoassay as a supplemental test for HCV infection. Clin Chim Acta. 2002;325:39–46. doi: 10.1016/s0009-8981(02)00245-0. [DOI] [PubMed] [Google Scholar]
- 19.Hudson L, Hay FC. Antibody as a probe. In: Practical Immunology, third edition, Blackwell Scientific Publications Oxford London; 1989. pp. 34–43. [Google Scholar]
- 20.Löhr HF, Goergen B, Meyer zum Büschenfelde KH, Gerken G. HCV replication in mononuclear cells stimulates anti-HCV-secreting B cells and reflects nonresponsiveness to interferon-alpha. J Med Virol. 1995;46:314–320. doi: 10.1002/jmv.1890460405. [DOI] [PubMed] [Google Scholar]
- 21.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 22.Crotty PL, Staggs RA, Porter PT, Killeen AA, McGlennen RC. Quantitative analysis in molecular diagnostics. Hum Pathol. 1994;25:572–579. doi: 10.1016/0046-8177(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 23.Reischl U, Kochanowski B. Quantitative PCR. A survey of the present technology. Mol Biotechnol. 1995;3:55–71. doi: 10.1007/BF02821335. [DOI] [PubMed] [Google Scholar]
- 24.Wang JT, Sheu JC, Lin JT, Wang TH, Chen DS. Detection of replicative form of hepatitis C virus RNA in peripheral blood mononuclear cells. J Infect Dis. 1992;166:1167–1169. doi: 10.1093/infdis/166.5.1167. [DOI] [PubMed] [Google Scholar]
- 25.Taliani G, Badolato C, Lecce R, Poliandri G, Bozza A, Duca F, Pasquazzi C, Clementi C, Furlan C, De Bac C. Hepatitis C virus RNA in peripheral blood mononuclear cells: relation with response to interferon treatment. J Med Virol. 1995;47:16–22. doi: 10.1002/jmv.1890470105. [DOI] [PubMed] [Google Scholar]
- 26.Zignego AL, De Carli M, Monti M, Careccia G, La Villa G, Giannini C, D'Elios MM, Del Prete G, Gentilini P. Hepatitis C virus infection of mononuclear cells from peripheral blood and liver infiltrates in chronically infected patients. J Med Virol. 1995;47:58–64. doi: 10.1002/jmv.1890470112. [DOI] [PubMed] [Google Scholar]
- 27.Cribier B, Schmitt C, Bingen A, Kirn A, Keller F. In vitro infection of peripheral blood mononuclear cells by hepatitis C virus. J Gen Virol. 1995;76(Pt10):2485–2491. doi: 10.1099/0022-1317-76-10-2485. [DOI] [PubMed] [Google Scholar]
- 28.Gong GZ, Lai LY, Jiang YF, He Y, Su XS. HCV replication in PBMC and its influence on interferon therapy. World J Gastroenterol. 2003;9:291–294. doi: 10.3748/wjg.v9.i2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El-Awady MK, Ismail SM, El-Sagheer M, Sabour YA, Amr KS, Zaki EA. Assay for hepatitis C virus in peripheral blood mononuclear cells enhances sensitivity of diagnosis and monitoring of HCV-associated hepatitis. Clin Chim Acta. 1999;283:1–14. doi: 10.1016/s0009-8981(99)00007-8. [DOI] [PubMed] [Google Scholar]
- 30.El-Awady MK, Abdel Rahman MM, Ismail SM, Amr KS, Omran M, Mohamed SA. Prediction of relapse after interferon therapy in hepatitis C virus-infected patients by the use of triple assay. J Gastroenterol Hepatol. 2003;18:68–73. doi: 10.1046/j.1440-1746.2003.02919.x. [DOI] [PubMed] [Google Scholar]
- 31.Gabrielli A, Manzin A, Candela M, Caniglia ML, Paolucci S, Danieli MG, Clementi M. Active hepatitis C virus infection in bone marrow and peripheral blood mononuclear cells from patients with mixed cryoglobulinaemia. Clin Exp Immunol. 1994;97:87–93. doi: 10.1111/j.1365-2249.1994.tb06584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouffard P, Hayashi PH, Acevedo R, Levy N, Zeldis JB. Hepatitis C virus is detected in a monocyte/macrophage subpopulation of peripheral blood mononuclear cells of infected patients. J Infect Dis. 1992;166:1276–1280. doi: 10.1093/infdis/166.6.1276. [DOI] [PubMed] [Google Scholar]
- 33.Chang TT, Young KC, Yang YJ, Lei HY, Wu HL. Hepatitis C virus RNA in peripheral blood mononuclear cells: comparing acute and chronic hepatitis C virus infection. Hepatology. 1996;23:977–981. doi: 10.1002/hep.510230506. [DOI] [PubMed] [Google Scholar]
- 34.Müller HM, Pfaff E, Goeser T, Kallinowski B, Solbach C, Theilmann L. Peripheral blood leukocytes serve as a possible extrahepatic site for hepatitis C virus replication. J Gen Virol. 1993;74(Pt 4):669–676. doi: 10.1099/0022-1317-74-4-669. [DOI] [PubMed] [Google Scholar]
- 35.Mihm S, Hartmann H, Ramadori G. A reevaluation of the association of hepatitis C virus replicative intermediates with peripheral blood cells including granulocytes by a tagged reverse transcriptase/polymerase chain reaction technique. J Hepatol. 1996;24:491–497. doi: 10.1016/s0168-8278(96)80171-1. [DOI] [PubMed] [Google Scholar]
- 36.Lerat H, Berby F, Trabaud MA, Vidalin O, Major M, Trépo C, Inchauspé G. Specific detection of hepatitis C virus minus strand RNA in hematopoietic cells. J Clin Invest. 1996;97:845–851. doi: 10.1172/JCI118485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sansonno D, Iacobelli AR, Cornacchiulo V, Iodice G, Dammacco F. Detection of hepatitis C virus (HCV) proteins by immunofluorescence and HCV RNA genomic sequences by non-isotopic in situ hybridization in bone marrow and peripheral blood mononuclear cells of chronically HCV-infected patients. Clin Exp Immunol. 1996;103:414–421. doi: 10.1111/j.1365-2249.1996.tb08296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen L, Chen P, Fan G, Li L, Liu C. Localization of hepatitis C virus core protein in the nucleus of peripheral blood mononuclear cells of hepatitis C patients. Zhonghua ShiYan He LinChuang BingDuXue ZaZhi. 2002;16:37–39. [PubMed] [Google Scholar]