

Abstract
Infiltrating lobular breast cancer (ILC) is the most common special breast cancer subtype. With mutational or epigenetic inactivation of the cell adhesion molecule E-cadherin (CDH1) being confined almost exclusively to ILC, this tumor entity stands out from all other types of breast cancers. The molecular basis of ILC is linked to loss of E-cadherin, as evidenced by human CDH1 germline mutations and conditional knockout mouse models. A better understanding of ILC beyond the level of descriptive studies depends on physiologically relevant and functional tools. This review provides a detailed overview on ILC models, including well-characterized cell lines, xenograft tumors and genetically engineered mouse models. We consider advantages and limitations of these models and evaluate their representativeness for human ILC. The still incompletely defined mechanisms by which loss of E-cadherin drives malignant transformation are discussed based on recent findings in these models. Moreover, candidate genes and signaling pathways potentially involved in ILC development and progression as well as anticancer drug and endocrine resistance are highlighted.
Introduction
Infiltrating lobular breast cancer (ILC) is the most common special breast cancer (BC) subtype and accounts for 10 to 15% of all mammary carcinomas. ILCs are defined by histomorphological characteristics, such as small, discohesive and nonpolarized tumor cells with little nuclear atypia and a single-file invasion pattern (Figure 1A). Although first termed ILCs in the 1940s [1], these tumors had been recognized as a histologically distinct entity (scirrhous spheroidal cell carcinoma) long before the terminus ILC became established [2].
Figure 1.
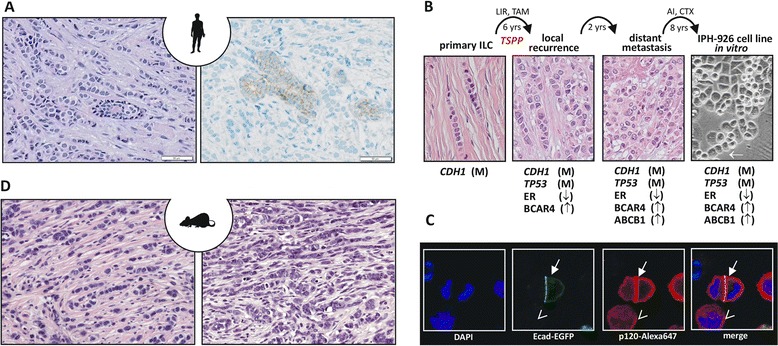
Infiltrating lobular breast cancer, a human infiltrating lobular breast cancer cell line and a genetically engineered mouse model for infiltrating lobular breast cancer. (A) Representative photomicrographs of infiltrating lobular breast cancer (ILC) stained with hematoxylin and eosin (left) or subjected to immunohistochemistry for E-cadherin (right). Note the E-cadherin-positive normal mammary gland duct surrounded by E-cadherin-negative ILC cells. (B) Molecular evolution of the IPH-926 ILC cell line. Photomicrographs show the histomorphology of the corresponding clinical tumor specimens and the IPH-926 cell line in vitro. Arrow highlights a single file linear cord reminiscent of primary ILC. AI, aromatase inhibitor; CTX, various conventional chemotherapies; LIR, local irradiation; TAM, tamoxifen; TSPP, transition to a secondary pleomorphic phenotype; yrs, years; M, mutational inactivation; ↑, overexpression; ↓, loss of expression. (C) Reconstitution of E-cadherin in ILC cells induces relocation of p120-catenin (p120) to the cell membrane. Shown are fluorescence images of IPH-926 cells transiently transfected with an E-cadherin–enhanced green fluorescent protein (Ecad-EGFP) expression construct and stained with an anti-p120-Alexa647 antibody. Closed arrow, cells with ectopic expression of Ecad-EGFP; open arrow, a cell without Ecad-EGFP. Note the accentuated membranous p120 staining in cells expressing Ecad-EGFP. DAPI, 4′,6-diamidino-2-phenylindole. (D) Mouse ILC from genetically engineered mouse models. Left, a tumor reminiscent of classic ILC; right, a pleomorphic mouse ILC. Both micrographs generated from the WAPcre;Cdh1 F/F ;Trp53 F/F mouse ILC model.
In the 1980s, uvomorulin (E-cadherin) was discovered as a transmembrane glycoprotein that mediates cis and trans calcium-dependent homotypic cell adhesion in epithelial tissues, thereby controlling cell polarity and proper blastocyst formation during embryogenesis [3]. Together with the associated catenins, E-cadherin forms the adherens junction (AJ) on the apical side of the cell, where it links the plasma membrane to the actin and microtubule cytoskeleton [4]. E-cadherin is of pivotal relevance for two special tumor entities, namely ILC and diffuse gastric cancer (DGC). In the 1990s, it was reported that nearly all ILCs and their adjacent intraepithelial precursor lesions, termed lobular carcinoma in situ (LCIS), lacked E-cadherin expression [5,6]. E-cadherin is encoded by the CDH1 gene on chromosome 16q22. Loss of E-cadherin in LCIS and ILC is due to somatic CDH1 frameshift mutation and loss of heterozygosity or aberrant CDH1 promoter methylation [7-9]. CDH1 germline mutations are associated with the hereditary DGC syndrome [10] and ILC belongs to the tumor spectrum in these patients [11]. Cases of LCIS and ILC associated with CDH1 germline mutation without gastric cancer are also increasingly recognized [12]. In addition to genetically engineered mouse (GEM) models (see below), these findings from medical genetics have provided evidence that E-cadherin functions as a tumor suppressor and that its inactivation underpins ILC etiology.
The molecular impact of E-cadherin inactivation has been studied extensively in the context of a biological process referred to as the epithelial-to-mesenchymal transition. While these studies have provided vast insight into the epigenetic mechanisms that can silence E-cadherin and their functional consequences, they also indicated that E-cadherin inactivation through transcriptional repressors is not the main driver of ILC development. Based on immunohistochemical and genetic studies, it is now established that loss of E-cadherin triggers secondary changes affecting several cadherin/catenin complex molecules. These changes include loss of β-catenin and aberrant cytoplasmic and/or nuclear localization of p120-catenin (p120) [13,14].
Almost all ILCs are estrogen receptor (ER)-positive and belong to the luminal or normal-like molecular subtype [15]. Their proliferation is slow and mostly estrogen dependent [16]. Overexpression or amplification of the ERBB2 oncogene is rare, although somatic activating mutations have been reported [17]. In contrast, mutational activation of the PIK3CA oncogene is a dominant feature in ILC [18]. TP53 mutations are rare [15], except for a more aggressive ILC variant termed pleomorphic ILC, which is more often ER-negative and occasionally ERBB2-positive [19,20]. Based on these findings and various genomic profiling studies [15,21,22], it became evident that ILC represents a biologically distinct entity.
A better understanding of ILC beyond the descriptive level of genetic and histopathological studies depends on clinically relevant models. This review provides an overview of mouse and human ILC models and their relevance for understanding ILC biology.
Review
Human ILC cell lines are a rare resource
Human BC cell lines are a powerful experimental tool. In many instances, information derived from in vitro models with BC cell lines has improved the understanding of cancer [23]. In other studies, potentially misleading data have been generated because cell lines were not representative for the tumor type investigated. Numerous studies have aimed to (re)classify BC cell lines in terms of their tumor origin and molecular properties. This revealed a lack of ILC cell lines [23,24]. Among more than 100 BC cell lines established to date, only seven can be tracked back to histologically confirmed or suspected primary ILCs (Table 1) [25-31]. Importantly, an ILC origin cannot be concluded simply based on the lack of E-cadherin expression. This is because in vitro culturing can induce epithelial-to-mesenchymal transition and a subsequent epigenetic silencing of E-cadherin [32]. In particular, this applies to BC cell lines with a basal molecular subtype that have most probably undergone epithelial-to-mesenchymal transition in culture. A renowned example is MDA-MB-231. The MDA-MB-231 cell line lacks E-cadherin due to hypermethylation, but is hardly comparable with ILC given its fast proliferation, its basal-like expression profile and its actual origin from infiltrating ductal BC [32]. The list of human BC cell lines similar to MDA-MB-231 is long. While the majority of in vitro studies related to the function of E-cadherin in BC have been conducted with cell lines such as MDA-MB-231, these cell lines are inappropriate as models for ILC because they have not based their tumor evolution on E-cadherin loss. Authentic human ILC cell lines are rare and therefore studying E-cadherin function in bona fide ILC cells is just in its beginnings [14,33]. The following section describes human ILC cell lines and their molecular properties.
Table 1.
Human infiltrating lobular breast cancer cell models
| Protein expression | Mutational status | Molecular subtype | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor origin | Tissue | Ecad | ER | PR | ERBB2 | CDH1 | TP53 | CTNNA1 | References | ||
| Cell lines | |||||||||||
| MDA-MB-134 | Unknown (ILC?) | A | Neg | Pos | Neg | Neg | muta | wt/mutd | wt | lum | [25,30,34-40,45] |
| MDA-MB-330 | ILC | P | Pos | Neg | Neg | Pos | wt | mute | muti | ErbB2-pos | [26,46] |
| MA-11 | Mixed ITC/ILC | BM | Pos | Neg | Neg | Neg | NA | NA | NA | NA | [27] |
| SUM-44PE | Unknown (ILC?) | P | Neg | Pos | Pos | Neg | mutb | mutf | wt | lum | [28,33,36-38] |
| HCC-2185 | ILC | P | NA | Neg | Neg | Pos | NA | NA | NA | lum | [24,29] |
| IPH-926 | ILC | A | Neg | Neg | Neg | Neg | mutc | mutg | wt | lum | [30,33,41-43,70] |
| BCK-4 | Unknown (muc-ILC?) | P | Neg | Pos | Pos | Neg | na | na | NA | NA | [31] |
| Xenograft tumors | |||||||||||
| HBCX-7 | ILC | PT | NA | Neg | Neg | Neg | NA | wt | NA | NA | [48] |
| HBCX-19 | ILC | PT | NA | Neg | Neg | Neg | NA | muth | NA | NA | [48] |
| HBCX-36 | ILC | PT | NA | Pos | Pos | Pos | NA | NA | NA | NA | [49] |
| HCI-005 | Mixed ILC/IDC | P | Pos | Pos | Pos | Pos | NA | NA | NA | NA | [47] |
| HCI-013 | ILC | P | Neg | Pos | NA | NA | NA | NA | NA | NA | [36] |
All information compiled from the literature; molecular subtype determined by microarray expression profiling in different studies. A, malignant ascites; BM, bone metastasis; Ecad, E-cadherin; ER, estrogen receptor; IDC, infiltrating ductal breast cancer; ILC, infiltrating lobular cancer; ITC, infiltrating tubular cancer; lum, luminal; muc-ILC, ILC with extracellular mucin; mut, mutated; NA, not assessed; neg, negative; P, malignant pleural effusion; pos, positive; PR, progesterone receptor; PT, primary breast tumor; wt, wild-type. ac.688del145; p.L230EfsX4. bc.1269delT; p.F423LfsX8. cc.241ins4;p.V82fsX93. dc.853G > A; p.E285K. Different mutational status in different studies; possibly a TP53-mutated subclone exists. ec.659C > T; p.Y220C. fc.82_84delinsCA; pE28fsX16. gc.853G > A; p.E285K. hSequence unknown. ic.1322C > G; p.S441X.
In vitro models based on human infiltrating lobular breast cancer cell lines
The MDA-MB-134 cell line was initially reported to derive from infiltrating ductal BC [25]. Reis-Filho and colleagues reclassified this cell line as ILC (Table 1) [34]. MDA-MB-134 is E-cadherin-negative and ER-positive and belongs to the luminal molecular subtype [24]. MDA-MB-134 harbors a homozygous deletion of CDH1 exon 6, which results in a frameshift and a premature stop codon [30,35]. Proliferation of MDA-MB-134 is moderately fast (doubling time of about 2 days) and depends on estrogenic stimulation [36,37]. MDA-MB-134 harbors a gain at chromosome 8p11-p12, an amplicon also common in primary ILCs [34]. MDA-MB-134 overexpresses FGFR1, which maps to chromosome 8p11-p12, and small interfering RNA-mediated silencing or inhibition of FGFR1 increases sensitivity to estrogen withdrawal or tamoxifen [36,38]. Accordingly, FGFR1 is thought to induce endocrine resistance in ILC. This is of relevance because endocrine control is the most important pharmacological treatment strategy for patients with ILC [16]. However, MDA-MB-134 cells also overexpress ZNF703, a newly identified oncogene involved in endocrine resistance. The ZNF703 gene is located <1 Mb upstream of FGFR1 and small interfering RNA-mediated silencing of ZNF703 also decreases viability of MDA-MB-134 [39]. Using MDA-MB-134 as a model, recent studies proposed that tamoxifen has a partially agonistic activity in ILC. According to these studies, ILC proliferation is induced rather than inhibited by tamoxifen, an effect attributed to ZNF703 [36,40]. An MDA-MB-134 subclone with an activating mutation of the KRAS oncogene and altered response to FGFR1 inhibition has also been reported [38].
The SUM-44PE cell line is another accepted ILC model (Table 1) [36,37]. SUM-44PE is E-cadherin-negative and ER-positive and was derived from a malignant pleural effusion. The corresponding primary tumor, presumably an ILC, remained uncharacterized [28]. Compared with MDA-MB-134, SUM-44PE has a shorter doubling time (approximately 1 day), which may be due to amplification of cyclin D1 (CCND1), and is also responsive to steroid hormones. SUM-44PE harbors homozygous frameshift mutations in the CDH1 and TP53 tumor suppressor genes [35]. The SUM-44LCCTam subclone was established by chronic in vitro selection of SUM-44PE against tamoxifen. SUM-44-LCCTam cells overexpress ERRγ, an orphan nuclear receptor, which induces endocrine resistance [37]. Like MDA-MB-134, SUM-44PE cells overexpress FGFR1. Contrary to MDA-MB-134, silencing of FGFR1 only modestly increases sensitivity to estrogen withdrawal or tamoxifen [38].
The IPH-926 cell line was derived from malignant ascites of a metastatic ILC [30]. The corresponding primary tumor, a grade 1 ER-positive ILC, was diagnosed 16 years before establishment of the cell line (Figure 1B). The patient had undergone breast-conserving surgery and adjuvant tamoxifen therapy but experienced local and distant recurrences. The tumor recurrences had converted to an ER-negative status and histological grade 3, corresponding to a secondary pleomorphic phenotype [41]. Further treatment included conventional chemotherapies. The IPH-926 cell line was established from the endocrine-resistant and chemotherapy-resistant progressive disease [30]. In vitro, IPH-926 cells grow in loosely adherent grape-like clusters, but also form some single-file linear cords reminiscent of primary ILC (Figure 1B, arrow). IPH-926 harbors a unique homozygous CDH1 frameshift mutation and lacks E-cadherin. Detection of the same CDH1 mutation in archival tissue of the original ER-positive breast tumor verified the clonal origin of IPH-926 from ILC [30]. p120 relocates to the cell membrane upon reconstitution of E-cadherin in IPH-926 (Figure 1C) [33]. IPH-926 cells are ER/progesterone receptor (PR)/ERBB2 (triple)-negative, but retain a luminal subtype, as defined by microarray profiling [42]. IPH-926 has also retained a chemoresistant phenotype, which depends on an endogenous overexpression of the ABCB1/MDR1 xenobiotic transporter [43]. Cell proliferation of IPH-926 is slow (doubling time of 14 days) and independent from estrogenic stimulation. This seems related to an overexpression of BCAR4, a mediator of endocrine resistance [43,44]. Like MDA-MB-134 and SUM-44PE, IPH-926 harbors a gain at chromosome 8p12-p11. However, it lacks overexpression of FGFR1 and is not sensitive to FGFR1 inhibition [30]. In their in vivo clonal ancestry, IPH-926 cells acquired an additional TP53 mutation [41]. The p53 mutant expressed in IPH-926, E285K, has temperature-sensitive loss of function characteristics. Interestingly, activation or inactivation of p53 has little impact on cell cycle distribution or apoptosis in these cells. Instead, restoration of p53 activity results in a metabolic suppression. Microarray analyses identified p53-regulated genes associated with this metabolic suppression, one of which is the AKT-inhibitor PHLDA3 [41]. Notably, p53 E285K is also evident in a subclone of MDA-MB-134 [35,45] and has repeatedly been detected in therapy-refractory ILC [17].
Few other cell lines from ILC have ever been reported (Table 1) [26,27,29,31]. The MDA-MB-330 cell line expresses wild-type but dysfunctional E-cadherin due to a biallelic mutation in α-catenin (CTNNA1), which may represent an alternate mechanism to impair E-cadherin function [46]. The BCK-4 cell line was derived from an ILC with extracellular mucin, an exceptionally rare ILC variant [31].
The three most intensively investigated models (MDA-MB-134, SUM-44PE and IPH-926) have some features in common. These commonalities include a metastatic origin, mutation of CDH1 and TP53, a luminal molecular subtype and amplification of chromosome 8p12-p11. All three cell lines lack PIK3CA hot-spot mutations common in primary ILC. As stated above, TP53 mutations are rare in primary ILC, except for the pleomorphic variant. The accumulation of TP53 mutations in the few available ILC cell lines may suggest a selection bias. Indeed, p53-deficient tumor cells are notorious for their superior in vitro growth. Establishment of a cell line from human nonmetastatic ILC with wild-type p53 and an activating PIK3CA mutation has not been achieved. Hence, human ILC cell lines have limitations regarding their representativeness for primary ILC, but have provided insight into mechanisms of endocrine resistance, chemoresistance and tumor progression.
Infiltrating lobular breast cancer xenograft models
Engraftment of human tumor tissues into immunodeficient mice promises exact phenocopying of BC subtypes [47]. However, only few ILC xenografts have ever been described (Table 1) [36,47-49]. Tumor take rates are generally low for ER-positive BCs (approximately 2 to 4%) [48,49]. Cottu and colleagues, using Swiss nude mice as hosts, reported that tumor take was a modest 1/59 (1.7%) for ER-positive ILC [49]. The ER-positive ILC that did engraft was ERBB2-positive, which is uncharacteristic for ILC.
The low tumor take rate of ILC could occur for several reasons. First, correct macroscopic identification of ILC areas in human breast resection specimens is challenging. This is due to the often sparse cellularity of ILC. Hence, it is nearly impossible to control for the number of tumor cells transplanted. Second, the slow proliferation of ILC is probably not compatible with xenograft models. Development of a large tumor from a small tissue fragment may take several years and extend beyond the host’s lifespan.
Nonetheless, Sikora and colleagues established HCI-013 ILC xenograft tumors in nonobese diabetic/severe combined immunodeficient mice. Estrogen withdrawal decreased the time to tumor detection in this model [36]. Hence, HCI-013 recapitulates estrogen-dependent growth in vivo. Finally, ILC xenografts have also been generated by orthotopic or subcutaneous inoculation of human ILC cell lines. IPH-926 xenografts show histomorphological features reminiscent of human primary ILC [30]. BCK-4 xenografts switch from mucinous to lobular histology when supplemented with estrogens [31]. However, due to low tumor take rates, ILC xenograft models are currently of limited utility for ILC research.
Sporadic infiltrating lobular breast cancer in animals
Before discussing GEM models, it is reasonable to ask whether ILC occurs as a sporadic tumor in animals. Sporadic BCs are well studied in dogs and cats, which, as pet animals, undergo surgical tumor resections. Current veterinary classification systems for canine, feline and rodent mammary tumors do not cover ILC as a naturally occurring entity [50,51]. However, Ressel and colleagues reviewed nearly 4,000 canine BCs and identified three cases of ILC [52]. Canine ILCs were E-cadherin-negative but were also ER/PR-negative, suggesting species-specific differences in hormonal growth control. E-cadherin-deficient LCIS has been reported in primates, but ILC is not known [53]. Hence, ILC is primarily a human disease and is very rare in domestic or free-ranging animals.
E-cadherin knockout is lethal in conventional genetically engineered mouse models
GEM models have revolutionized cancer research [54]. There are three reasons for the success of GEM tumor models. First, mice are mammals. Second, mice share genetic similarities with humans. Third, the mouse germline can be easily manipulated to induce overexpression or knockout of target genes.
Early conventional GEM tumor models were based on the activation or inactivation of a single gene in the germline or large tissue compartments. This only crudely mimicked human tumorigenesis. Furthermore, embryonic lethality was a main drawback of the conventional GEM models. This is exemplified by knockout of Cdh1 in the mouse germline. Homozygous loss of E-cadherin (Cdh1−/−) is lethal due to defective blastocyst formation [55]. Heterozygous mice (Cdh1+/−) develop normally and show no increased tumor incidence, suggesting that either E-cadherin haploinsufficiency does not induce tumors, E-cadherin loss is not tolerated and/or that the lifespan of the mouse is not sufficient to allow evolutionary loss of heterozygosity [55].
Although conventional Cdh1 knockout was of little immediate value for elucidating the tumor suppressor function of E-cadherin, it inspired many decisive studies on the important roles of E-cadherin in murine embryonic stem cells and embryonic development [56]. Heterozygous mice (Cdh1+/−) have also been employed to establish a model resembling gastric signet ring cell carcinoma (a form of DGC) [57]. Exposure to carcinogenic N-methyl-N-nitrosourea via drinking water induced a 10-fold increase of E-cadherin-negative signet ring cell carcinomas in heterozygous mice (Cdh1+/−) compared with wild-type mice (Cdh1+/+) (Table 2) [57]. This study clearly implicated loss of E-cadherin as a second and collaborating hit in tumor formation and provided a compelling example of how genetic and environmental factors cooperate in the initiation of distinct tumors. The lack of an equivalent ILC model based on heterozygous (Cdh1+/−) mice may be for several reasons. One aspect is that carcinogens involved in gastric tumorigenesis are well defined, while environmental factors associated with BC are complex and cannot easily be adopted for laboratory animals.
Table 2.
Genetically engineered mouse models related to infiltrating lobular breast cancer
| Promoter element | Recombinase/transgene | Targeted tissues | Conditional knockout | Tumors | Tumor spectrum (mammary gland or stomach) | References |
|---|---|---|---|---|---|---|
| Mammary gland | ||||||
| MMTV | Cre | MGa, SALG, SV | Cdh1 F/F | No | x | [59] |
| K14 | Cre | MGa, SALG, EPD, ESO | Cdh1 F/F | No | x | [60] |
| K14 | Cre | MGa, SALG, EPD, ESO | Cdh1 F/F | No | x | [61] |
| WAP | Cre | MGa | Cdh1 F/F | No | x | [62] |
| WAP | Cre | MGa | Cdh1 F/F | No | x | [63] |
| K14 | Cre | MGa, SALG, EPD, ESO | Cdh1 F/F ; Trp53 F/F | Yes | mILC (54%), CS (27%), sAC (23%) | [61] |
| WAP | Cre | MGa | Cdh1 F/F ; Trp53 F/F | Yes | mILC (60%), sC/CS (67%), AC (2%) | [63] |
| Stomach | ||||||
| x | Cdh1 +/− | None | x | No | x | [57] |
| x | Cdh1 +/− (+MNU) | None | x | Yes | tubAd (75%), SRCC (45%) | [57] |
| ATP4B | Cre | GPC | Cdh1 F/F | No | (noninvasive signet ring cell clusters) | [64] |
| ATP4B | Cre | GPC | Cdh1 F/F ; Trp53 F/F | Yes | DGC | [65] |
| PDX1 | Cre | GMC, DMC, PIC | Cdh1 F/+ ; Smad4 F/F ; Trp53 F/F | Yes | DGC (85%, Ecad-negative), DDAC (26% Ecad-positive), SCC (24%) | [66] |
All information compiled from the literature. AC, adenocarcinoma; CS, carcinosarcoma; DGC, diffuse gastric cancer; DDAC, duodenal adenocarcinoma; DMC, duodenal mucosal cells; Ecad, E-cadherin; EPD, epidermis; ESO, esophageal mucosa; GMC, gastric mucosal cells; GPC, gastric parietal cells; MG, mammary gland; mILC, murine infiltrating lobular carcinoma; MNU, carcinogenic N-methyl-N-nitrosourea given with the drinking water; sAC, solid adenocarcinoma; sC, solid carcinoma; PIC, pancreatic islet cells; SCC, squamous cell carcinoma; SALG, salivary gland; SRCC, signet ring cell carcinoma; SV, seminal vesicle; tubAd, tubular adenoma. aLuminal and myoepithelial cells in virgin and pregnant female mice.
E-cadherin knockout is not tumorigenic in conditional genetically engineered mouse models
To study all properties of BC pathology, GEM models are needed that mimic not only human tumor phenotypes but also their initiation from individual cells in adult tissues. Conditional GEM tumor models based on site-specific recombination systems, such as Cre/loxP from bacteriophage P1, allow for somatic and stochastic mutation of target genes in defined tissues of a wild-type background [58]. A number of different approaches have been employed, using different promoter elements driving Cre recombinase expression to cell type-specific Cdh1 ablation in the mouse mammary gland and gastrointestinal tract [59-66] (Table 2).
The common denominator of these studies is that mice fail to develop BC upon conditional knockout of E-cadherin using either K14, WAP or MMTV as Cre recombinase drivers. The underlying reason for this phenomenon is the fact that loss of E-cadherin is not tolerated in the luminal epithelial compartment of the mouse mammary gland. Depending on the promoter driving Cre, E-cadherin ablation will result in massive apoptosis (MMTV) or nearly undetectable clearance of E-cadherin-deficient luminal cells (K14) [59,61-63]. Intriguingly, human E-cadherin-negative LCIS can subsist for years without regression or progression. Based on data from mouse models, this implies either that a primary oncogenic hit allowing E-cadherin loss is already present in human ILC or, in contrast to the mouse, human luminal mammary cells can cope with E-cadherin loss. The latter option may be explained by redundancy mechanisms. Cdh1 knockout in the basal stratified and follicular epidermal cells of the skin induces a compensatory upregulation of P-cadherin (Cdh3) that rescues epithelial integrity in the basal layer of the epidermis, but not in the hair follicle [60]. Since luminal epithelial cells exclusively express E-cadherin and myoepithelial cells express P-cadherin, this seems an unlikely scenario for the mammary gland. Cdh1 ablation in the gastric mucosa also did not result in gastric cancer, although noninvasive E-cadherin-negative cell aggregates occurred [64]. Together, conditional GEM models imply that additional oncogenic hits are compulsory in the mammary gland before Cdh1 inactivation can be tolerated and unleash its full tumorigenic potential.
Compound genetically engineered mouse models provide insight into infiltrating lobular breast cancer biology
Given that somatic inactivation of E-cadherin as a primary hit is not tolerated in the breast, compound conditional GEM models have been developed based on concomitant inactivation of p53 and E-cadherin. Somatic inactivation of p53 also allowed study of the E-cadherin function in tumor progression, because mammary-specific inactivation of p53 alone resulted in nonmetastatic, locally expansive tumors [61,63]. In contrast, dual conditional knockout of E-cadherin and p53 using either K14cre or WAPcre synergized with p53 loss and induced a dramatic shift from expansive to infiltrating growth. Homozygous loss of E-cadherin led to the formation of carcinomas that phenocopied human ILC. While from a cytopathological perspective most of these tumors showed extensive nuclear pleomorphism, due to their striking similarity to human ILC invasion patterns they were designated as mouse ILC (Figure 1D). Based on mRNA profiling and expression of cytokeratin 8, mouse ILC is luminal in character but does not express ER/PR during the later stages of tumor progression. Since conditional mouse BC models in general show absence of ER and PR expression, this probably reflects species-specific physiological differences. Of importance, however, is the finding that mouse ILC shows a dissemination spectrum similar to human ILC, with specific metastasis to the gastrointestinal tract and peritoneum and common sites such as the lung and bone marrow [61,63]. Chemotherapy prolongs survival but does not eradicate metastases in this model [67]. While antihormone therapy can keep human metastatic ILC under control, endocrine-resistant ILC is notorious for poor response to chemotherapy [68]. Mouse ILC models are therefore better categorized as models for endocrine-resistant and chemorefractory metastatic ILC.
Availability of the mouse ILC models has provided new opportunities to study ILC cell biology. Reconstitution of E-cadherin in p53-deficient mouse ILC cell lines abolished their ability to proliferate under anchorage-independent conditions, showing causality for the loss of E-cadherin in this process [61]. Follow-up studies showed that, unlike β-catenin, p120 is not proteosomically degraded in ILC but instead resides in the cytosol and nucleus upon E-cadherin loss. Cytosolic p120 (a distinguishing feature of human ILC) controls Rho/Rock-dependent anoikis resistance of ILC by binding and inhibition of the Rho antagonist Mrip [14]. While it is still unclear how p120 triggers anchorage-independent survival distal from Rho and Rock, future answers may come from the ability of p120 to inhibit transcriptional repression by Kaiso [69]. ILC is characterized by a decrease in nuclear Kaiso and relief from p120-dependent Kaiso repression [70]. Our unpublished data have also identified noncanonical Wnt11 as a Kaiso target driving autocrine Rho-dependent anoikis resistance (van de Ven RAH, unpublished data), suggesting that p120 is a multifaceted oncoprotein in ILC. These findings furthermore denote options for future intervention because ILC progression depends on Rho/Rock signaling, a pathway that is susceptible to pharmacological inhibition.
Interestingly, indirect mammary-specific loss of E-cadherin function through ablation of p120 did not induce murine ILC. In this context, p120 functioned as a tumor suppressor, and its loss in WAPcre;Ctnnd1F/F;Trp53F/F female mice led to dismantling of the E-cadherin-dependent cell–cell adhesion and formation of metastatic tumors that resembled metaplastic triple-negative BC [71]. These studies also showed that inactivation of the AJ partially controls anchorage independence through hypersensitization of endogenous growth factor receptors. This phenomenon appeared independent of the phenotypic outcome, but was dependent on the absence of cadherin-based AJs [71]. These data may provide an explanation for the prevalence of oncogenic events that lead to activation of PI3K/AKT-dependent cues in ILC. Moreover, they suggest that ILC patients may be eligible for clinical interventions using therapies that target growth factor receptor signaling, especially in the absence of activating mutations or amplifications.
Comparable compound conditional GEM models have also been established for gastric cancer. Inactivation of E-cadherin and p53 in ATP4Bcre;Cdh1F/F;Trp53F/F mice resulted in the progression of E-cadherin-negative gastric mucosal cell aggregates to invasive and metastatic tumors resembling human DGC [65] (Table 2). In another compound model, E-cadherin-deficient DGCs developed in PDX1cre;Cdh1F/+;Trp53F/FSmad4F/F mice, suggesting a selection pressure for spontaneous inactivation of a remaining wild-type Cdh1 allele during gastric tumorigenesis [66].
Taken together, several conditional compound GEM models involving somatic knockout of Cdh1 recapitulate ILC-like or DGC-like tumors (Table 2). Follow-up studies showed that Cdh1 inactivation releases p120 from AJs to the cytosol and nucleus, where it controls tumor progression through distinct and druggable signaling pathways. Blocking these pathways may therefore be a rational strategy for the design of targeted therapies to better treat metastatic ILC.
Conclusions
ILC is the most common special BC subtype. With mutational or epigenetic inactivation of E-cadherin being confined almost exclusively to ILC and LCIS, this tumor entity stands out from all other kinds of BCs. The molecular basis of ILC is clearly linked to loss of E-cadherin, as evidenced by hereditary cases associated with CDH1 germline mutation and conditional knockout mouse models.
What is the big picture we obtain from ILC models? ILC is difficult to study on a functional level. Human ILC cell lines are rare. All of them are of metastatic origin and harbor mutant p53. Their biology reflects end-stage ILC progression. Primary ILCs show almost no tumor take in xenograft models. Currently established GEM models phenocopy ILC but lack ER expression. Nonetheless, ILC models have advanced our understanding of this disease tremendously. Several new candidate genes and signaling pathways have come to the fore of ILC biology. These include FGFR1, ZNF703, ERRγ, BCAR4 and ABCB1/MDR1 as mediators of therapy resistance and mutant p53 as the gatekeeper towards pleomorphic ILC. Furthermore, p120 has been confirmed to mediate multiple oncogenic signals through Rho-Rock signaling upon inactivation of E-cadherin.
What are the most important questions that remain to be explored? It is still unclear how loss of E-cadherin impacts on gene expression patterns and differentiation of developing ILC. Microarray analyses following E-cadherin reconstitution in ILC cells might be instructive. Our own profiling of IPH-926 and MDA-MB-134 reconstituted with E-cadherin has so far revealed little or no reorganization of the transcriptome (Karch I, unpublished observation). This makes the mystery surrounding E-cadherin even more intriguing.
Regarding mechanisms of endocrine resistance, collaboration between basic scientist, histopathologists and clinicians is warranted [72]. Current clinical trials addressing pretreatment/post-treatment biomarker changes in BC patients receiving neoadjuvant endocrine therapy establish an invaluable resource of tumor tissues informative for endocrine responsiveness. Future translational research studies will take advantage of this resource to determine the predictability of clinical endocrine resistance using surrogate markers from ILC models, such as FGFR1, ZNF703 and BCAR4.
In the field of GEM models, conditional inactivation of Cdh1 combined with activation of latent mutant Pik3caH1047R promises tumors with unique, perhaps ILC-like properties. The most important challenge, however, is to understand and target the mechanisms that counteract the proapoptotic signals upon loss of E-cadherin in LCIS and ILC. These mechanisms might be heterogeneous and could predestinate for metachronous cancer development in the one LCIS patient but not in another. The role of a recently identified ILC-specific single nucleotide polymorphism on chromosome 7q34 will also be of interest in this context [73]. Definition of prognostically favorable and unfavorable LCIS would be a major achievement, since LCIS is common and the risk of progression to invasive ILC is as yet entirely unpredictable on an individual patient basis.
Abbreviations
- AJ
Adherens junction
- BC
Breast cancer
- DGC
Diffuse gastric cancer
- ER
Estrogen receptor
- GEM
Genetically engineered mouse
- ILC
Infiltrating lobular breast cancer
- LCIS
Lobular carcinoma in situ
- p120
p120-catenin
- PR
Progesterone receptor
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MC and PWBD both wrote the manuscript.
Contributor Information
Matthias Christgen, Email: Christgen.Matthias@MH-Hannover.de.
Patrick WB Derksen, Email: pderksen@umcutrecht.nl.
References
- 1.Foote FW, Stewart FW. Lobular carcinoma in situ: a rare form of mammary cancer. Am J Pathol. 1941;17:491–6. doi: 10.3322/canjclin.32.4.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willis RA. Epithelial tumours of the breast. In: Willis RA, editor. Pathology of tumours. London: Butterworth Co. Ltd; 1948. pp. 208–57. [Google Scholar]
- 3.Hyafil F, Babinet C, Jacob F. Cell–cell interactions in early embryogenesis: a molecular approach to the role of calcium. Cell. 1981;26:447–54. doi: 10.1016/0092-8674(81)90214-2. [DOI] [PubMed] [Google Scholar]
- 4.Niessen CM, Gottardi CJ. Molecular components of the adherens junction. Biochim Biophys Acta. 2008;1778:562–71. doi: 10.1016/j.bbamem.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moll R, Mitze M, Frixen UH, Birchmeier W. Differential loss of E-cadherin expression in infiltrating ductal and lobular breast carcinomas. Am J Pathol. 1993;143:1731–42. [PMC free article] [PubMed] [Google Scholar]
- 6.Vos CB, Cleton-Jansen AM, Berx G, de Leeuw WJ, ter Haar NT, van Roy F, Cornelisse CJ, Peterse JL, van de Vijver MJ. E-cadherin inactivation in lobular carcinoma in situ of the breast: an early event in tumorigenesis. Br J Cancer. 1997;76:1131–3. doi: 10.1038/bjc.1997.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, van Roy F. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. Embo J. 1995;14:6107–15. doi: 10.1002/j.1460-2075.1995.tb00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Droufakou S, Deshmane V, Roylance R, Hanby A, Tomlinson I, Hart IR. Multiple ways of silencing E-cadherin gene expression in lobular carcinoma of the breast. Int J Cancer. 2001;92:404–8. doi: 10.1002/ijc.1208. [DOI] [PubMed] [Google Scholar]
- 9.Ellis MJ, Perou CM. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013;3:27–34. doi: 10.1158/2159-8290.CD-12-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–5. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- 11.Benusiglio PR, Malka D, Rouleau E, De Pauw A, Buecher B, Nogues C, Fourme E, Colas C, Coulet F, Warcoin M, Grandjouan S, Sezeur A, Laurent-Puig P, Molière D, Tlemsani C, Di Maria M, Byrde V, Delaloge S, Blayau M, Caron O. CDH1 germline mutations and the hereditary diffuse gastric and lobular breast cancer syndrome: a multicentre study. J Med Genet. 2013;50:486–9. doi: 10.1136/jmedgenet-2012-101472. [DOI] [PubMed] [Google Scholar]
- 12.Petridis C, Shinomiya I, Kohut K, Gorman P, Caneppele M, Shah V, Troy M, Pinder SE, Hanby A, Tomlinson I, Trembath RC, Roylance R, Simpson MA, Sawyer EJ. Germline CDH1 mutations in bilateral lobular carcinoma in situ. Br J Cancer. 2014;110:1053–7. doi: 10.1038/bjc.2013.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarrio D, Perez-Mies B, Hardisson D, Moreno-Bueno G, Suarez A, Cano A, Martin-Perez J, Gamallo C, Palacios J. Cytoplasmic localization of p120ctn and E-cadherin loss characterize lobular breast carcinoma from preinvasive to metastatic lesions. Oncogene. 2004;23:3272–83. doi: 10.1038/sj.onc.1207439. [DOI] [PubMed] [Google Scholar]
- 14.Schackmann RC, van Amersfoort M, Haarhuis JH, Vlug EJ, Halim VA, Roodhart JM, Vermaat JS, Voest EE, van der Groep P, van Diest PJ, Jonkers J, Derksen PW. Cytosolic p120-catenin regulates growth of metastatic lobular carcinoma through Rock1-mediated anoikis resistance. J Clin Invest. 2011;121:3176–88. doi: 10.1172/JCI41695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertucci F, Orsetti B, Negre V, Finetti P, Rouge C, Ahomadegbe JC, Bibeau F, Mathieu MC, Treilleux I, Jacquemier ᅟ, Ursule L, Martinec A, Wang Q, Bénard J, Puisieux A, Birnbaum D, Theillet C. Lobular and ductal carcinomas of the breast have distinct genomic and expression profiles. Oncogene. 2008;27:5359–72. doi: 10.1038/onc.2008.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rakha EA, El-Sayed ME, Powe DG, Green AR, Habashy H, Grainge MJ, Robertson JF, Blamey R, Gee J, Nicholson RI, Lee AH, Ellis IO. Invasive lobular carcinoma of the breast: response to hormonal therapy and outcomes. Eur J Cancer. 2008;44:73–83. doi: 10.1016/j.ejca.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 17.Ross JS, Wang K, Sheehan CE, Boguniewicz AB, Otto G, Downing SR, Sun J, He J, Curran JA, Ali S, Yelensky R, Lipson D, Palmer G, Miller VA, Stephens PJ. Relapsed classic E-cadherin (CDH1)-mutated invasive lobular breast cancer shows a high frequency of HER2 (ERBB2) gene mutations. Clin Cancer Res. 2013;19:2668–76. doi: 10.1158/1078-0432.CCR-13-0295. [DOI] [PubMed] [Google Scholar]
- 18.Christgen M, Noskowicz M, Schipper E, Christgen H, Heil C, Krech T, Langer F, Kreipe H, Lehmann U. Oncogenic PIK3CA mutations in lobular breast cancer progression. Genes Chromosomes Cancer. 2013;52:69–80. doi: 10.1002/gcc.22007. [DOI] [PubMed] [Google Scholar]
- 19.Simpson PT, Reis-Filho JS, Lambros MB, Jones C, Steele D, Mackay A, Iravani M, Fenwick K, Dexter T, Jones A, Reid L, Da Silva L, Shin SJ, Hardisson D, Ashworth A, Schmitt FC, Palacios J, Lakhani SR. Molecular profiling pleomorphic lobular carcinomas of the breast: evidence for a common molecular genetic pathway with classic lobular carcinomas. J Pathol. 2008;215:231–44. doi: 10.1002/path.2358. [DOI] [PubMed] [Google Scholar]
- 20.Ercan C, van Diest PJ, van der Ende B, Hinrichs J, Bult P, Buerger H, van der Wall E, Derksen PW. p53 mutations in classic and pleomorphic invasive lobular carcinoma of the breast. Cell Oncol (Dordr) 2012;35:111–8. doi: 10.1007/s13402-012-0071-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korkola JE, DeVries S, Fridlyand J, Hwang ES, Estep AL, Chen YY, Chew KL, Dairkee SH, Jensen RM, Waldman FM. Differentiation of lobular versus ductal breast carcinomas by expression microarray analysis. Cancer Res. 2003;63:7167–75. [PubMed] [Google Scholar]
- 22.Weigelt B, Geyer FC, Natrajan R, Lopez-Garcia MA, Ahmad AS, Savage K, Kreike B, Reis-Filho JS. The molecular underpinning of lobular histological growth pattern: a genome-wide transcriptomic analysis of invasive lobular carcinomas and grade- and molecular subtype-matched invasive ductal carcinomas of no special type. J Pathol. 2010;220:45–57. doi: 10.1002/path.2629. [DOI] [PubMed] [Google Scholar]
- 23.Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13:215. doi: 10.1186/bcr2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo WL, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–27. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cailleau R, Young R, Olive M, Reeves WJ., Jr Breast tumor cell lines from pleural effusions. J Natl Cancer Inst. 1974;53:661–74. doi: 10.1093/jnci/53.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engel LW, Young NA. Human breast carcinoma cells in continuous culture: a review. Cancer Res. 1978;38:4327–39. [PubMed] [Google Scholar]
- 27.Rye PD, Norum L, Olsen DR, Garman-Vik S, Kaul S, Fodstad O. Brain metastasis model in athymic nude mice using a novel MUC1-secreting human breast-cancer cell line, MA11. Int J Cancer. 1996;68:682–7. doi: 10.1002/(SICI)1097-0215(19961127)68:5<682::AID-IJC20>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 28.Ethier SP, Mahacek ML, Gullick WJ, Frank TS, Weber BL. Differential isolation of normal luminal mammary epithelial cells and breast cancer cells from primary and metastatic sites using selective media. Cancer Res. 1993;53:627–35. [PubMed] [Google Scholar]
- 29.Gazdar AF, Kurvari V, Virmani A, Gollahon L, Sakaguchi M, Westerfield M, Kodagoda D, Stasny V, Cunningham HT, Wistuba II, Tomlinson G, Tonk V, Ashfaq R, Leitch AM, Minna JD, Shay JW. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int J Cancer. 1998;78:766–74. doi: 10.1002/(SICI)1097-0215(19981209)78:6<766::AID-IJC15>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 30.Christgen M, Bruchhardt H, Hadamitzky C, Rudolph C, Steinemann D, Gadzicki D, Hasemeier B, Romermann D, Focken T, Krech T, Ballmaier M, Schlegelberger B, Kreipe H, Lehmann U. Comprehensive genetic and functional characterization of IPH-926: a novel CDH1-null tumour cell line from human lobular breast cancer. J Pathol. 2009;217:620–32. doi: 10.1002/path.2495. [DOI] [PubMed] [Google Scholar]
- 31.Jambal P, Badtke MM, Harrell JC, Borges VF, Post MD, Sollender GE, Spillman MA, Horwitz KB, Jacobsen BM. Estrogen switches pure mucinous breast cancer to invasive lobular carcinoma with mucinous features. Breast Cancer Res Treat. 2012;137:431–48. doi: 10.1007/s10549-012-2377-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hollestelle A, Peeters JK, Smid M, Timmermans M, Verhoog LC, Westenend PJ, Heine AA, Chan A, Sieuwerts AM, Wiemer EA, Klijn JG, van der Spek PJ, Foekens JA, Schutte M, den Bakker MA, Martens JW. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res Treat. 2013;138:47–57. doi: 10.1007/s10549-013-2415-3. [DOI] [PubMed] [Google Scholar]
- 33.Karch I, Schipper E, Christgen H, Kreipe H, Lehmann U, Christgen M. Is upregulation of BCL2 a determinant of tumor development driven by inactivation of CDH1/E-cadherin? PLoS One. 2013;8:e73062. doi: 10.1371/journal.pone.0073062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reis-Filho JS, Simpson PT, Turner NC, Lambros MB, Jones C, Mackay A, Grigoriadis A, Sarrio D, Savage K, Dexter T, Iravani M, Fenwick K, Weber B, Hardisson D, Schmitt FC, Palacios J, Lakhani SR, Ashworth A. FGFR1 emerges as a potential therapeutic target for lobular breast carcinomas. Clin Cancer Res. 2006;12:6652–62. doi: 10.1158/1078-0432.CCR-06-1164. [DOI] [PubMed] [Google Scholar]
- 35.Hollestelle A, Nagel JH, Smid M, Lam S, Elstrodt F, Wasielewski M, Ng SS, French PJ, Peeters JK, Rozendaal MJ, Riaz M, Koopman DG, Ten Hagen TL, de Leeuw BH, Zwarthoff EC, Teunisse A, van der Spek PJ, Klijn JG, Dinjens WN, Ethier SP, Clevers H, Jochemsen AG, den Bakker MA, Foekens JA, Martens JW, Schutte M. Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res Treat. 2010;121:53–64. doi: 10.1007/s10549-009-0460-8. [DOI] [PubMed] [Google Scholar]
- 36.Sikora MJ, Cooper KL, Bahreini A, Luthra S, Wang G, Chandran UR, Davidson NE, Dabbs DJ, Welm AL, Oesterreich S. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res. 2014;74:1463–74. doi: 10.1158/0008-5472.CAN-13-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riggins RB, Lan JP, Klimach U, Zwart A, Cavalli LR, Haddad BR, Chen L, Gong T, Xuan J, Ethier SP, Clarke R. ERRgamma mediates tamoxifen resistance in novel models of invasive lobular breast cancer. Cancer Res. 2008;68:8908–17. doi: 10.1158/0008-5472.CAN-08-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A, Gillett C, Grigoriadis A, Tutt A, Reis-Filho JS, Ashworth A. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–94. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sircoulomb F, Nicolas N, Ferrari A, Finetti P, Bekhouche I, Rousselet E, Lonigro A, Adelaide J, Baudelet E, Esteyries S, Wicinski J, Audebert S, Charafe-Jauffret E, Jacquemier J, Lopez M, Borg JP, Sotiriou C, Popovici C, Bertucci F, Birnbaum D, Chaffanet M, Ginestier C. ZNF703 gene amplification at 8p12 specifies luminal B breast cancer. EMBO Mol Med. 2011;3:153–66. doi: 10.1002/emmm.201100121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Mu X, Huang O, Xie Z, Jiang M, Geng M, Shen K. Luminal breast cancer cell lines overexpressing ZNF703 are resistant to tamoxifen through activation of Akt/mTOR signaling. PLoS One. 2013;8:e72053. doi: 10.1371/journal.pone.0072053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christgen M, Noskowicz M, Heil C, Schipper E, Christgen H, Geffers R, Kreipe H, Lehmann U. IPH-926 lobular breast cancer cells harbor a p53 mutant with temperature-sensitive functional activity and allow for profiling of p53-responsive genes. Lab Invest. 2012;92:1635–47. doi: 10.1038/labinvest.2012.126. [DOI] [PubMed] [Google Scholar]
- 42.Christgen M, Geffers R, Kreipe H, Lehmann U. IPH-926 lobular breast cancer cells are triple-negative but their microarray profile uncovers a luminal subtype. Cancer Sci. 2013;104:1726–30. doi: 10.1111/cas.12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krech T, Scheuerer E, Geffers R, Kreipe H, Lehmann U, Christgen M. ABCB1/MDR1 contributes to the anticancer drug-resistant phenotype of IPH-926 human lobular breast cancer cells. Cancer Lett. 2012;315:153–60. doi: 10.1016/j.canlet.2011.09.038. [DOI] [PubMed] [Google Scholar]
- 44.Meijer D, van Agthoven T, Bosma PT, Nooter K, Dorssers LC. Functional screen for genes responsible for tamoxifen resistance in human breast cancer cells. Mol Cancer Res. 2006;4:379–86. doi: 10.1158/1541-7786.MCR-05-0156. [DOI] [PubMed] [Google Scholar]
- 45.Keimling M, Wiesmuller L. DNA double-strand break repair activities in mammary epithelial cells – influence of endogenous p53 variants. Carcinogenesis. 2009;30:1260–8. doi: 10.1093/carcin/bgp117. [DOI] [PubMed] [Google Scholar]
- 46.Hollestelle A, Elstrodt F, Timmermans M, Sieuwerts AM, Klijn JG, Foekens JA, den Bakker MA, Schutte M. Four human breast cancer cell lines with biallelic inactivating alpha-catenin gene mutations. Breast Cancer Res Treat. 2010;122:125–33. doi: 10.1007/s10549-009-0545-4. [DOI] [PubMed] [Google Scholar]
- 47.DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R, Matsen C, Milash BA, Nelson E, Neumayer L, Randall RL, Stijleman IJ, Welm BE, Welm AL. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011;17:1514–20. doi: 10.1038/nm.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, de Plater L, Guyader C, De Pinieux G, Judde JG, Rebucci M, Tran-Perennou C, Sastre-Garau X, Sigal-Zafrani B, Delattre O, Diéras V, Poupon MF. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res. 2007;13:3989–98. doi: 10.1158/1078-0432.CCR-07-0078. [DOI] [PubMed] [Google Scholar]
- 49.Cottu P, Marangoni E, Assayag F, de Cremoux P, Vincent-Salomon A, Guyader C, de Plater L, Elbaz C, Karboul N, Fontaine JJ, Chateau-Joubert S, Boudou-Rouquette P, Alran S, Dangles-Marie V, Gentien D, Poupon MF, Decaudin D. Modeling of response to endocrine therapy in a panel of human luminal breast cancer xenografts. Breast Cancer Res Treat. 2012;133:595–606. doi: 10.1007/s10549-011-1815-5. [DOI] [PubMed] [Google Scholar]
- 50.Goldschmidt M, Pena L, Rasotto R, Zappulli V. Classification and grading of canine mammary tumors. Vet Pathol. 2011;48:117–31. doi: 10.1177/0300985810393258. [DOI] [PubMed] [Google Scholar]
- 51.Cardiff RD, Wellings SR. The comparative pathology of human and mouse mammary glands. J Mammary Gland Biol Neoplasia. 1999;4:105–22. doi: 10.1023/A:1018712905244. [DOI] [PubMed] [Google Scholar]
- 52.Ressel L, Millanta F, Poli A. Canine invasive lobular carcinoma of the mammary gland: morphological and immunohistochemical characterizations of three cases. J Comp Pathol. 2011;144:303–7. doi: 10.1016/j.jcpa.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 53.Wood CE, Usborne AL, Starost MF, Tarara RP, Hill LR, Wilkinson LM, Geisinger KR, Feiste EA, Cline JM. Hyperplastic and neoplastic lesions of the mammary gland in macaques. Vet Pathol. 2006;43:471–83. doi: 10.1354/vp.43-4-471. [DOI] [PubMed] [Google Scholar]
- 54.Jonkers J, Derksen PW. Modeling metastatic breast cancer in mice. J Mammary Gland Biol Neoplasia. 2007;12:191–203. doi: 10.1007/s10911-007-9050-8. [DOI] [PubMed] [Google Scholar]
- 55.Riethmacher D, Brinkmann V, Birchmeier C. A targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc Natl Acad Sci U S A. 1995;92:855–9. doi: 10.1073/pnas.92.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Redmer T, Diecke S, Grigoryan T, Quiroga-Negreira A, Birchmeier W, Besser D. E-cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep. 2011;12:720–6. doi: 10.1038/embor.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Humar B, Blair V, Charlton A, More H, Martin I, Guilford P. E-cadherin deficiency initiates gastric signet-ring cell carcinoma in mice and man. Cancer Res. 2009;69:2050–6. doi: 10.1158/0008-5472.CAN-08-2457. [DOI] [PubMed] [Google Scholar]
- 58.Gu H, Zou YR, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73:1155–64. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- 59.Boussadia O, Kutsch S, Hierholzer A, Delmas V, Kemler R. E-cadherin is a survival factor for the lactating mouse mammary gland. Mech Dev. 2002;115:53–62. doi: 10.1016/S0925-4773(02)00090-4. [DOI] [PubMed] [Google Scholar]
- 60.Tinkle CL, Lechler T, Pasolli HA, Fuchs E. Conditional targeting of E-cadherin in skin: insights into hyperproliferative and degenerative responses. Proc Natl Acad Sci U S A. 2004;101:552–7. doi: 10.1073/pnas.0307437100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, van Beijnum JR, Griffioen AW, Vink J, Krimpenfort P, Peterse JL, Cardiff RD, Berns A, Jonkers J. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10:437–49. doi: 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 62.Kotb AM, Hierholzer A, Kemler R. Replacement of E-cadherin by N-cadherin in the mammary gland leads to fibrocystic changes and tumor formation. Breast Cancer Res. 2011;13:R104. doi: 10.1186/bcr3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Derksen PW, Braumuller TM, van der Burg E, Hornsveld M, Mesman E, Wesseling J, Krimpenfort P, Jonkers J. Mammary-specific inactivation of E-cadherin and p53 impairs functional gland development and leads to pleomorphic invasive lobular carcinoma in mice. Dis Model Mech. 2011;4:347–58. doi: 10.1242/dmm.006395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mimata A, Fukamachi H, Eishi Y, Yuasa Y. Loss of E-cadherin in mouse gastric epithelial cells induces signet ring-like cells, a possible precursor lesion of diffuse gastric cancer. Cancer Sci. 2011;102:942–50. doi: 10.1111/j.1349-7006.2011.01890.x. [DOI] [PubMed] [Google Scholar]
- 65.Shimada S, Mimata A, Sekine M, Mogushi K, Akiyama Y, Fukamachi H, Jonkers J, Tanaka H, Eishi Y, Yuasa Y. Synergistic tumour suppressor activity of E-cadherin and p53 in a conditional mouse model for metastatic diffuse-type gastric cancer. Gut. 2012;61:344–53. doi: 10.1136/gutjnl-2011-300050. [DOI] [PubMed] [Google Scholar]
- 66.Park JW, Jang SH, Park DM, Lim NJ, Deng C, Kim DY, Green JE, Kim HK. Cooperativity of E-cadherin and Smad4 loss to promote diffuse-type gastric adenocarcinoma and metastasis. Mol Cancer Res. 2014;12:1088–99. doi: 10.1158/1541-7786.MCR-14-0192-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doornebal CW, Klarenbeek S, Braumuller TM, Klijn CN, Ciampricotti M, Hau CS, Hollmann MW, Jonkers J, de Visser KE. A preclinical mouse model of invasive lobular breast cancer metastasis. Cancer Res. 2013;73:353–63. doi: 10.1158/0008-5472.CAN-11-4208. [DOI] [PubMed] [Google Scholar]
- 68.Tubiana-Hulin M, Stevens D, Lasry S, Guinebretiere JM, Bouita L, Cohen-Solal C, Cherel P, Rouesse J. Response to neoadjuvant chemotherapy in lobular and ductal breast carcinomas: a retrospective study on 860 patients from one institution. Ann Oncol. 2006;17:1228–33. doi: 10.1093/annonc/mdl114. [DOI] [PubMed] [Google Scholar]
- 69.Daniel JM, Reynolds AB. The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol Cell Biol. 1999;19:3614–23. doi: 10.1128/mcb.19.5.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vermeulen JF, van de Ven RA, Ercan C, van der Groep P, van der Wall E, Bult P, Christgen M, Lehmann U, Daniel J, van Diest PJ, Derksen PW. Nuclear Kaiso expression is associated with high grade and triple-negative invasive breast cancer. PLoS One. 2012;7:e37864. doi: 10.1371/journal.pone.0037864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schackmann RC, Klarenbeek S, Vlug EJ, Stelloo S, van Amersfoort M, Tenhagen M, Braumuller TM, Vermeulen JF, van der Groep P, Peeters ᅟ, van der Wall E, van Diest PJ, Jonkers J, Derksen PW. Loss of p120-catenin induces metastatic progression of breast cancer by inducing anoikis resistance and augmenting growth factor receptor signaling. Cancer Res. 2013;73:4937–49. doi: 10.1158/0008-5472.CAN-13-0180. [DOI] [PubMed] [Google Scholar]
- 72.Arthur LM, Turnbull AK, Webber VL, Larionov AA, Renshaw L, Kay C, Thomas JS, Dixon JM, Sims AH. Molecular changes in lobular breast cancers in response to endocrine therapy. Cancer Res. 2014;74:5371–6. doi: 10.1158/0008-5472.CAN-14-0620. [DOI] [PubMed] [Google Scholar]
- 73.Sawyer E, Roylance R, Petridis C, Brook MN, Nowinski S, Papouli E, Fletcher O, Pinder S, Hanby A, Kohut K, Gorman P, Caneppele M, Peto J, Dos Santos SI, Johnson N, Swann R, Dwek M, Perkins KA, Gillett C, Houlston R, Ross G, De Ieso P, Southey MC, Hopper JL, Provenzano E, Apicella C, Wesseling J, Cornelissen S, Keeman R, Fasching PA, et al. Genetic predisposition to in situ and invasive lobular carcinoma of the breast. PLoS Genet. 2014;10:e1004285. doi: 10.1371/journal.pgen.1004285. [DOI] [PMC free article] [PubMed] [Google Scholar]