

Abstract
Oxidative stress (OS) has been characterized by an imbalance between the production of reactive oxygen species (ROS) and a biological system’s ability to repair oxidative damage or to neutralize the reactive intermediates including peroxides and free radicals. High ROS production has been associated with significant decrease in antioxidant defense mechanisms leading to protein, lipid and DNA damage and subsequent disruption of cellular functions. In humans, OS has been reported to play a role in the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease, Huntington’s disease, Lou Gehrig’s disease, multiple sclerosis and Parkinson’s disease, as well as atherosclerosis, autism, cancer, heart failure, and myocardial infarction. Although OS has been linked to the etiology and development of chronic diseases, many chemotherapeutic drugs have been shown to exert their biologic activity through induction of OS in affected cells. This review highlights the controversial role of OS in the development and progression of leukemia cancer and the therapeutic application of increased OS and antioxidant approaches to the treatment of leukemia patients.
Electronic supplementary material
The online version of this article (doi:10.1186/s13046-014-0106-5) contains supplementary material, which is available to authorized users.
Keywords: Oxidative stress, Leukemia, Antioxidants, Reactive oxygen species, Treatment, Apoptosis
Introduction
Despite the numerous scientific studies on leukemia, there are still gaps on information about the risk factors and actual causes and such knowledge are necessary for effective treatment and prevention strategies to be established. OS is a cellular environmental condition that results from excessive production of ROS with reduced or lack of antioxidants production to maintain homeostasis [1]. Although OS is known to induce cancer, it has also been reported to have beneficial attributes. For instance it induces apoptosis which is a mechanism in cancer treatment [2]. ROS are essential signaling molecules which play different important roles in cellular processes such as promoting health and longevity [3] and antimicrobial phagocytosis by cells of the innate immune system [4]. Over production of reactive oxygen species without adequate response by the innate antioxidant system to maintain the balance leads to an OS environment. Some of the ways OS and free radicals are created are through chemotherapeutic agents and radiation therapy which generate reactive ROS in patients during cancer therapy. The interaction between growing cancer cells and the host immune response also generate OS. The effect of OS to the cells may either be acute or chronic. Chronic OS results from little oxidative damage which accumulates during the life cycle of the cell and subsequently disrupts essential cellular functions and triggers many cancers [5]-[7], including solid tumors such as prostate carcinoma [8], melanoma [9], and several hematopoietic malignancies such as acute lymphoblastic leukemia (ALL), [10] myelodysplastic syndrome (MDS), [11] and myeloid leukemia; chronic myeloid leukemia (CML) and acute myeloid leukemia (AML) [12]. OS has also been implicated in other human diseases including acute respiratory distress syndrome [13], aging [14], Alzheimer [15], atherosclerosis [16], cardiovascular diseases, and amyotrophic lateral sclerosis [17], diabetes [18], inflammation [19], inflammatory joint disease [20], neurological disease [21], obesity [22], Parkinson [23], pulmonary fibrosis [24], Rheumatoid arthritis [25], and vascular disease [26].
In some cancers ROS facilitate carcinogenesis by protecting the cell from apoptosis and promoting cell survival [27],[28], inducing cell proliferation [29], migration and metastasis [30] and drug-resistance [31]. The regulation of OS is an important factor in both tumor development and responses to anticancer therapies. Many signalling pathways that are linked to tumorigenesis can also regulate the metabolism of reactive oxygen species (ROS) through direct or indirect mechanisms [32]. Depending on the condition and environment, OS may influence the role of ROS to either benefit malignant cancer formation or cancer treatment. This review explores the role of OS in leukemogenesis and highlights its importance in leukemia treatment. Understanding the molecular mechanisms of oxidative will help in developing new and reliable treatment and preventive measures for the different types of leukemia.
Oxidative stress
OS results when there is an imbalance between the generation of oxygen-free radicals or reactive oxygen species (ROS) and response from the antioxidant defense systems [33]. Free radicals are molecules or molecular fragments that contain one or more unpaired electrons which make them highly reactive [34]. Under normal circumstances the effect of reactive species is balanced by the antioxidant action of enzymatic and non-enzymatic antioxidants. Essential cellular functions, such as gene expression, are influenced by the balance between pro- and antioxidant conditions [35]. Antioxidant defenses are extremely important as they represent the direct removal of free radicals (pro-oxidants), providing maximal protection for biological sites. Physiological processes of the cell including cellular proliferation and host defense may be interrupted when the ROS exceed or antioxidants fall below the homeostatic set point as illustrated in Figure 1. Increased ROS could be detrimental and cause cell death or accelerate ageing and age-related diseases. ROS contribute to cellular dysfunction and cell death by damaging proteins, lipids, and DNA. They may also serve as stress signals which activate specific redosensitive signaling pathways [36]. Production of ROS, reactive oxygen intermediate (ROI) and reactive nitrogen intermediate (RNI) are part of human body’s physiological processes [37]. Both ROI and RNI activities affect cells in similar ways, thus, the concept of OS has been expanded to include; hydroxyl and superoxide radicals, hydrogen peroxide and singlet oxygen, reactive nitrogen species (RNS) or RNI including nitric oxide (NO), peroxynitrite and, S-nitrosothiols [38].
Figure 1.
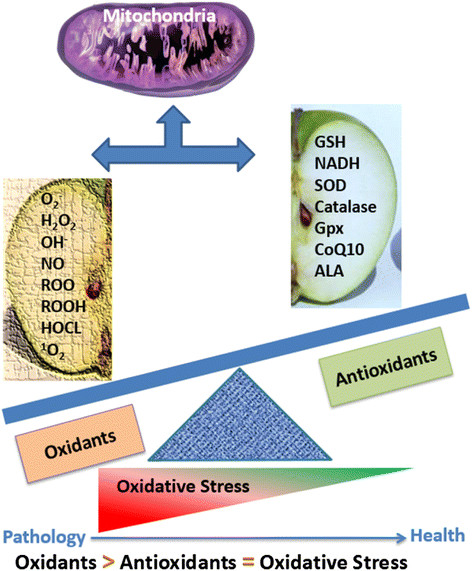
Types of oxidants and antioxidants which imbalance will lead to oxidative stress (OS). OS is described as an excess production of ROS compared to antioxidant defense.
Naturally, the human body maintains the redox balance by the combined action of antioxidant enzymes which include superoxide dismutase (SOD), catalase (CAT), glutathione (GSH), glutathione peroxidase (GPx) and glutathione reductase, monoamine oxidase (MAO), ascorbate, α-tocopherol, cysteine, thioredoxin, and vitamins [33],[39]. Coenzyme Q10 (CoQ10) is a very important endogenous antioxidant [40]. Also crucial are the peroxiredoxins (Prx), a family of small non-seleno peroxidases that regulate cellular ROS [41]. In their study, Valko and co-workers observed that ROS generated by the mitochondria, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases impacted on cell-cycle progression, cell motility, and growth factor signaling in a variety of normal cell types [4]. More ROS are produced when there is irregularity with the body’s antioxidant defense system due to disease other negative environmental conditions [39]. For example, ROS production spikes as cells transition from normal tissue to invasive carcinoma caused by the increase in metabolic aberrations in the transforming cells [42]. Also, levels of oxidants increase with corresponding decrease in levels of antioxidants in leukemia [43]. This view is supported by a study on ALL patients which showed that a tilt in ROS/antioxidant balance led to increase in serum level of markers of OS such as thiobarbituric acid reactive substances (TBARS), and serum protein carbonylation in ALL patients than in normal controls [10]. Literature is replete with reports on the harmful effects of ROS, however, some of the harmful concept are being exploited as potential targets for drug development [34]. For example low concentrations of ROS was observed to induce mitogenic response and trigger a series of cellular responses during noxia which helps to fight against infectious. But higher concentrations of ROS may induce damage of cell structures, including lipids and membranes, proteins and nucleic acids [44].
Sources/Activators of Reactive Oxygen Species (ROS)
ROS production can be triggered by both endogenous and exogenous factors. The endogenous sources are from cellular metabolism especially during mitochondria-catalyzed electron transport reactions, cytochrome P450 metabolism, and activities of peroxisomes, neutrophils, eosinophils and macrophages during inflammation [45]. Activated macrophages can initiate an increase in oxygen uptake which triggers increases in different reactive oxygen species, such as superoxide anion, nitric oxide and hydrogen peroxide [46]. Animal Cytochrome P450 enzymes is suspected to have a dual role of providing protection against natural toxic chemicals from plants and conversely inducing the production of reactive oxygen species especially superoxide anion and hydrogen peroxide after the breakdown or uncoupling of the P450 catalytic cycle [47]. Exogenous sources may include exposure to various xenobiotics, irradiation by UV light, X-rays and gamma-rays. And metal-catalyzed reactions that produce free radicals, chlorinated compounds, and barbiturates [48]. Mitochondria are regarded as the most important physiological source of radicals in living organisms because it generates approximately 2–3 nmol of superoxide/min per mg of protein [45]. Superoxide radical is considered as the stoichiometric precursor for hydrogen peroxide (H2O2) [49]. Ubisemiquinone is a reductant of oxygen in mitochondrial membranes [45]. Other endogenous sources of superoxide radical besides mitochondria include xanthine oxidase (XO), an enzyme that is found within the various tissues of mammals and which can be acquired from bacteria [50]. XO catalyzes the conversion of hypoxanthine to xanthine and xanthine to uric acid reducing molecular oxygen to form superoxide anion which subsequently produces hydrogen peroxide [34]. During their normal catalytic process, enzymes found in the peroxisomes can produce hydrogen peroxide (H2O2), superoxide (O2•−), or nitric oxide (NO•) which can readily react to form other ROS and RNS such as peroxynitrite (ONOO−), hydroxyl radical (•OH), and alkyl peroxides (ROOH). Peroxisomes play vital roles in fat metabolism especially in the liver and other organs leading to the production of H2O2, but not O2•− mostly after prolonged starvation [51],[52]. Superoxide may act as a reductant or an oxidant and is a key molecule in several subsequent physiologic reactions. Most of the superoxide generated in vivo is converted into H2O2 primarily by the actions of superoxide dismutases, which exist in cytosolic (SOD1), mitochondrial (SOD2), and extracellular (SOD3) isoform [51]. Activators and inhibitors of ROS are illustrated in Figure 2.
Figure 2.
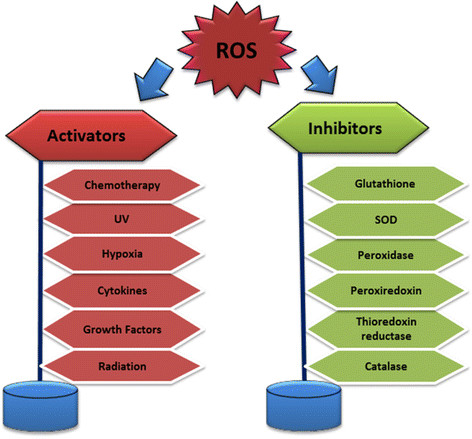
Schematic representation of various activators and inhibitors of reactive oxygen species production[53].
Mechanisms of action of ROS
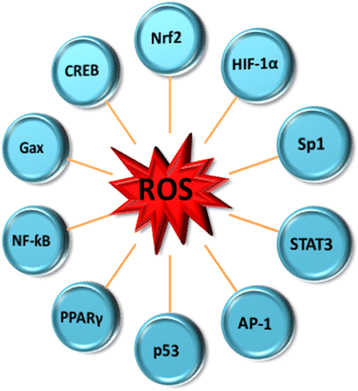
There are different mechanisms through which ROS cause cell damage. For instance endogenous damage occurs when intermediates of oxygen (dioxygen) reduction - oxygen-free radicals (OFR) attack the bases and the deoxyribosyl backbone of DNA. This endogenous DNA damage demonstrates the genotoxic, carcinogenic, and mutations induction properties of ROS [34]. Another important mechanism is the perturbation of transcription factors activities. Understanding the complex redox regulation of the transcription of specific eukaryotic genes will give more insight into the role of redox-sensitive transcription factors in this process. Figure 3 shows a schematic diagram of transcription factors that are modulated by ROS. The hypoxia-inducible transcription factor HIF-1 mediates upregulation of plasminogen activator inhibitor-1 (PAI-1) expression under low oxygen tension (hypoxia) and promotes angiogenesis [54]. Self-renewal of hematopoietic stem cells (HSCs), leukemia-initiating cells (LICs) and myeloproliferative neoplasms occurs under hypoxic condition [55]. There are contrary opinions on the role of HIF-1 in leukemia pathogenesis but it induces genes that facilitate adaptation and survival of cells and the whole organism when the environment changes from normoxia (∼21% O2) to hypoxia (∼1% O2). Cells and tissues may also be protected from oxidative damage by NF-E2 related factor 2 (Nrf2), a member of the cap‘n’collar family of basic leucine zipper transcription factors which induces activation of genes encoding detoxifying and antioxidant enzymes [56]. Activator protein 1 (AP-1) regulates diverse biological functions, including cell proliferation, protein synthesis, apoptosis and secretion of inflammatory and profibrotic factors. AP-1 also has a double edge function in tumorigenesis in that it can have both oncogenic or tumor suppressive activity depending on the cell context and the genetic background of the tumor [57],[58]. Other important genes involved in the linkage between OS and leukemia are matrix metallopeptidase 1 (MMP-1) and endothelin-1 which are expressed in response to Angiotensin II (Ang II) [59]. The growth arrest homeobox gene, mesenchyme homeobox 2 (Gax or MEOX2) regulates cell differentiation, proliferation, and migration and Gax is likely to have a regulatory function in the G0-to-G1 transition of the cell cycle in vascular smooth muscle cells [60],[61]. Cyclic AMP response element-binding protein (CREB) may have pro-survival effects on APL cells by protecting blasts from APL patients with acute promyelocytic leukemia (APL) against death induced by first-line anti-leukemic anthracyclines like daunorubicin (DNR) [62].
Figure 3.
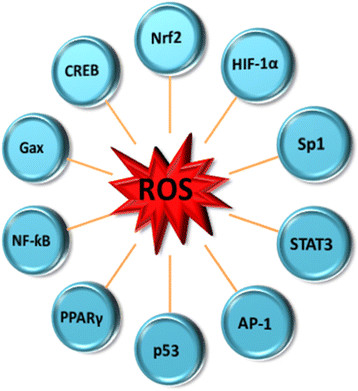
Transcription factors that are modulated by reactive oxygen species[53],[54].
Peroxisome proliferator-activated receptor gamma (PPARγ) is involved in pathological processes of different disease and it is a potential therapeutic target for the treatment of a diverse array of disorders including AML, in which supplementary treatment is suggested to include deregulation of the PPARγ signaling [63]. The ability of the tumor suppressor p53 under stress to differentially regulate its target genes which regulate cell-cycle arrest, apoptosis or senescence of hematopoietic stem cell (HSC) could be a possible treatment target for leukemia [64]. However, there is progress in the development of drugs based on OS mechanism. Some are on clinical trial stage, for instance a potent NF-kB inhibitor, Deferasirox is on clinical trial for the treatment of AML and MDS [65]. Another target of interest is the antiapoptotic transcription factor, signal transducer and activator of transcription 3 (STAT3) which is modulated by OS. STAT3 is activated when spleen tyrosine kinase (SYK) is induced in an ecdysone-inducible mammalian expression system. The link between SYK and STAT3 is being explored as a potential target for therapeutic purposes in human B-lineage leukemia/lymphoma cells [66].
Sources/activators of antioxidants
To counter the deleterious effects of free radicals and OS in the organelle, mitochondria also harbor antioxidants including GSH and enzymes, (superoxide dismutase (SOD) and glutathione peroxidase (GPx),) which are present on both sides of their membranes [67]. Antioxidants could be either endogenous or exogenous. The endogenous antioxidants produced mostly in the mitochondria include superoxide dismutase, (SOD), alpha lipoic acid (ALA), Coenzyme Q10 (CoQ10), catalase (CAT), and glutathione peroxidase (Gpx), glutathion (GSH), ferritin, uric acid, bilirubin, metallothioneine, L-carnitine and melatonin [68]. Exogenous antioxidants are acquired from diet such as vitamin E (α-tocopherol) which is present in vegetable oil and wheat germ. Vitamin E can prevent lipid peroxidation of plasma membrane because it is lipid soluble [69]. Other important exogenous antioxidants found in plants (fruits, vegetables, medicinal herbs) include phenolic compounds (phenolic acids, flavonoids, quinones, coumarins, lignans, stilbenes, tannins etc.), nitrogen compounds (alkaloids, amines, betalains), vitamins, and terpenoids (including carotenoids) [70],[71].
Mechanism of action of antioxidants
The most efficient enzymatic antioxidants are superoxide dismutase (SOD) and catalase (CAT). SOD activity protects cells from free radicals induced injury by catalyzing the dismutation of O2•− to O2 and to the less-reactive species hydrogen peroxide (H2O2) [72]. The enzyme CAT on the other hand catalyzes the conversion of H2O2 to water and molecular oxygen. A cervical cancer based study to examine the relationship between OS and enzymatic antioxidant status in the erythrocytes of adult cervical cancer patients and healthy subjects showed a significant increase in lipid peroxidation and impaired enzymatic antioxidant activities in the erythrocytes of cervical cancer patients [73]. Non-enzymatic antioxidants such as thiol antioxidants and vitamin E also play vital antioxidant roles. Vitamin E prevents lipid peroxidation and aging of cells [69]. Non-protein thiols have a variety of functions in bioreduction and detoxification processes. Intracellular redox homeostasis is regulated by thiol-containing molecules, such as glutathione and thioredoxin [74]. Catalina et al. evaluated the apoptotic effects of Cellfood™ (CF), a nutritional supplement containing deuterium sulphate, minerals, amino acids, and enzymes, on three leukemic cell lines including Jurkat, U937, and K562 cells, and reported that this natural anti-oxidant extracted from the red algae Lithothamnion calcareum, modulates cell signalling and apoptosis in cancer cells by activating caspase 3, inducing nucleosomal DNA frangmentation, and altering cell metabolism through down-regulation of HIF-1α and GLUT-1 expression [75]. In a study of the mitochondrial pathway of apoptosis in HepG2 cells, Chang et al. [76] reported that Norcantharidin, the demethylated analog of cantharidin derived from a traditional Chinese medicine, Mylabris, exerts its anti-cancer effects through induction of oxidative stress leading to loss of mitochondrial membrane potential, release of cytochrome c from the mitochondria to the cytosol, down-regulation of Bcl-2, up-regulation of Bax , caspase 3 and caspase 9, and subsequent cleavage of PARP.
Leukemia
Leukemia is one of the top 10 cancers affecting all races in the United States [77]. Leukemia remains a public health problem though there is a decline in annual death rate when compared with the death rate in 1991 [78]. According to the American Cancer Society estimates, about 1,660,290 new cancer cases and 580,350 cancer deaths occurred in the United States in 2013 and Leukemia account for 48,610 new cases and about 23,720 deaths [78]. Leukemia is cancer of the blood or bone marrow characterized by an abnormal proliferation and circulation of immature clonal hematopoietic cells. Diseases associated with leukemia are commonly referred to as hematological neoplasms and they constitute one of the 10 killer cancers in the US and the most commonly diagnosed cancers and leading causes of cancer death in children aged 0–19 years [79],[80]. In 2011 according to the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) program report, about 302,800 people were living with leukemia in the United States. About 52,380 new cases are estimated in 2014, which accounts for 3.1% of all new cancer cases. Also, 24,090 deaths are expected to occur in the US in 2014 due to leukemia which represents 4.1% of all cancer deaths [81]. Global deaths due to leukemia in 2010 were about 281,500 [82]. In 2000, leukemia accounted for about 3% of the seven million deaths due to cancer and about 0.35% of all deaths from any cause around the world [83]. There is a racial divide in the prevalence of leukemia; white American children are almost twice more likely to develop leukemia than black American children and boys are more likely to be affected than girls. Hispanics under 20 years of age are at the highest risk for leukemia, while whites, Native Americans, Asians, and Alaska Natives are at higher risk than blacks. Around 30 percent more men than women are diagnosed with leukemia and die from the disease [78]. But more than 90% of all leukemias are diagnosed in adults with the peak incidence between 40 and 60 years [84].
Leukemogenesis
Leukemia develops when hematopoietic stem cells lose the capacity to differentiate normally to mature blood cells at different stages of their maturation and differentiation [85]. Under normal process, all blood cells are originate from blood stem cells with the myeloid pathway producing red blood cells, platelets, and white blood cells, and the lymphoid pathway generating different types of lymphocytes [80]. This hematopoietic system produces the required amount of blood cells during an individual’s lifetime and it involves a carefully balanced mechanism of differentiation, proliferation and self-renewal [86]. The perturbation of this balance due to different cellular conditions and external pressures may lead to irregular differentiation of cell and premature generation of immature/abnormal cells and presence of heterogeneous populations of genetically distinct sub-clones in circulation causing the adverse effects associated with different forms of tumors [87],[88].
Types of leukemia
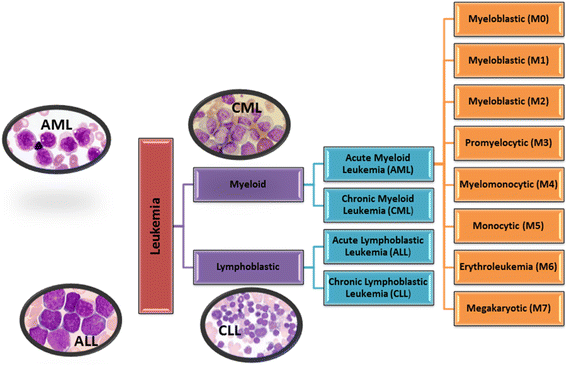
Leukemia are classified based on; onset (acute or chronic), the affected blood cell type (lymphoblastic/lymphocytic or myeloid/myelogenous), the maturity stage of the blood cell and phenotypic expression of the disease. There are four major clinical types of leukemia and there also different pathological subtypes as shown in Table 1. The common types of leukemia are; acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), chronic myeloid leukemia (CML), chronic lymphocytic leukemia (CLL), and acute promyelocytic leukemia (APL) [87] as illustrated in Figure 4. AML is further divided into eight subtypes based on the cell the leukemia developed from: Myeloblastic (M0), Myeloblastic (M1) - without maturation, Myeloblastic (M2) - with maturation, Promyeloctic (M3), Myelomonocytic (M4), Monocytic (M5), Erythroleukemia (M6) and Megakaryocytic (M7) [89]. Other sub-types include; hairy cell leukemia, chronic myelomonocytic leukemia (CMML), juvenile myelomonocytic leukemia (JMML) is seen mostly in children and it could be cured using hematopoietic stem cell transplantation (HSCT) [79],[90],[91].
Table 1.
Differentiating characteristics of the four types of leukemia
| Type/Age at onset (Yr.) | Gender predilection | Racial predilection | Cell of origin | Specific markers |
|---|---|---|---|---|
| Acute Lymphocytic (ALL) <15 | Males | Caucasian | B-cell | CALLA+ |
| Hyperdiploidy | ||||
| TDT+ | ||||
| Acute Myelogenous (AML) 15 - 39 | Equal incidence | None | Myeloblast | TDT- |
| Myelocyte | t(9;22) | |||
| Promyelocyte | t(15;17) | |||
| Myelomonocyte | ||||
| Chronic Myelogenous (CML) > 50 | Males | None | Myeloid cell | Ph1 chromosome |
Figure 4.

Major types of leukemia: AML- acute myeloid leukemia; ALL- acute lymphoblastic leukemia; CML- chronic myeloid leukemia; CLL- chronic lymphoblastic leukemia.
Acute myeloid leukemia
Acute myeloid leukemia (AML) known by different names such as acute myelogenous leukemia, acute granulocyctic leukemia or acute non-lymphocytic leukemia is a malignant neoplasm that originates in the cells within the bone marrow. AML can be divided into five main types: AML with genetic abnormalities (e.g. t[8;21], t(15;17), t(9;22) ), AML with FLT3 mutation, AML with multilineage dysplasia, therapy-related AML, and uncategorized AML [84]. However, World Health Organization (WHO) compresses AML types into three subgroups: (1) AML with recurrent genetic abnormalities, (2) AML with multilineage dysplasia, and (3) AML and MDS (myelodysplastic syndromes), therapy related [92]. AML is the second most common type of leukemia in children [93] and it affects heterogeneous group of tumor cell populations including myeloblast, myelocyte, promyelocyte, and myelomonocyte. It is described as acute because AML progresses quickly without treatment. Primitive myeloid cells are not well developed and cannot carry out the normal functions of the blood cell [79]. As a result cellular and molecular activities malfunction leading to DNA damage, increased proliferation, deficient cell death, genetic instability and ROS-induced OS [10],[94],[95]. MDS are malignant tumors that closely resemble AML and may develop to AML [96]. MDS are characterized by cytopenias which predisposes to infection, bleeding, and death [96]. Most AML can be characterized by the expression of the common acute lymphoblastic leukemia antigen (CALLA) which is a 749-amino acid type II integral membrane protein [97]. WHO recommends using the following cytogenetics parameters to define AML to avoid ambiguity, mischaracterization and misdiagnosis of AML; patients with the specific recurring cytogenetic abnormalities t(8;21)(q22;q22), inv(16)(p13q22) or t(16;16)(p13;q22), and t(15;17)(q22;q12) regardless of the blast percentage [84]. Both genetic predisposition and environmental risk factors such as radiation, cigarette smoking and exposure to other environmental carcinogens have been linked to AML pathogenesis. A previous study by Zhuo et al. [98] pointed out that CYP1A1 MspI polymorphism might be a possible risk factor for AML in Asian populations. Prognosis of AML is generally poor but best prognosis is with bone marrow transplant [99]. Acute leukemia may also have blast cells that are Sudan Black and terminal deoxynucleotidyl transferase (TdT) positive [100]. The survival rate of AML is very slim necessitating development of more effective treatment therapies [101],[102]. Treatment for AML may include chemotherapy, radiation therapy, stem cell transplant and/or immunotherapy [81],[89].
Chronic Myelogenous Leukemia (CML)
CML also referred to as chronic myeloid, chronic myelocytic or chronic granulocytic leukemia, is a clonal myeloproliferative disorder in which transformed, primitive hematopoietic progenitor cells are pushed into circulation. CML is characterized by increased proliferation of granulocytic cells without loss in their capacity to differentiate. CML is characterized by the Philadelphia (Ph1) chromosome and it is the first human disease malignancy associated with a gene translocation in which a specific abnormality of the karyotype could be linked to pathogenic processes of leukemogenesis [103]. The Philadelphia (Ph1) chromosome is created from the bcr-abl fusion gene, which is a result of the reciprocal translocation between the abl oncogene on the long arm of chromosome 9 and the bcr region on the long arm of chromosome 22, t(9;22)(q34;q11). The bcr-abl fusion gene is seen in more than 90% of CML cases [104],[105]. Prognosis is generally poor and it is worse if there is no Ph1 chromosome. In CML the chronic phase is often followed by an accelerated blastic phase, a more acute disease phase, which is generally fatal [103]. In a study of CML pathogenesis, Long et al. [106] evaluated the role of the Hedgehog (Hh) signaling pathway, and reported that Hh-related genes such as Sonic hedgehog (Shh), Smoothened (Smo), and Gli1 genes were significantly upregulated in CML patients when compared with normal people. They concluded that Hh signalling maybe associated with CML progression [106]. Treatment for CML may include radiation therapy, chemotherapy, stem cell transplant and/or immunotherapy. A common treatment for chronic leukemias is oral chemotherapy such as Gleevec (imatinib), Sprycel (dasatinib) and Tasigna (nilotinib) [89].
Chronic Myelomonocytic Leukemia (CMML)
CMML is an aggressive malignancy characterized by ineffective hematopoiesis and peripheral monocytosis. It was previously classified as a subtype of the myelodysplastic syndromes (MDSs) but was recently demonstrated to be a distinct entity with distinct characteristics [107]. However, it is placed under mixed myelodysplastic/myeloproliferative disease in the WHO classification [108]. About 20 to 40 percent of CMML patients have chromosomal abnormalities with 1 to 4 percent having translocation involving the PDGFR-β and TEL genes [90]. Chemotherapy with imatinib has been successful in the treatment or patients with PDGFR-β and TEL gene mutation [109].
Acute Promyelocytic Leukemia (APL)
APL is a form of acute myeloid leukemia in which abnormal promyelocytes predominate and it can affect both adults and children but mostly children [110]. The over production of promyelocytes leads to a shortage of normal white blood cells, red blood cells and platelets in circulation, which causes many of the signs and symptoms observed in APL. General signs and symptoms may occur as fever, loss of appetite, and weight loss but disseminated intravascular coagulation is a common symptom and could be life-threatening. Other signs of the malignancy include leukopenia, susceptibility to developing bruises, small red dots under the skin (petechiae), nosebleeds, bleeding from the gums, blood in the urine (hematuria), or excessive menstrual bleeding [111], low number of red blood cells (anemia), and excessive tiredness (fatigue). Some patients experience bones and joints pains when the leukemic cells spread to the bones and joints [110]. Genetic studies show that cells from most patients have a balanced reciprocal translocation between chromosomes 15 and 17 [112], which generates a fusion transcript joining the PML(promyelocyte) and RAR-α (retinoic acid receptor-α) genes [113]. The promyelocytic leukemia gene (PML) involved in the t(15;17) (q22;q12) translocation is a growth suppressor and pro-apoptotic factor [114],[115]. Disruption of the PML gene by the t(15:17) translocation in APL could be critical in leukemogenesis because accelerated cell proliferation was observed when the PML gene was knocked-out [116],[117]. APL is most often diagnosed around age 40, although it can be diagnosed at any age. Prognosis is poorer in adults than in children but prognosis is better than in AML and it is curable in children [118]. A combination of All-trans retinoic acid (ATRA) and arsenic trioxide (ATO) has been effective in treating APL especially in newly diagnosed patients. However, ATRA with anthracycline-based chemotherapy for induction and consolidation and additional use of low dose maintenance ATRA is considered as the standard treatment protocol [110]. ATRA has been reported to exert its therapeutic action against APL cancer through induction of cell differentiation via mechanisms that include degradation of PML-RARA gene [119] and inhibition of arachidonic acid metabolic pathway in other cancer cells [119].
Acute Lymphoblastic Leukemia (ALL)
ALL is a disease characterized by uncontrolled proliferation and maturation arrest of lymphoid progenitor cells in bone marrow resulting in an excess of malignant cells. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells leading to varying degrees of anemia, thrombocytopenia, and neutropenia [120]. ALL is the most common cancer found in children and it accounts for more than 50% of childhood hematopoietic malignancies. But it is relatively rare in adults, accounting for only 2–3% of hematopoietic malignancies [120]. Abnormal expression of genes, which is usually a result of chromosomal translocations, is suggested as one of the causes of ALL. The examination of the cytogenetic lesion in Ph(+) in ALL shows that the translocation of most cases of ALL with break point in the minor cluster region (m-BCR) give rise to (P190) fusion protein [121]. A previous in-vitro study using inhibitors of glycogen synthase kinase-3β (GSK-3β) found that it significantly accumulates in the nuclei of ALL cells compared to control cells; leading to a downregulation of NF-κB-target Survivin gene and promotion of apoptosis in ALL cells in vitro [122]. It is a curable disease with an expected long term survival rate of at least 70%, when treated with modern therapeutic regimens. A common chemotherapy treatment for ALL begins with induction chemotherapy, in which a combination of drugs is used to destroy as many leukemia cells as possible and bring blood counts to normal [123]. This is followed by consolidation chemotherapy, to destroy any remaining leukemia cells that cannot be seen in the blood or bone marrow. Patients with ALL may also receive maintenance chemotherapy. This less intensive course of chemotherapy is used to reduce the risk of the disease recurring after treatment has finished [123].
Chronic Lymphocytic Leukemia (CLL)
CLL starts in B-cells in the bone marrow before invading the blood. Leukemia cells take a long time to accumulate in the peripheral blood, bone marrow, and lymphoid tissues and may take a few years before symptoms start showing up in many people [124]. In the United States about 15,720 new cases of CLL and 4,600 deaths will occur in 2014 [125]. Most of CLL patients are the elderly, with a median age at diagnosis of 72 years with more males than females being affected. Also, more Caucasians than African-Americans have CLL and rarely seen among Hispanics, Native Americans, and Asian populations [126]. The prognosis is poor and it is the only leukemia with a possible genetic predisposition [124],[126]. Features of CLL include hypogammaglobulinemia and recurrent infection due to compromised humoral and cellular immunity [127]. Another feature observed in CLL is Trisomy 12 and it is one of chromosomal abnormalities frequently seen in B-cell CLL [128]. OS has been recognized to play a role in CLL development and treatment response. A recent study evaluating ROS-related damages in lymphocytes from patients with monoclonal B-lymphocytosis and CLL reported increased levels of oxidatively modified DNA and lipids in the sera of untreated CLL patients due to increased oxidative phosphorylation in CLL cells. Furthermore, CLL cells adapted to intrinsic OS by up-regulating the stress-responsive heme-oxygenase-1 (HO-1) [117]. Ibrutinib is approved for the treatment of patients with CLL who have received at least one prior therapy. It is also approved for relapsed/refractory mantle-cell lymphoma patients [129]. Hairy cell leukemia is a type of chronic lymphocytic leukemia [130].
Leukemia and reactive oxygen species
It is well recognized that oxidants play a role in several stages of carcinogenesis [53]. OS is associated with numerous pathological phenomena, including infection, inflammation, ultraviolet- and γ-irradiation, increased mutation frequency [79] and acute promyelocytic leukemia [131]. Increased free radical generation, especially superoxide anion in leukemia patients and increased antioxidant defense enzymes, which is an adaptive protective response, are indicative of mild OS [43]. Several studies have implicated OS as a factor in carcinogenesis [132],[133]. Some endogenous oxidants are considered as important naturally occurring carcinogens and may contribute to several stages of malignant transformation [134],[135]. ROS can induce genetic mutations as well as chromosomal alterations and thus contribute to cancer development in multistep carcinogenesis [133],[136]. ROS also can trigger intercellular secondary messengers and thus modulate various aspects of cellular functions including proliferation, apoptosis, and gene expression [137]. ROS initiate carcinogenesis by activating kinases, denaturing DNA through induction of poly ADP-ribosylation of chromosomal proteins [134]. Cell damage from oxygen free radicals (OFR) is ubiquitous and may be significant in the expansion of tumor clones and the acquisition of malignant properties [135]. Stress-activated signaling cascades are affected by altered redox potential due to ROS formed by exogenous genotoxic agents such as irradiation, inflammatory cytokines and chemical carcinogens. ROS and altered redox potential can be considered as the primary intracellular changes which regulate protein kinases, thus serving as an important cellular component linking external stimuli with signal transduction in stress response [137]. As observed earlier, various antioxidants are decreased in cancer, SOD and CAT activities were decreased in ALL patients. Reduced CAT activity was observed in just diagnosed patients and patients in both treatment groups suggesting a disturbance of the protective role of these enzymes against free radicals in ALL and chronic lymphocytic leukemia (CLL) [10],[138]. Nishiura et al. reported elevated serum SOD activity in acute leukemia and indicated that regression of the leukemia was accompanied by a decrease in the serum level of SOD [139]. Likewise, there is GSH depletion in lymphocytes isolated from the blood of patients with CLL [140]. These findings suggest that there are alterations in the enzymatic antioxidant defenses, which can interfere in the direct removal of free radicals (pro-oxidants) and in the protection for biological sites [141].
Oxidative stress in leukemia treatment
Traditionally, the appropriate treatment for any leukemia disease depends mainly on the type of leukemia, age, and general health of the patient [89]. The current cytotoxic drugs used in standard leukemia therapy are designed to attack DNA replication process within malignant cells, and it does not discriminate between malignant and non-malignant cells since it targets cell proliferation [142]. Adding to the toxic side effects of standard chemotherapy is the issues of drug resistance from quiescent clones [143]. These make discovery of new treatments measures for leukemia very imperative. Targeting ROS level could be a novel approach since ROS levels are higher in malignant cell than in non-malignant cells. Despite the negative effect of oxidants some of their mechanisms of action have found application in the treatment of malignant diseases [6]. Conversely, there are reports showing that antioxidative conditions promote some carcinogenic processes [144]. Both antioxidant and oxidants processes are being explored for treatment of hematologic malignancy. Lau and co-authors in their review discussed the potential applications of ROS in leukemia treatment [6]. As illustrated in Figure 5, there two approaches applied in using ROS for leukemia treatment; oxidant treatment and anti-oxidant treatment. The oxidant approaches may cause cell death by: increasing ROS, lipid peroxidation, protein oxidation and mutation, increasing mitochondria stress, apoptosis, and activation of a G2/M phase cell cycle checkpoint [145]. The antioxidant mechanism uses the following reactions to balance the negative effect of the oxidant processes: reduced ROS signalling, reduced proliferative drive, and suppression of cell cycle which may reduce tumor burden and buffer nonmalignant cells from oxidative damage [146]. A combination of antioxidants and chemotherapeutic agents have promising synergistic effect, however, some argue that suppression of cell cycle and antagonism of chemotherapy-induced ROS may also affect treatment efficacy [147]. Vitamin antioxidants have been shown to reduce the risk of various cancers by suppressing the state of OS [148],[149]. Likewise, antioxidant vitamins C and E from supplements have shown promises in reducing risk of ovarian cancer [149].
Figure 5.
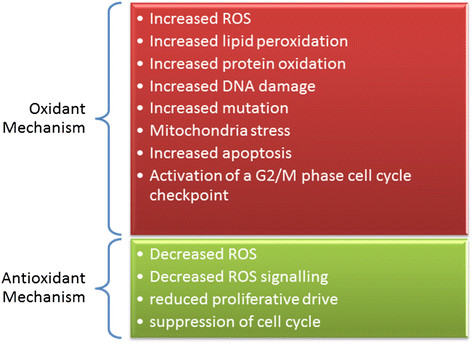
Antioxidant treatment versus pro-oxidant treatment as a therapy for hematologic malignancy.
Pro-oxidant treatment may cause depletion of antioxidant defenses leading to more production of ROS beyond the level produced by the malignant cell [31]. Although it may be difficult it is very important to determine the optimum level of ROS that will be most efficient and effective in the treatment of Leukemia, only a thin line separates the beneficial level and deleterious level of ROS [150]. For instance when the intrinsic stress that is already present in malignant cells is doubled by the treatment-induced OS malignant cells can be sensitized to mainstay treatments or initiate the apoptotic pathway [31]. It can conversely trigger lipid peroxidation, oxidation of redox-sensitive residues within proteins, and DNA oxidation leading to increased tumor burden [147],[150]. As observed with proxidants, extramitochondrial antioxidants such as vitamin C (ascorbic acid) can act both as prooxidant and as antioxidant [144].
Bortezomib-induced oxidative cell injury function at a proximal point in the cell death cascade to attack and disrupt cytoprotective ERK1/2 signaling. The JNK pathway, is activated followed by induction of mitochondrial dysfunction, caspase activation, and apoptotic cell death [151]. Anthracycline daunorubicin is widely used in the treatment of acute nonlymphocytic leukemia and oxidative stress is one of the triggers of the body’s response to this drug [152]. In a study from our laboratory we demonstrated that dietary supplement such as garlic induces cytotoxicity and apoptosis in HL-60 cells through phosphatidylserine externalization, caspase-3 activation, and nucleosomal DNA fragmentation associated with the formation of malondialdehyde, a by-product of lipid peroxidation and biomarker of OS [153].
Application of antioxidant principles may illicit same effect, for example inhibition of intracellular antioxidants such as GSH [154] and heme oxygenase-1 (HO-1) [155]. Isothiocyanates and adaphostine are other pro-oxidant drugs. Isothiocyanates act by depleting GSH pools, and efficiently kill fludarabine-resistant chronic lymphocytic leukemia (CLL) cells [156] and imatinib-resistant CML cells, [157] selectively without attacking normal hematopoietic cells. Adaphostine, a tyrphostin kinase inhibitor is able to induce up-regulation of ROS and cause DNA damage-induced apoptosis in BCR-ABL–expressing CML [158]. Other leukemic drugs that have pro-oxidant properties include arsenic trioxide (ATO) which is currently used for treatment of relapsed APL which may function by inhibiting thioredoxin [159], inducing ROS production [160] or NOX activation [161]. However, Jeanne and co-researchers suggest that ATO-induced ROS production plays a critical role in degradation of PML-RARα fusion proteins in ATO-treated APL cells [162]. Isothiocyanates when used together with ATO were effective in killing CML- and AML-derived cell lines in vitro [163]. In a recent in vitro study, we demonstrated that OS plays a key role in ATO-induced mitochondrial pathway of apoptosis in HL-60 cells. We discovered that this apoptotic signaling is associated with DNA damage, change in mitochondrial membrane potential, activation and translocation of pro-apoptotic proteins, and down regulation of anti-apoptotic proteins [2]. These findings are in support of previous research from our laboratory reporting phosphatidylserine externalization and caspase 3 activation and nucleosomal DNA fragmentation in HL-60 cells exposed to ATO [164]. A recent study from our laboratory has also demonstrated that vitamin D3 (Vit D3) potentiates the antitumor effect of ATO in HL-60 cells. This potentiation is mediated at least in part, through induction of phosphatidylserine externalization and nucleosomal DNA fragmentation [165]. Also, we previously reported similar potentiation of ATO effect by vitamin C [166],[167]. These findings highlight the potential impact of Vit C and Vit D3 in promoting the pharmacological effect of ATO, suggesting a possible future role of Vit C/Vit D3/ATO combination therapy in patients with acute promyelocytic leukemia.
Antioxidant molecules can be used to suppress the high levels of ROS observed in some cancer cells. For example targeting NOX4-derived ROS which promotes survival in some pancreatic cancers could be effective in growth arrest [168]. Also, antioxidant treatment may alleviate chemotherapy-related toxicity, reducing the requirement for dose reduction in some patients and allowing an increased proportion of patients to complete their therapy [169]. Another study demonstrated a trend of longer clinical progression-free survival and overall survival in CML patients when they were treated with vitamin A in combination with standard chemotherapy, although this trend was not significant [170]. Figure 5 illustrates the involvement of oxidant and antioxidant treatment in the control of leukemia cancer.
Conclusions
OS has both beneficial and negative effect on leukemogenesis. Most of the genes that regulate the redox processes have double edged sword activities. This makes it difficult to determine the optimum point to harness the therapeutic attributes because only a very thin line separates the oncogenic properties from the tumor suppressive activity. A better understanding of the relationship between OS and leukemogenesis will give more insight on how to ameliorate its deleterious effect and how to tap unto the beneficial attributes. It is known that ROS causes nonspecific oxidative damage to biomolecules in myeloid cells, under this condition there is a persistent increase in ROS level and depletion of the cell’s antioxidant defenses culminating in cancer induction. Another process is the hyperactivation of ROS signaling pathway. Significant progress has been made in developing therapeutic measures for various types of leukemia and some can be cured while treatment for some such as, myeloid leukemia are still difficult. New effective therapeutic strategies are needed. Increased OS in some myeloid leukemia may be a promising therapeutic target. Despite the negative effect of OS to the cell, tapping into the possible application of its activities for therapeutic capabilities is worth pursuing because even the current leukemia treatment drugs have cytotoxic effects as well. Further studies are required to determine the source and species of ROS generated by leukemic cells and whether the ROS that has therapeutic effects originate from the normal cell metabolism or from the malignant cell population. The knowledge that OS induces lipid peroxidation and protein carbonylation by inactivating antioxidant enzymes could be used to determine an efficient and effective way to boost endogenous production and incorporation of antioxidants into diets to neutralize the free radical produced by cellular metabolisms. This may serve as potent prophylaxis for various leukemia since most cancers develop under OS environment. It is heartening that some drugs based on oxidative mechanisms are on clinical trial stage. Further studies are needed to understand the effectiveness, long time effect and consequences of using such drugs.
Acknowledgement
This work was supported by NIH Grant # G12MD007581.
Abbreviations
- ALA
Alpha lipoic acid
- ALL
Acute lymphoblastic leukemia
- AML
Acute myeloid leukemia
- Ang II
Angiotensin II
- ATO
Arsenic trioxide
- ATRA
All-trans retinoic acid
- CALLA
Common acute lymphoblastic leukemia antigen
- CAT
Catalase
- CLL
Chronic lymphoblastic leukemia
- CML
Chronic myeloid leukemia
- CMML
Chronic myelomonocytic leukemia
- CoQ10
Coenzyme Q10
- GPx
Glutathione peroxidase
- GSH
Glutathion
- GSK-3β
Glycogen synthase kinase-3β
- Hh
Hedgehog
- HO-1
Heme-oxygenase-1
- HSC
Hematopoietic stem cell
- HSCT
Hematopoietic stem cell transplantation
- JMML
Juvenile myelomonocytic leukemia
- MBL
Monoclonal B-cell lymphocytosis
- MDS
Myelodysplastic syndromes
- MMP-1
Matrix metallopeptidase 1
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NCI
National Cancer Institute
- OFR
Oxygen free radicals
- OS
Oxidative stress
- PML
Promyelocytic leukemia gene
- PPARγ
Peroxisome proliferator-activated receptor gamma
- RNI
Reactive nitrogen intermediate
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- SEER
Ssurveillance, epidemiology, and end results
- Shh
Sonic hedgehog
- SOD
Superoxide dismutase
- STAT3
Signal transducer and activator of transcription 3
- SYK
Spleen tyrosine kinase
- TBARS
Thiobarbituric acid reactive substances
- TdT
Deoxynucleotidyl transferase
- WHO
World Health Organization
- XO
Xanthine oxidase
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
UKU and PBT conceived the research. UKU collected the literature. PBT designed the manuscript. Both authors wrote and edited the manuscript. Both authors read and approved the final manuscript.
Contributor Information
Udensi K Udensi, Email: udensi.k.udensi@jsums.edu.
Paul B Tchounwou, Email: paul.b.tchounwou@jsums.edu.
References
- 1.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/S0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 2.Kumar S, Yedjou CG, Tchounwou PB. Arsenic trioxide induces oxidative stress, DNA damage, and mitochondrial pathway of apoptosis in human leukemia (HL-60) cells. J Exp Clin Cancer Res. 2014;33:42. doi: 10.1186/1756-9966-33-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ristow M, Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis) Exp Gerontol. 2010;45:410–418. doi: 10.1016/j.exger.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 4.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Weinberg F, Chandel NS. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol Life Sci. 2009;66:3663–3673. doi: 10.1007/s00018-009-0099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau AT, Wang Y, Chiu JF. Reactive oxygen species: current knowledge and applications in cancer research and therapeutic. J Cell Biochem. 2008;104:657–667. doi: 10.1002/jcb.21655. [DOI] [PubMed] [Google Scholar]
- 7.Renschler MF. The emerging role of reactive oxygen species in cancer therapy. Eur J Cancer. 2004;40:1934–1940. doi: 10.1016/j.ejca.2004.02.031. [DOI] [PubMed] [Google Scholar]
- 8.Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008;68:1777–1785. doi: 10.1158/0008-5472.CAN-07-5259. [DOI] [PubMed] [Google Scholar]
- 9.Fried L, Arbiser JL. The reactive oxygen-driven tumor: relevance to melanoma. Pigment Cell Melanoma Res. 2008;21:117–122. doi: 10.1111/j.1755-148X.2008.00451.x. [DOI] [PubMed] [Google Scholar]
- 10.Battisti V, Maders LD, Bagatini MD, Santos KF, Spanevello RM, Maldonado PA, Brulé AO, Araújo Mdo C, Schetinger MR, Morsch VM. Measurement of oxidative stress and antioxidant status in acute lymphoblastic leukemia patients. Clin Biochem. 2008;41:511–518. doi: 10.1016/j.clinbiochem.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 11.Farquhar MJ, Bowen DT. Oxidative stress and the myelodysplastic syndromes. Int J Hematol. 2003;77:342–350. doi: 10.1007/BF02982641. [DOI] [PubMed] [Google Scholar]
- 12.Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008;270:1–9. doi: 10.1016/j.canlet.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 13.Wilson JN, Pierce JD, Clancy RL. Reactive oxygen species in acute respiratory distress syndrome. Heart Lung. 2001;30:370–375. doi: 10.1067/mhl.2001.118298. [DOI] [PubMed] [Google Scholar]
- 14.Dugan LL, Quick KL. Reactive oxygen species and aging: evolving questions. Sci Aging Knowledge Environ. 2005;2005:e20. doi: 10.1126/sageke.2005.26.pe20. [DOI] [PubMed] [Google Scholar]
- 15.Hensley K, Butterfield DA, Hall N, Cole P, Subramaniam R, Mark R, Mattson MP, Markesbery WR, Harris ME, Aksenov M, Aksenova M, Wu JF, Carney JM. Reactive oxygen species as causal agents in the neurotoxicity of the Alzheimer’s disease-associated amyloid beta peptide. Ann N Y Acad Sci. 1996;786:120–134. doi: 10.1111/j.1749-6632.1996.tb39057.x. [DOI] [PubMed] [Google Scholar]
- 16.Halliwell B. Free radicals, reactive oxygen species and human disease: a critical evaluation with special reference to atherosclerosis. Br J Exp Pathol. 1989;70:737–757. [PMC free article] [PubMed] [Google Scholar]
- 17.Touyz RM. Reactive oxygen species and angiotensin II signaling in vascular cells – implications in cardiovascular disease. Braz J Med Biol Res. 2004;37:1263–1273. doi: 10.1590/S0100-879X2004000800018. [DOI] [PubMed] [Google Scholar]
- 18.Muhammad S, Bierhaus A, Schwaninger M. Reactive oxygen species in diabetes-induced vascular damage, stroke, and Alzheimer’s disease. J Alzheimers Dis. 2009;16:775–785. doi: 10.3233/JAD-2009-0982. [DOI] [PubMed] [Google Scholar]
- 19.Di VF. New pathways for reactive oxygen species generation in inflammation and potential novel pharmacological targets. Curr Pharm Des. 2004;10:1647–1652. doi: 10.2174/1381612043384727. [DOI] [PubMed] [Google Scholar]
- 20.Moulton PJ. Inflammatory joint disease: the role of cytokines, cyclooxygenases and reactive oxygen species. Br J Biomed Sci. 1996;53:317–324. [PubMed] [Google Scholar]
- 21.Bolanos JP, Moro MA, Lizasoain I, Almeida A. Mitochondria and reactive oxygen and nitrogen species in neurological disorders and stroke: Therapeutic implications. Adv Drug Deliv Rev. 2009;61:1299–1315. doi: 10.1016/j.addr.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tieu K, Ischiropoulos H, Przedborski S. Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life. 2003;55:329–335. doi: 10.1080/1521654032000114320. [DOI] [PubMed] [Google Scholar]
- 24.Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med. 2005;172:417–422. doi: 10.1164/rccm.200501-017PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelderman KA, Hultqvist M, Olsson LM, Bauer K, Pizzolla A, Olofsson P, Holmdahl R. Rheumatoid arthritis: the role of reactive oxygen species in disease development and therapeutic strategies. Antioxid Redox Signal. 2007;9:1541–1567. doi: 10.1089/ars.2007.1569. [DOI] [PubMed] [Google Scholar]
- 26.Jeremy JY, Shukla N, Muzaffar S, Handley A, Angelini GD. Reactive oxygen species, vascular disease and cardiovascular surgery. Curr Vasc Pharmacol. 2004;2:229–236. doi: 10.2174/1570161043385691. [DOI] [PubMed] [Google Scholar]
- 27.Maraldi T, Prata C, Vieceli Dalla Sega F, Caliceti C, Zambonin L, Fiorentini D, Hakim G. NAD(P)H oxidase isoform Nox2 plays a prosurvival role in human leukaemia cells. Free Radic Res. 2009;43:1111–1121. doi: 10.1080/10715760903186132. [DOI] [PubMed] [Google Scholar]
- 28.Naughton R, Quiney C, Turner SD, Cotter TG. Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway. Leukemia. 2009;23:1432–1440. doi: 10.1038/leu.2009.49. [DOI] [PubMed] [Google Scholar]
- 29.Arnold RS, He J, Remo A, Ritsick D, Yin-Goen Q, Lambeth JD, Datta MW, Young AN, Petros JA. Nox1 expression determines cellular reactive oxygen and modulates c-fos-induced growth factor, interleukin-8, and Cav-1. Am J Pathol. 2007;171:2021–2032. doi: 10.2353/ajpath.2007.061144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu WS, Wu JR, Hu CT. Signal cross talks for sustained MAPK activation and cell migration: the potential role of reactive oxygen species. Cancer Metastasis Rev. 2008;27:303–314. doi: 10.1007/s10555-008-9112-4. [DOI] [PubMed] [Google Scholar]
- 31.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 32.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 33.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/B:MCBI.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 35.Arrigo AP. Gene expression and the thiol redox state. Free Radic Biol Med. 1999;27:936–944. doi: 10.1016/S0891-5849(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 36.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 37.Nathan C. Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J Clin Invest. 2003;111:769–778. doi: 10.1172/JCI200318174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kroncke KD. Nitrosative stress and transcription. Biol Chem. 2003;384:1365–1377. doi: 10.1515/BC.2003.153. [DOI] [PubMed] [Google Scholar]
- 39.Halliwell B, Gutteridge J. Free radicals in biology and medicine. 3. Oxford: Clarendon; 1999. [DOI] [PubMed] [Google Scholar]
- 40.Niklowitz P, Wiesel T, Andler W, Menke T. Coenzyme Q10 concentration in the plasma of children suffering from acute lymphoblastic leukaemia before and during induction treatment. Biofactors. 2007;29:83–89. doi: 10.1002/biof.552029208. [DOI] [PubMed] [Google Scholar]
- 41.Liu CX, Zhou HC, Yin QQ, Wu YL, Chen GQ. Targeting peroxiredoxins against leukemia. Exp Cell Res. 2013;319:170–176. doi: 10.1016/j.yexcr.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 42.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 43.Devi GS, Prasad MH, Saraswathi I, Raghu D, Rao DN, Reddy PP. Free radicals antioxidant enzymes and lipid peroxidation in different types of leukemias. Clin Chim Acta. 2000;293:53–62. doi: 10.1016/S0009-8981(99)00222-3. [DOI] [PubMed] [Google Scholar]
- 44.Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Oxidative stress and cell signalling. Curr Med Chem. 2004;11:1163–1182. doi: 10.2174/0929867043365323. [DOI] [PubMed] [Google Scholar]
- 45.Inoue M, Sato EF, Nishikawa M, Park AM, Kira Y, Imada I, Utsumi K. Mitochondrial generation of reactive oxygen species and its role in aerobic life. Curr Med Chem. 2003;10:2495–2505. doi: 10.2174/0929867033456477. [DOI] [PubMed] [Google Scholar]
- 46.Conner EM, Grisham MB. Inflammation, free radicals, and antioxidants. Nutrition. 1996;12:274–277. doi: 10.1016/S0899-9007(96)00000-8. [DOI] [PubMed] [Google Scholar]
- 47.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci U S A. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cadenas E. Biochemistry of oxygen toxicity. Annu Rev Biochem. 1989;58:79–110. doi: 10.1146/annurev.bi.58.070189.000455. [DOI] [PubMed] [Google Scholar]
- 49.Loschen G, Flohe L, Chance B. Respiratory chain linked H(2)O(2) production in pigeon heart mitochondria. FEBS Lett. 1971;18:261–264. doi: 10.1016/0014-5793(71)80459-3. [DOI] [PubMed] [Google Scholar]
- 50.Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol. 2002;282:C227–C241. doi: 10.1152/ajpcell.00112.2001. [DOI] [PubMed] [Google Scholar]
- 51.Gupta M, Dobashi K, Greene EL, Orak JK, Singh I. Studies on hepatic injury and antioxidant enzyme activities in rat subcellular organelles following in vivo ischemia and reperfusion. Mol Cell Biochem. 1997;176:337–347. doi: 10.1023/A:1006829902442. [DOI] [PubMed] [Google Scholar]
- 52.Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim Biophys Acta. 1822;2012:1363–1373. doi: 10.1016/j.bbadis.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 53.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Görlach A, Berchner-Pfannschmidt U, Wotzlaw C, Cool RH, Fandrey J, Acker H, Jungermann K, Kietzmann T. Reactive oxygen species modulate HIF-1 mediated PAI-1 expression: involvement of the GTPase Rac1. Thromb Haemost. 2003;89:926–935. [PubMed] [Google Scholar]
- 55.Velasco-Hernandez T, Hyrenius-Wittsten A, Rehn M, Bryder D, Cammenga J. HIF-1alpha can act as a tumor suppressor gene in murine Acute Myeloid Leukemia. Blood. 2014;124(24):3597–3607. doi: 10.1182/blood-2014-04-567065. [DOI] [PubMed] [Google Scholar]
- 56.Surh YJ, Kundu JK, Na HK, Lee JS. Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J Nutr. 2005;135:2993S–3001S. doi: 10.1093/jn/135.12.2993S. [DOI] [PubMed] [Google Scholar]
- 57.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 58.Verde P, Casalino L, Talotta F, Yaniv M, Weitzman JB. Deciphering AP-1 function in tumorigenesis: fra-ternizing on target promoters. Cell Cycle. 2007;6:2633–2639. doi: 10.4161/cc.6.21.4850. [DOI] [PubMed] [Google Scholar]
- 59.Browatzki M, Larsen D, Pfeiffer CA, Gehrke SG, Schmidt J, Kranzhofer A, Katus HA, Kranzhofer R. Angiotensin II stimulates matrix metalloproteinase secretion in human vascular smooth muscle cells via nuclear factor-kappaB and activator protein 1 in a redox-sensitive manner. J Vasc Res. 2005;42:415–423. doi: 10.1159/000087451. [DOI] [PubMed] [Google Scholar]
- 60.Saito T, Itoh H, Yamashita J, Doi K, Chun TH, Tanaka T, Inoue M, Masatsugu K, Fukunaga Y, Sawada N, Sakaguchi S, Arai H, Tojo K, Tajima N, Hosoya T, Nakao K. Angiotensin II suppresses growth arrest specific homeobox (Gax) expression via redox-sensitive mitogen-activated protein kinase (MAPK) Regul Pept. 2005;127:159–167. doi: 10.1016/j.regpep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 61.Gorski DH, LePage DF, Patel CV, Copeland NG, Jenkins NA, Walsh K. Molecular cloning of a diverged homeobox gene that is rapidly down-regulated during the G0/G1 transition in vascular smooth muscle cells. Mol Cell Biol. 1993;13:3722–3733. doi: 10.1128/MCB.13.6.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gausdal G, Wergeland A, Skavland J, Nguyen E, Pendino F, Rouhee N, McCormack E, Herfindal L, Kleppe R, Havemann U, Schwede F, Bruserud O, Gjertsen BT, Lanotte M, Ségal-Bendirdjian E, Døskeland SO. Cyclic AMP can promote APL progression and protect myeloid leukemia cells against anthracycline-induced apoptosis. Cell Death Dis. 2013;4:e516. doi: 10.1038/cddis.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Faber K, Bullinger L, Ragu C, Garding A, Mertens D, Miller C, Martin D, Walcher D, Döhner K, Döhner H, Claus R, Plass C, Sykes SM, Lane SW, Scholl C, Fröhling S. CDX2-driven leukemogenesis involves KLF4 repression and deregulated PPARgamma signaling. J Clin Invest. 2013;123:299–314. doi: 10.1172/JCI64745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y, Elf SE, Asai T, Miyata Y, Liu Y, Sashida G, Huang G, Di Giandomenico S, Koff A, Nimer SD. The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle. 2009;8:3120–3124. doi: 10.4161/cc.8.19.9627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Breccia M, Alimena G. Efficacy and safety of deferasirox in myelodysplastic syndromes. Ann Hematol. 2013;92:863–870. doi: 10.1007/s00277-013-1703-7. [DOI] [PubMed] [Google Scholar]
- 66.Uckun FM, Qazi S, Ma H, Tuel-Ahlgren L, Ozer Z. STAT3 is a substrate of SYK tyrosine kinase in B-lineage leukemia/lymphoma cells exposed to oxidative stress. Proc Natl Acad Sci U S A. 2010;107:2902–2907. doi: 10.1073/pnas.0909086107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/S0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 68.Rizzo AM, Berselli P, Zava S, Montorfano G, Negroni M, Corsetto P, Berra B. Endogenous antioxidants and radical scavengers. Adv Exp Med Biol. 2010;698:52–67. doi: 10.1007/978-1-4419-7347-4_5. [DOI] [PubMed] [Google Scholar]
- 69.Burton GW, Ingold KU. Vitamin E as an in vitro and in vivo antioxidant. Ann N Y Acad Sci. 1989;570:7–22. doi: 10.1111/j.1749-6632.1989.tb14904.x. [DOI] [PubMed] [Google Scholar]
- 70.Cai Y, Sun M, Corke H. Antioxidant activity of betalains from plants of the amaranthaceae. J Agric Food Chem. 2003;51:2288–2294. doi: 10.1021/jf030045u. [DOI] [PubMed] [Google Scholar]
- 71.Zheng W, Wang SY. Antioxidant activity and phenolic compounds in selected herbs. J Agric Food Chem. 2001;49:5165–5170. doi: 10.1021/jf010697n. [DOI] [PubMed] [Google Scholar]
- 72.Oberley LW, Buettner GR. Role of superoxide dismutase in cancer: a review. Cancer Res. 1979;39:1141–1149. [PubMed] [Google Scholar]
- 73.Manoharan S, Kolanjiappan K, Kayalvizhi M. Enhanced lipid peroxidation and impaired enzymic antioxidant activities in the erythrocytes of patients with cervical carcinoma. Cell Mol Biol Lett. 2004;9:699–707. [PubMed] [Google Scholar]
- 74.Oktyabrsky ON, Smirnova GV. Redox regulation of cellular functions. Biochemistry (Mosc) 2007;72:132–145. doi: 10.1134/S0006297907020022. [DOI] [PubMed] [Google Scholar]
- 75.Catalani S, Carbonaro V, Palma F, Arshakyan M, Galati R, Nuvoli B, Battistelli S, Canestrari F, Benedetti S. Metabolism modifications and apoptosis induction after Cellfood administration to leukemia cell lines. J Exp Clin Cancer Res. 2013;32:63. doi: 10.1186/1756-9966-32-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chang C, Zhu YQ, Mei JJ, Liu SQ, Luo J. Involvement of mitochondrial pathway in NCTD-induced cytotoxicity in human hepG2 cells. J Exp Clin Cancer Res. 2010;29:145. doi: 10.1186/1756-9966-29-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997–2002. Cancer Causes Control. 2008;19:379–390. doi: 10.1007/s10552-007-9097-2. [DOI] [PubMed] [Google Scholar]
- 78.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 79.Chung YJ, Robert C, Gough SM, Rassool FV, Aplan PD. Oxidative stress leads to increased mutation frequency in a murine model of myelodysplastic syndrome. Leuk Res. 2014;38:95–102. doi: 10.1016/j.leukres.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.CDC: Cancer Among Children, Cancer Prevention and Control. http://www.cdc.gov/cancer/dcpc/data/children.htm. Retrieved 6/5/2014. 2014.,
- 81.NCI: Surveillance, Epidemiology, and End Results (SEER) Program, Stat Fact Sheets: Leukemia. http://seer.cancer.gov/statfacts/html/leuks.html. Retrieved 6/5/2014. 2014. National Cancer Institute.,
- 82.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mathers CD, Shibuya K, Boschi-Pinto C, Lopez AD, Murray CJ. Global and regional estimates of cancer mortality and incidence by site: I. Application of regional cancer survival model to estimate cancer mortality distribution by site. BMC Cancer. 2002;2:36. doi: 10.1186/1471-2407-2-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–2302. doi: 10.1182/blood-2002-04-1199. [DOI] [PubMed] [Google Scholar]
- 85.Vyas P, Jacobsen SE. Clever leukemic stem cells branch out. Cell Stem Cell. 2011;8:242–244. doi: 10.1016/j.stem.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 86.Kennedy JA, Barabe F. Investigating human leukemogenesis: from cell lines to in vivo models of human leukemia. Leukemia. 2008;22:2029–2040. doi: 10.1038/leu.2008.206. [DOI] [PubMed] [Google Scholar]
- 87.NCI: Leukemia, National Cancer Institute (NCI). http://www.cancer.gov/cancertopics/wyntk/leukemia/page1. Retrieved 6/5/2014. 2014.,
- 88.Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, Ma J, Minden MD, Downing JR, Dick JE. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469:362–367. doi: 10.1038/nature09733. [DOI] [PubMed] [Google Scholar]
- 89.CTCA: Leukemia types. http://www.cancercenter.com/leukemia/types/tab/chronic-myeloid-leukemia/, Cancer Treatment Centers of America (CTCA). Retrieved 8/04/2014.,
- 90.Orazi A, Germing U. The myelodysplastic/myeloproliferative neoplasms: myeloproliferative diseases with dysplastic features. Leukemia. 2008;22:1308–1319. doi: 10.1038/leu.2008.119. [DOI] [PubMed] [Google Scholar]
- 91.Niemeyer CM, Kratz CP, Hasle H. Pediatric myelodysplastic syndromes. Curr Treat Options Oncol. 2005;6:209–214. doi: 10.1007/s11864-005-0004-3. [DOI] [PubMed] [Google Scholar]
- 92.Germing U, Gattermann N, Strupp C, Aivado M, Aul C. Validation of the WHO proposals for a new classification of primary myelodysplastic syndromes: a retrospective analysis of 1600 patients. Leuk Res. 2000;24:983–992. doi: 10.1016/S0145-2126(00)00088-6. [DOI] [PubMed] [Google Scholar]
- 93.Bhatia S, Neglia JP. Epidemiology of childhood acute myelogenous leukemia. J Pediatr Hematol Oncol. 1995;17:94–100. doi: 10.1097/00043426-199505000-00002. [DOI] [PubMed] [Google Scholar]
- 94.Austin C. Does oxidative damage contribute to the generation of leukemia? Leuk Res. 2009;33:1297. doi: 10.1016/j.leukres.2009.04.038. [DOI] [PubMed] [Google Scholar]
- 95.Zhou F, Zhang W, Wei Y, Zhou D, Su Z, Meng X, Hui L, Tian W. The changes of oxidative stress and human 8-hydroxyguanine glycosylase1 gene expression in depressive patients with acute leukemia. Leuk Res. 2007;31:387–393. doi: 10.1016/j.leukres.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 96.Troy JD, Atallah E, Geyer JT, Saber W. Myelodysplastic syndromes in the United States: an update for clinicians. Ann Med. 2014;46(5):283–289. doi: 10.3109/07853890.2014.898863. [DOI] [PubMed] [Google Scholar]
- 97.Shipp MA, Vijayaraghavan J, Schmidt EV, Masteller EL, D’Adamio L, Hersh LB, Reinherz EL. Common acute lymphoblastic leukemia antigen (CALLA) is active neutral endopeptidase 24.11 (“enkephalinase”): direct evidence by cDNA transfection analysis. Proc Natl Acad Sci U S A. 1989;86:297–301. doi: 10.1073/pnas.86.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhuo W, Zhang L, Wang Y, Zhu B, Chen Z. CYP1A1 MspI polymorphism and acute myeloid leukemia risk: meta-analyses based on 5018 subjects. J Exp Clin Cancer Res. 2012;31:62. doi: 10.1186/1756-9966-31-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Linker CA. Autologous stem cell transplantation for acute myeloid leukemia. Bone Marrow Transplant. 2003;31:731–738. doi: 10.1038/sj.bmt.1704020. [DOI] [PubMed] [Google Scholar]
- 100.Marcus RE, Matutes E, Drysdale H, Catovsky D. Phenotypic conversion of TdT+ adult AML to CALLA+ ALL. Scand J Haematol. 1985;35:343–347. doi: 10.1111/j.1600-0609.1985.tb01717.x. [DOI] [PubMed] [Google Scholar]
- 101.Smits EL, Berneman ZN, Van Tendeloo VF. Immunotherapy of acute myeloid leukemia: current approaches. Oncologist. 2009;14:240–252. doi: 10.1634/theoncologist.2008-0165. [DOI] [PubMed] [Google Scholar]
- 102.Marbello L, Ricci F, Nosari AM, Turrini M, Nador G, Nichelatti M, Tedeschi A, Vismara E, Morra E. Outcome of hyperleukocytic adult acute myeloid leukaemia: a single-center retrospective study and review of literature. Leuk Res. 2008;32:1221–1227. doi: 10.1016/j.leukres.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 103.Faderl S, Kantarjian HM, Talpaz M. Chronic myelogenous leukemia: update on biology and treatment. Oncology (Williston Park) 1999;13:169–180. [PubMed] [Google Scholar]
- 104.Bartram CR, de Klein A, Hagemeijer A, van Agthoven T, Geurts van Kessel A, Bootsma D, Grosveld G, Ferguson-Smith MA, Davies T, Stone M, Heisterkamp N, Stephenson JR, Groffen J. Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1983;306:277–280. doi: 10.1038/306277a0. [DOI] [PubMed] [Google Scholar]
- 105.Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 106.Long B, Zhu H, Zhu C, Liu T, Meng W. Activation of the Hedgehog pathway in chronic myelogeneous leukemia patients. J Exp Clin Cancer Res. 2011;30:8. doi: 10.1186/1756-9966-30-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Padron E, Komrokji R, List AF. The clinical management of chronic myelomonocytic leukemia. Clin Adv Hematol Oncol. 2014;12:172–178. [PubMed] [Google Scholar]
- 108.Emanuel PD. Juvenile myelomonocytic leukemia and chronic myelomonocytic leukemia. Leukemia. 2008;22:1335–1342. doi: 10.1038/leu.2008.162. [DOI] [PubMed] [Google Scholar]
- 109.Pardanani A, Tefferi A. Imatinib targets other than bcr/abl and their clinical relevance in myeloid disorders. Blood. 2004;104:1931–1939. doi: 10.1182/blood-2004-01-0246. [DOI] [PubMed] [Google Scholar]
- 110.Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood. 2009;114:5126–5135. doi: 10.1182/blood-2009-07-216457. [DOI] [PubMed] [Google Scholar]
- 111.Paulson K, Serebrin A, Lambert P, Bergeron J, Everett J, Kew A, Jones D, Mahmud S, Meloche C, Sabloff M, Sharif I, Storring J, Turner D, Seftel MD. Acute promyelocytic leukaemia is characterized by stable incidence and improved survival that is restricted to patients managed in leukaemia referral centres: a pan-Canadian epidemiological study. Br J Haematol. 2014;166(5):660–666. doi: 10.1111/bjh.12931. [DOI] [PubMed] [Google Scholar]
- 112.Rowley JD, Golomb HM, Dougherty C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet. 1977;1:549–550. doi: 10.1016/S0140-6736(77)91415-5. [DOI] [PubMed] [Google Scholar]
- 113.Grignani F, Ferrucci PF, Testa U, Talamo G, Fagioli M, Alcalay M, Mencarelli A, Grignani F, Peschle C, Nicoletti I, Pelicci PG. The acute promyelocytic leukemia-specific PML-RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell. 1993;74:423–431. doi: 10.1016/0092-8674(93)80044-F. [DOI] [PubMed] [Google Scholar]
- 114.Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, Pandolfi PP. PML is essential for multiple apoptotic pathways. Nat Genet. 1998;20:266–272. doi: 10.1038/3030. [DOI] [PubMed] [Google Scholar]
- 115.Quignon F, De BF, Koken M, Feunteun J, Ameisen JC, de Thé H. PML induces a novel caspase-independent death process. Nat Genet. 1998;20:259–265. doi: 10.1038/3068. [DOI] [PubMed] [Google Scholar]
- 116.Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ, International Workshop on Chronic Lymphocytic Leukemia Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, Saul D, Eckart MJ, Mackensen A, Mougiakakos D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood. 2014;123:2663–2672. doi: 10.1182/blood-2013-10-532200. [DOI] [PubMed] [Google Scholar]
- 118.Lo CF, Nervi C, Avvisati G, Mandelli F. Acute promyelocytic leukemia: a curable disease. Leukemia. 1998;12:1866–1880. doi: 10.1038/sj.leu.2401230. [DOI] [PubMed] [Google Scholar]
- 119.Chlapek P, Redova M, Zitterbart K, Hermanova M, Sterba J, Veselska R. Enhancement of ATRA-induced differentiation of neuroblastoma cells with LOX/COX inhibitors: an expression profiling study. J Exp Clin Cancer Res. 2010;29:45. doi: 10.1186/1756-9966-29-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Faderl S, Jeha S, Kantarjian HM. The biology and therapy of adult acute lymphoblastic leukemia. Cancer. 2003;98:1337–1354. doi: 10.1002/cncr.11664. [DOI] [PubMed] [Google Scholar]
- 121.Espérou H, Boiron JM, Cayuela JM, Blanchet O, Kuentz M, Jouet JP, Milpied N, Cahn JY, Faucher C, Bourhis JH, Michallet M, Tanguy ML, Vernant JP, Gabert J, Bordigoni P, Ifrah N, Baruchel A, Dombret H, French Bone Marrow Transplantation A potential graft-versus-leukemia effect after allogeneic hematopoietic stem cell transplantation for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: results from the French Bone Marrow Transplantation Society. Bone Marrow Transplant. 2003;31:909–918. doi: 10.1038/sj.bmt.1703951. [DOI] [PubMed] [Google Scholar]
- 122.Hu Y, Gu X, Li R, Luo Q, Xu Y. Glycogen synthase kinase-3beta inhibition induces nuclear factor-kappaB-mediated apoptosis in pediatric acute lymphocyte leukemia cells. J Exp Clin Cancer Res. 2010;29:154. doi: 10.1186/1756-9966-29-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bassan R, Rossi G, Pogliani EM, Di Bona E, Angelucci E, Cavattoni I, Lambertenghi-Deliliers G, Mannelli F, Levis A, Ciceri F, Mattei D, Borlenghi E, Terruzzi E, Borghero C, Romani C, Spinelli O, Tosi M, Oldani E, Intermesoli T, Rambaldi A. Chemotherapy-phased imatinib pulses improve long-term outcome of adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: Northern Italy Leukemia Group protocol 09/00. J Clin Oncol. 2010;28:3644–3652. doi: 10.1200/JCO.2010.28.1287. [DOI] [PubMed] [Google Scholar]
- 124.Chavez JC, Sahakian E, Pinilla-Ibarz J. Ibrutinib: an evidence-based review of its potential in the treatment of advanced chronic lymphocytic leukemia. Core Evid. 2013;8:37–45. doi: 10.2147/CE.S34068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 126.Gribben JG. How I treat CLL up front. Blood. 2010;115:187–197. doi: 10.1182/blood-2009-08-207126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hamblin AD, Hamblin TJ. The immunodeficiency of chronic lymphocytic leukaemia. Br Med Bull. 2008;87:49–62. doi: 10.1093/bmb/ldn034. [DOI] [PubMed] [Google Scholar]
- 128.Hjalmar V, Kimby E, Matutes E, Sundstrom C, Wallvik J, Hast R. Atypical lymphocytes in B-cell chronic lymphocytic leukemia and trisomy 12 studied by conventional staining combined with fluorescence in situ hybridization. Leuk Lymphoma. 2000;37:571–576. doi: 10.3109/10428190009058509. [DOI] [PubMed] [Google Scholar]
- 129.Parmar S, Patel K, Pinilla-Ibarz J. Ibrutinib (imbruvica): a novel targeted therapy for chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2014;39:483–519. [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang X, Machii T, Matsumura I, Ezoe S, Kawasaki A, Tanaka H, Ueda S, Sugahara H, Shibayama H, Mizuki M, Kanakura Y. Constitutively activated Rho guanosine triphosphatases regulate the growth and morphology of hairy cell leukemia cells. Int J Hematol. 2003;77:263–273. doi: 10.1007/BF02983784. [DOI] [PubMed] [Google Scholar]
- 131.Wrede JE, Sundram U, Kohler S, Cherry AM, Arber DA, George TI. Fluorescence in situ hybridization investigation of cutaneous lesions in acute promyelocytic leukemia. Mod Pathol. 2005;18:1569–1576. doi: 10.1038/modpathol.3800465. [DOI] [PubMed] [Google Scholar]
- 132.Moriya K, Nakagawa K, Santa T, Shintani Y, Fujie H, Miyoshi H, Tsutsumi T, Miyazawa T, Ishibashi K, Horie T, Imai K, Todoroki T, Kimura S, Koike K. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001;61:4365–4370. [PubMed] [Google Scholar]
- 133.Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313(Pt 1):17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cerutti PA, Trump BF. Inflammation and oxidative stress in carcinogenesis. Cancer Cells. 1991;3:1–7. [PubMed] [Google Scholar]
- 135.Dreher D, Junod AF. Role of oxygen free radicals in cancer development. Eur J Cancer. 1996;32A:30–38. doi: 10.1016/0959-8049(95)00531-5. [DOI] [PubMed] [Google Scholar]
- 136.Cerutti PA. Oxy-radicals and cancer. Lancet. 1994;344:862–863. doi: 10.1016/S0140-6736(94)92832-0. [DOI] [PubMed] [Google Scholar]
- 137.Adler V, Yin Z, Tew KD, Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18:6104–6111. doi: 10.1038/sj.onc.1203128. [DOI] [PubMed] [Google Scholar]
- 138.Oltra AM, Carbonell F, Tormos C, Iradi A, Saez GT. Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic Biol Med. 2001;30:1286–1292. doi: 10.1016/S0891-5849(01)00521-4. [DOI] [PubMed] [Google Scholar]
- 139.Nishiura T, Suzuki K, Kawaguchi T, Nakao H, Kawamura N, Taniguchi M, Kanayama Y, Yonezawa T, Iizuka S, Taniguchi N. Elevated serum manganese superoxide dismutase in acute leukemias. Cancer Lett. 1992;62:211–215. doi: 10.1016/0304-3835(92)90098-G. [DOI] [PubMed] [Google Scholar]
- 140.Silber R, Farber CM, Papadopoulos E, Nevrla D, Liebes L, Bruck M, Brown R, Canellakis ZN. Glutathione depletion in chronic lymphocytic leukemia B lymphocytes. Blood. 1992;80:2038–2043. [PubMed] [Google Scholar]
- 141.Flora SJ. Structural, chemical and biological aspects of antioxidants for strategies against metal and metalloid exposure. Oxid Med Cell Longev. 2009;2:191–206. doi: 10.4161/oxim.2.4.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 143.Moore N, Lyle S. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J Oncol. 2011;2011:Article ID: 396076. doi: 10.1155/2011/396076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Augustin W, Wiswedel I, Noack H, Reinheckel T, Reichelt O. Role of endogenous and exogenous antioxidants in the defence against functional damage and lipid peroxidation in rat liver mitochondria. Mol Cell Biochem. 1997;174:199–205. doi: 10.1023/A:1006804423627. [DOI] [PubMed] [Google Scholar]
- 145.Guachalla LM, Rudolph KL. ROS induced DNA damage and checkpoint responses: influences on aging? Cell Cycle. 2010;9:4058–4060. doi: 10.4161/cc.9.20.13577. [DOI] [PubMed] [Google Scholar]
- 146.Block KI, Koch AC, Mead MN, Tothy PK, Newman RA, Gyllenhaal C. Impact of antioxidant supplementation on chemotherapeutic toxicity: a systematic review of the evidence from randomized controlled trials. Int J Cancer. 2008;123:1227–1239. doi: 10.1002/ijc.23754. [DOI] [PubMed] [Google Scholar]
- 147.Hole PS, Darley RL, Tonks A. Do reactive oxygen species play a role in myeloid leukemias? Blood. 2011;117:5816–5826. doi: 10.1182/blood-2011-01-326025. [DOI] [PubMed] [Google Scholar]
- 148.Schuurman AG, Goldbohm RA, Brants HA, van den Brandt PA. A prospective cohort study on intake of retinol, vitamins C and E, and carotenoids and prostate cancer risk (Netherlands) Cancer Causes Control. 2002;13:573–582. doi: 10.1023/A:1016332208339. [DOI] [PubMed] [Google Scholar]
- 149.Fleischauer AT, Olson SH, Mignone L, Simonsen N, Caputo TA, Harlap S. Dietary antioxidants, supplements, and risk of epithelial ovarian cancer. Nutr Cancer. 2001;40:92–98. doi: 10.1207/S15327914NC402_3. [DOI] [PubMed] [Google Scholar]
- 150.Rodrigues MS, Reddy MM, Sattler M. Cell cycle regulation by oncogenic tyrosine kinases in myeloid neoplasias: from molecular redox mechanisms to health implications. Antioxid Redox Signal. 2008;10:1813–1848. doi: 10.1089/ars.2008.2071. [DOI] [PubMed] [Google Scholar]
- 151.Yu C, Rahmani M, Dent P, Grant S. The hierarchical relationship between MAPK signaling and ROS generation in human leukemia cells undergoing apoptosis in response to the proteasome inhibitor Bortezomib. Exp Cell Res. 2004;295:555–566. doi: 10.1016/j.yexcr.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 152.Laurent G, Jaffrezou JP. Signaling pathways activated by daunorubicin. Blood. 2001;98:913–924. doi: 10.1182/blood.V98.4.913. [DOI] [PubMed] [Google Scholar]
- 153.Yedjou CG, Tchounwou PB. In vitro assessment of oxidative stress and apoptotic mechanisms of garlic extract in the treatment of acute promyelocytic leukemia. J Cancer Sci Ther. 2012;2012:6. doi: 10.4172/1948-5956.S3-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Townsend DM, Findlay VL, Tew KD. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005;401:287–307. doi: 10.1016/S0076-6879(05)01019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Miyazaki T, Kirino Y, Takeno M, Samukawa S, Hama M, Tanaka M, Yamaji S, Ueda A, Tomita N, Fujita H, Ishigatsubo Y. Expression of heme oxygenase-1 in human leukemic cells and its regulation by transcriptional repressor Bach1. Cancer Sci. 2010;101:1409–1416. doi: 10.1111/j.1349-7006.2010.01550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Trachootham D, Zhang H, Zhang W, Feng L, Du M, Zhou Y, Chen Z, Pelicano H, Plunkett W, Wierda WG, Keating MJ, Huang P. Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood. 2008;112:1912–1922. doi: 10.1182/blood-2008-04-149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhang H, Trachootham D, Lu W, Carew J, Giles FJ, Keating MJ, Arlinghaus RB, Huang P. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia. 2008;22:1191–1199. doi: 10.1038/leu.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chandra J, Tracy J, Loegering D, Flatten K, Verstovsek S, Beran M, Gorre M, Estrov Z, Donato N, Talpaz M, Sawyers C, Bhalla K, Karp J, Sausville E, Kaufmann SH. Adaphostin-induced oxidative stress overcomes BCR/ABL mutation-dependent and -independent imatinib resistance. Blood. 2006;107:2501–2506. doi: 10.1182/blood-2005-07-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, Keating MJ, Huang P. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem. 2003;278:37832–37839. doi: 10.1074/jbc.M301546200. [DOI] [PubMed] [Google Scholar]
- 161.Wang J, Li L, Cang H, Shi G, Yi J. NADPH oxidase-derived reactive oxygen species are responsible for the high susceptibility to arsenic cytotoxicity in acute promyelocytic leukemia cells. Leuk Res. 2008;32:429–436. doi: 10.1016/j.leukres.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 162.Jeanne M, Lallemand-Breitenbach V, Ferhi O, Koken M, Le BM, Duffort S, Peres L, Berthier C, Soilihi H, Raught B, de Thé H. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 2010;18:88–98. doi: 10.1016/j.ccr.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 163.Doudican NA, Bowling B, Orlow SJ. Enhancement of arsenic trioxide cytotoxicity by dietary isothiocyanates in human leukemic cells via a reactive oxygen species-dependent mechanism. Leuk Res. 2010;34:229–234. doi: 10.1016/j.leukres.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yedjou C, Tchounwou P, Jenkins J, McMurray R. Basic mechanisms of arsenic trioxide (ATO)-induced apoptosis in human leukemia (HL-60) cells. J Hematol Oncol. 2010;3:28. doi: 10.1186/1756-8722-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rogers CS, Yedjou CG, Sutton DJ, Tchounwou PB. Vitamin D3 potentiates the antitumorigenic effects of arsenic trioxide in human leukemia (HL-60) cells. Exp Hematol Oncol. 2014;3:9. doi: 10.1186/2162-3619-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yedjou C, Thuisseu L, Tchounwou C, Gomes M, Howard C, Tchounwou P. Ascorbic acid potentiation of arsenic trioxide anticancer activity against acute promyelocytic leukemia. Arch Drug Inf. 2009;2:59–65. doi: 10.1111/j.1753-5174.2009.00022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yedjou CG, Rogers C, Brown E, Tchounwou PB. Differential effect of ascorbic acid and n-acetyl-L-cysteine on arsenic trioxide-mediated oxidative stress in human leukemia (HL-60) cells. J Biochem Mol Toxicol. 2008;22:85–92. doi: 10.1002/jbt.20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mochizuki T, Furuta S, Mitsushita J, Shang WH, Ito M, Yokoo Y, Yamaura M, Ishizone S, Nakayama J, Konagai A, Hirose K, Kiyosawa K, Kamata T. Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal-regulating kinase 1 pathway in pancreatic cancer PANC-1 cells. Oncogene. 2006;25:3699–3707. doi: 10.1038/sj.onc.1209406. [DOI] [PubMed] [Google Scholar]
- 169.Conklin KA. Cancer chemotherapy and antioxidants. J Nutr. 2004;134:3201S–3204S. doi: 10.1093/jn/134.11.3201S. [DOI] [PubMed] [Google Scholar]
- 170.Meyskens FL, Jr, Kopecky KJ, Appelbaum FR, Balcerzak SP, Samlowski W, Hynes H. Effects of vitamin A on survival in patients with chronic myelogenous leukemia: a SWOG randomized trial. Leuk Res. 1995;19:605–612. doi: 10.1016/0145-2126(95)00032-J. [DOI] [PubMed] [Google Scholar]