

Abstract
We report a synthetic biology strategy for rapid genetic manipulation of natural product biosynthetic pathways. Based on DNA assembler, this method synthesizes the entire expression vector containing the target biosynthetic pathway and the genetic elements required for DNA maintenance and replication in various hosts in a single-step manner through yeast homologous recombination, offering unprecedented flexibility and versatility in pathway manipulations.
Microorganisms and plants have evolved to produce a myriad array of complex molecules known as natural products or secondary metabolites that are of biomedical and biotechnological importance.1-3 For example, it has been estimated that 77% of antibacterial drugs and 78% of anticancer drugs are natural products or have been derived from natural products.4 While many effective drugs have been discovered in the past decades, a pressing need for the development of new therapeutic agents remains. This is mainly due to the rapid development of antibiotic resistance to existing drugs, the recent threat of bioterrorism, and the ever-increasing demand for new therapeutic agents for treatment of cancers and infectious diseases, to name a few.
Sequenced genomes and metagenomes represent a tremendously rich source for discovery of novel pathways involved in natural product biosynthesis.5, 6 Over the last decade, the complete genome sequences of more than 1500 organisms have been determined, with more than 7000 organisms in the pipeline (http://www.genomesonline.org/cgi-bin/GOLD/bin/gold.cgi). However, only a tiny fraction of these biosynthetic pathways have been characterized, and discovery and sustainable production of natural products are often hampered by our limited ability to manipulate the corresponding biosynthetic pathways.
Existing strategies for functional analysis of biosynthetic pathways can be broadly classified into two groups including the native host gene inactivation method and the heterologous host gene expression method.6 Compared to the native host-based method, the heterologous host-based method offers several advantages. First, most of native producers are not commonly cultivable in the laboratory and many microorganisms grow very slowly and produce minute amount of target compounds. It has been estimated that only 1% of bacteria and 5% of fungi have been cultivated in the laboratory.7-9 Second, maximizing expression of a biosynthetic pathway in a well-characterized organism is usually easier and comparison of metabolic profiles is often less difficult. Third, the genetic tools available for a well-characterized organism enables the overproduction of a target natural product through metabolic engineering and the generation of new derivatives of a target compound through combinatorial biosynthesis. The heterologous host-based method has been successfully demonstrated in discovery and characterization of many natural products, such as methylenomycin furans,10 phosphonic acids,11, 12 and polyketides.13, 14 However, this method has several limitations. Major drawbacks include but are not limited to, the difficult transfer and genetic manipulation of large biosynthetic pathways such as performing scar-less targeted gene disruption and introducing single/multiple point mutations, problems with promoter recognition and gene expression, limited availability of expression hosts, and low yield.15 Therefore, it is highly desirable to establish novel efficient modification, transfer, and expression technologies.
Due to its high efficiency and ease to work with, in vivo homologous recombination in Saccharomyces cerevisiae has been widely used for gene cloning and library creation.16-18 Larionov and coworkers developed a transformation-associated recombination (TAR) cloning method in S. cerevisiae and used it to isolate large chromosomal fragments or entire gene clusters from complex genomes.19-22 We developed the DNA assembler approach to assemble multiple gene expression cassettes into biochemical pathways either on a plasmid or on a chromosome in S. cerevisiae in a single-step fashion.23 The recent success of constructing the entire Mycoplasma genitalium and Mycoplasma mycoides genomes further demonstrated the efficacy of using S. cerevisiae as a robust factory to assemble DNA molecules.24, 25
Here we extended the DNA assembler approach, enabling it as a genomics-driven, synthetic biology-based method, for discovery, characterization, and engineering of natural product biosynthetic pathways. The strategy is illustrated in Scheme 1. Briefly, pathway fragments encoding the target biosynthetic pathway and helper fragments carrying the genetic elements needed for DNA maintenance and replication in S. cerevisiae, E. coli, and the target heterologous expression host are amplified from the genome of the native producer and the corresponding vectors, respectively, and subsequently co-transformed into S. cerevisiae. Since polymerase chain reaction (PCR) primers are designed to generate an overlap region between two adjacent fragments, these fragments will be assembled into a single DNA molecule in S. cerevisiae through homologous recombination. The isolated plasmids are transformed into E. coli for plasmid enrichment and verification, and the correct construct is transformed into the desired host for heterologous expression of the target biosynthetic pathway. Of special note, because the pathway fragments can be readily modified by PCR, various genetic manipulations such as site-directed mutagenesis and scar-less gene deletion and insertion can be easily introduced into the target pathway.
Scheme 1.

The DNA assembler based strategy for efficient manipulation of natural product biosynthetic pathways. Various genetic modifications are introduced in the pathway fragments to be assembled.
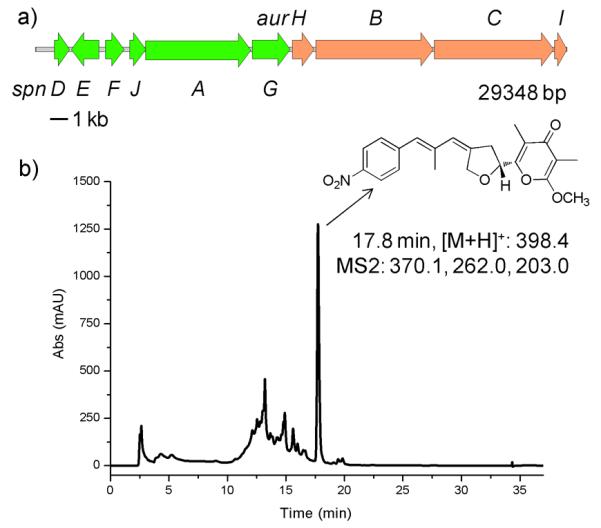
We sought to study two related biosynthetic pathways, the aureothin26, 27 (Fig. 1a) and the spectinabilin28 (Fig. 2a) biosynthetic pathways. Both pathways contain multiple type I polyketide synthases (PKSs) that are composed of domains including β-ketosynthase (KS), acyl transferase (AT), acyl carrier protein (ACP), ketoreductase (KR), dehydratase (DH), and enoylreductase (ER) domains. Because of the importance of polyketides in medicine and the modular, programmable nature of the biosynthetic machinery, PKSs have been extensively investigated and engineered to produce new derivatives.29 Aureothin is a rare nitroaryl-substituted polyketide (Fig. 1b, compound 1) from Streptomyces thioluteus, which exhibits antitumor, antifungal, and insecticidal activities.26, 27 Seven pathway fragments of 4-5 kb, were PCR-amplified from the genomic DNA, and co-transformed with the three helper fragments into S. cerevisiae, leading to their assembly into a single circular DNA molecule of 35.7 kb (29.1 kb from the aureothin biosynthetic pathway and 6.6 kb from the helper fragments). The details of designing the overlaps between adjacent fragments were described in Figure S1 in the Supplementary Information. Six out of the ten randomly picked constructs were shown to be correctly assembled, as verified by restriction digestion analysis, indicating a high assembly efficiency of 60%. Three of the correct constructs were subsequently conjugated to a heterologous host, Streptomyces lividans. Note that a Streptomyces phage-derived φC31 integrase gene, a φC31 recognition site, and an origin of conjugal transfer (Figure S2 in the Supplementary Information) were included in the Streptomyces helper fragment so that the target biosynthetic pathway would be transferred and integrated to the chromosome of S. lividans. As a result, all three S. lividans clones produced aureothin with a yield of 6.1 ± 0.2 mg/L (Fig. 1b and Figure S3 in the Supplementary Information), indicating that such a cluster of 29 kb has been successfully assembled and heterologously expressed.
Fig. 1.

Assembly of the wild type aureothin biosynthetic pathway and the mutant aureothin biosynthetic pathways for creation of new derivative. a) Scheme of the aureothin biosynthetic pathway. b) LC-MS analysis of the S. lividans clones carrying the wild type aureothin biosynthetic pathway and the mutant aureothin biosynthetic pathways.
Fig. 2.

Gene knock-out and inactivation studies on the spectinabilin biosynthetic pathways. a) Scheme of the spectinabilin biosynthetic pathway. b) LC-MS analysis of the S. lividans clones carrying the wild type spectinabilin biosynthetic pathway and the mutant spectinabilin biosynthetic pathways. The asterisks indicate the spectinabilin peaks. MutK, the mutant with a stop codon introduced to SpnK; ΔM, the mutant with SpnM deleted; ΔM+ΔL, the mutant with SpnM and SpnL deleted.
Based on this system, site-directed mutagenesis can now be readily used to generate new derivatives. Unlike the labor-intensive and time-consuming procedures used in the conventional methods,30-32 DNA assembler only requires adding site-specific mutation(s) into the PCR primers used to generate pathway fragments. As an example, the active sites of the DH domain of AurB were targeted. The motifs HXXXGXXXXP and DXXX(Q/H) were found to be conserved among the DH domains of PKSs and the histidine and the aspartic acid were identified as the catalytic residues33-35. Based on this information, AurB DH H964A mutant and AurB DH H964A/D1131A double mutant were generated. We expected them to produce compounds 2 (Fig. 1b) if the corresponding domain was completely inactivated, or a mixture of both compound 2 and aureothin if the activity was only reduced. The mutation led to significant changes in HPLC analysis in which several new peaks appeared and the aureothin peak was drastically decreased (Fig. 1b). The distinguishable peak at 15.8 min was confirmed to be compound 2 by its MS and MS2 patterns (Figure S4 in the Supplementary Information). Interestingly, it was found that the wild type pathway could also produce a minute amount of compound 2 due to the incomplete modification catalyzed by the AurB DH domain on the upstream intermediate. In order to demonstrate that the derivative was produced mainly due to the corresponding mutation(s), the relative yield, defined as the ratio between the level of compound 2 being produced and the level of the generated aureothin, was determined. The wild type pathway produced compound 2 at a relative yield of 0.023:1, while the AurB DH H964A mutant and the AurB DH H964A/D1131A mutant gave a relative yield of 1.03:1 and 26.3:1, respectively (Fig. 1b). From the evolutionary point of view, the wild type pathway should be most efficient for the synthesis of the product because almost no accumulation of intermediates was observed, whereas the modified pathways could accumulate some intermediates if the downstream enzymes cannot take those intermediates with modified structures as perfect substrates and only process them at a slower rate. On the other hand, the residual production of aureothin suggests that the corresponding mutation(s) either did not completely inactivate the enzymes, or completely inactivated the enzymes but another unknown enzyme with dehydratase activity played a role in the dehydration step. In addition, the wild type pathway can naturally synthesize a tiny amount of the aureothin analogue. This could take place when a small amount of an intermediate skips a certain enzymatic reaction and at the same time is processed by the downstream enzymes. Overall, our results have demonstrated that single/multiple site-directed mutagenesis can be easily performed without going through the complicated multi-step procedures used by other conventional approaches.30-32
Compared to aureothin, spectinabilin is a longer nitro-phenyl containing polyketide, which exhibits antimalarial and antiviral activities.36, 37 The biosynthetic pathway from Streptomyces spectabilis consists of 4 PKSs (Fig. 2a)28 and shares a very high sequence homology with the spectinabilin biosynthetic pathway from Streptomyces orinoci27 (Figure S5 in the Supplementary Information). However, only the biosynthetic pathway from S. spectabilis was successfully expressed in S. lividans,28 while the biosynthetic pathway from S. orinoci failed to produce any spectinabilin in the heterologous host.27, 38 Comparison of the two biosynthetic pathways revealed one major difference: the S. spectabilis cluster contains three extra genes, spnK, spnL, and spnM (Figure S5 in the Supplementary Information). To investigate their role in spectinabilin biosynthesis, we used DNA assembler to inactivate spnK by site-directed mutagenesis and completely delete spnM and spnM+spnL. Initially, we attempted to assemble the spectinabilin biosynthetic pathway using the same procedure as we did for the aureothin biosynthetic pathways. However, a low efficiency (<10%) was observed and most of the constructs underwent severe gene deletion(s), possibly due to the high sequence identity among PKS domains. To address this issue, a modified two-step assembly strategy was devised (Figure S6 in the Supplementary Information), through which we were able to assemble this biosynthetic pathway of 45 kb with an efficiency of 30%. To inactivate spnK, the codon TGC encoding Cys199 was changed to a stop codon TGA in the PCR primers. The genes spnM and spnM+spnL were individually removed from the biosynthetic pathway by redesigning the reverse primer for amplifying the last pathway fragment. All three mutants produced spectinabilin (Fig. 2b and Figure S7 in the Supplementary Information), although their yields were affected by the correponding mutations (Wild type, 31.8 ± 4.5 μg/L; MutK, 17.2 ± 0.5 μg/L; DelM, 21.2 ± 2.1 μg/L; DelML, 10.5 ± 1.6 μg/L), indicating that spnK, spnL, and spnM are not required for spectinabilin biosynthesis, supporting our previous bioinformatics study showing that they are only involved in up-regulating the substrate concentration and product transportation.28
To further demonstrate the versatility of our approach in manipulating large gene clusters, we created a hybrid biosynthetic pathway between the aureothin and the specitinabilin pathways. Because the aureothin pathway lacks the counterpart of the spnA’ gene in the spectinabilin biosynthetic pathway, we removed spnA’ and created a hybrid pathway comprising spnDEFJAG and aurHBCI (Fig. 3a). The analysis of the N-terminal sequences revealed that SpnA’ and AurB share a very high homology at their N-terminals while SpnA’ and SpnB are very different at this end (Figure S8 in the Supplementary Information), indicating SpnA could possibly recognize AurB rather than SpnB. As expected, the resulting construct produced aureothin in the heterologous host with a yield of 0.17 mg/L (Fig. 3b). The generation of aureothin by a hybrid pathway was achieved previously, but a much more complicated experimental protocol has to be used.27
Fig. 3.
Assembly of a hybrid biosynthetic pathway between the spectinabilin biosynthetic pathway and the aureothin biosynthetic pathway. a) Scheme of the hybrid biosynthetic pathway. b) LC-MS analysis of the S. lividans clone carrying the hybrid biosynthetic pathway.
Conclusions
In summary, we have demonstrated that DNA assembler enables sophisticated genetic manipulations such as point mutagenesis and scar-less gene substitution and deletion, which can be used to confirm the gene function, locate key amino acid residues, study the biosynthetic mechanism, express biosynthetic pathways heterologously, and generate new derivatives. Moreover, with little modification, DNA assembler may be used for many other applications such as promoter/regulator replacement, cryptic pathway activation, artificial operon construction, rapid library creation and pathway optimization. All these sophisticated genetic manipulations are difficult to accomplish by other conventional methods. Overall, because the DNA fragments to be assembled are completely mobile and amenable to all sorts of sophisticated genetic manipulations accessible to PCR or can be chemically synthesized de novo with optimized codons, this DNA assembler based strategy offers the ultimate versatility and flexibility in characterizing and engineering natural product biosynthetic pathways.
Supplementary Material
Acknowledgments
We thank Frances H. Arnold for critical reading of this manuscript. This work was supported by the National Academies Keck Futures Initiative on Synthetic Biology (NAKFI SB13) and the National Institutes of Health (GM077596).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental, supplementary figures and tables. See DOI: 10.1039/b000000x/
references
- 1.Dewick PM. Medical natural products. A biosynthetic approach. 2nd ed. John Wiley and Sons; Chichester, UK: 2002. [Google Scholar]
- 2.Herbert RB. The biosynthesis of secondary metabolites. 2nd ed. Chapman and Hall; London, UK: 1989. [Google Scholar]
- 3.Li JWH, Vederas JC. Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 4.Newman DJ, Cragg GM. J. Nat. Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 5.Challis GL. Microbiol-Sgm. 2008;154:1555–1569. doi: 10.1099/mic.0.2008/018523-0. [DOI] [PubMed] [Google Scholar]
- 6.Zerikly M, Challis GL. Chembiochem. 2009;10:625–633. doi: 10.1002/cbic.200800389. [DOI] [PubMed] [Google Scholar]
- 7.Bull AT, Goodfellow M, Slater JH. Annu Rev Microbiol. 1992;46:219–252. doi: 10.1146/annurev.mi.46.100192.001251. [DOI] [PubMed] [Google Scholar]
- 8.Demain AL. J. Ind. Microbiol. Biotechnol. 2006;33:486–495. doi: 10.1007/s10295-005-0076-x. [DOI] [PubMed] [Google Scholar]
- 9.Leadbetter JR. Curr. Opin. Microbiol. 2003;6:274–281. doi: 10.1016/s1369-5274(03)00041-9. [DOI] [PubMed] [Google Scholar]
- 10.Corre C, Song L, O’Rourke S, Chater KF, Challis GL. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17510–17515. doi: 10.1073/pnas.0805530105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woodyer RD, Shao Z, Thomas PM, Kelleher NL, Blodgett JA, Metcalf WW, van der Donk WA, Zhao H. Chem. Biol. 2006;13:1171–1182. doi: 10.1016/j.chembiol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 12.Eliot AC, Griffin BM, Thomas PM, Johannes TW, Kelleher NL, Zhao H, Metcalf WW. Chem. Biol. 2008;15:765–770. doi: 10.1016/j.chembiol.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C, Roege KE, Kelly WL. Chembiochem. 2009;10:1064–1072. doi: 10.1002/cbic.200800822. [DOI] [PubMed] [Google Scholar]
- 14.Wenzel SC, Bode HB, Kochems I, Muller R. Chembiochem. 2008;9:2711–2721. doi: 10.1002/cbic.200800456. [DOI] [PubMed] [Google Scholar]
- 15.Gross H. Appl. Microbiol. Biotechnol. 2007;75:267–277. doi: 10.1007/s00253-007-0900-5. [DOI] [PubMed] [Google Scholar]
- 16.Gunyuzlu PL, Hollis GF, Toyn JH. Biotechniques. 2001;31:1246–1248. doi: 10.2144/01316bm03. [DOI] [PubMed] [Google Scholar]
- 17.Raymond CK, Pownder TA, Sexson SL. Biotechniques. 1999;26:134–141. doi: 10.2144/99261rr02. [DOI] [PubMed] [Google Scholar]
- 18.Schaerer-Brodbeck C, Barberis A. Biotechniques. 2004;37:202–206. doi: 10.2144/04372BM05. [DOI] [PubMed] [Google Scholar]
- 19.Cocchia M, Kouprina N, Kim SJ, Larionov V, Schlessinger D, Nagaraja R. Nucleic Acids Res. 2000;28:E81. doi: 10.1093/nar/28.17.e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noskov V, Kouprina N, Leem SH, Koriabine M, Barrett JC, Larionov V. Nucleic Acids Res. 2002;30:E8. doi: 10.1093/nar/30.2.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noskov VN, Kouprina N, Leem SH, Ouspenski I, Barrett JC, Larionov V. BMC Genomics. 2003;4:16. doi: 10.1186/1471-2164-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng Z, Kim JH, Brady SF. J. Am. Chem. Soc. 2010;132:11902–11903. doi: 10.1021/ja104550p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shao Z, Zhao H, Zhao H. Nucleic Acids Res. 2009;37:e16. doi: 10.1093/nar/gkn991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibson DG, Benders GA, Axelrod KC, Zaveri J, Algire MA, Moodie M, Montague MG, Venter JC, Smith HO, Hutchison CA., 3rd Proc. Natl. Acad. Sci. U. S. A. 2008;105:20404–20409. doi: 10.1073/pnas.0811011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibson DG, Glass JI, Lartigue C, Noskov VN, Chuang RY, Algire MA, Benders GA, Montague MG, Ma L, Moodie MM, Merryman C, Vashee S, Krishnakumar R, Assad-Garcia N, Andrews-Pfannkoch C, Denisova EA, Young L, Qi ZQ, Segall-Shapiro TH, Calvey CH, Parmar PP, Hutchison CA, 3rd, Smith HO, Venter JC. Science. 2010;329:52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- 26.He J, Hertweck C. Chem. Biol. 2003;10:1225–1232. doi: 10.1016/j.chembiol.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 27.Traitcheva N, Jenke-Kodama H, He J, Dittmann E, Hertweck C. Chembiochem. 2007;8:1841–1849. doi: 10.1002/cbic.200700309. [DOI] [PubMed] [Google Scholar]
- 28.Choi YS, Johannes TW, Simurdiak M, Shao Z, Lu H, Zhao H. Mol. Biosyst. 6:336–338. doi: 10.1039/b923177c. [DOI] [PubMed] [Google Scholar]
- 29.Staunton J, Weissman KJ. Nat. Prod. Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 30.Ito T, Roongsawang N, Shirasaka N, Lu W, Flatt PM, Kasanah N, Miranda C, Mahmud T. Chembiochem. 2009;10:2253–2265. doi: 10.1002/cbic.200900339. [DOI] [PubMed] [Google Scholar]
- 31.Karray F, Darbon E, Nguyen HC, Gagnat J, Pernodet JL. J. Bacteriol. 192:5813–5821. doi: 10.1128/JB.00712-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blodgett JA, Thomas PM, Li G, Velasquez JE, van der Donk WA, Kelleher NL, Metcalf WW. Nat. Chem. Biol. 2007;3:480–485. doi: 10.1038/nchembio.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keatinge-Clay A. J Mol Biol. 2008;384:941–953. doi: 10.1016/j.jmb.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moriguchi T, Kezuka Y, Nonaka T, Ebizuka Y, Fujii I. J Biol Chem. 285:15637–15643. doi: 10.1074/jbc.M110.107391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pawlik K, Kotowska M, Chater KF, Kuczek K, Takano E. Arch. Microbiol. 2007;187:87–99. doi: 10.1007/s00203-006-0176-7. [DOI] [PubMed] [Google Scholar]
- 36.Kakinuma K, Hanson CA, Rinehart KL., Jr Tetrahedron. 1976;32:217–222. [Google Scholar]
- 37.Isaka M, Jaturapat A, Kramyu J, Tanticharoen M, Thebtaranonth Y. Antimicrob. Agents Chemother. 2002;46:1112–1113. doi: 10.1128/AAC.46.4.1112-1113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Traitcheva N. Leibniz-Institute for Natural Product Research and Infection Biology (HKI) 2006. Ph.D. Thesis. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.