

Abstract
The diverse immunomodulatory properties of mesenchymal stem/stromal cells (MSCs) may be exploited for treatment of a multitude of inflammatory conditions. MSCs have long been reported to be hypoimmunogenic or ‘immune privileged’; this property is thought to enable MSC transplantation across major histocompatibility barriers and the creation of off-the-shelf therapies consisting of MSCs grown in culture. However, recent studies describing generation of antibodies against and immune rejection of allogeneic donor MSCs suggest that MSCs may not actually be immune privileged. Nevertheless, whether rejection of donor MSCs influences the efficacy of allogeneic MSC therapies is not known, and no definitive clinical advantage of autologous MSCs over allogeneic MSCs has been demonstrated to date. Although MSCs may exert therapeutic function through a brief ‘hit and run’ mechanism, protecting MSCs from immune detection and prolonging their persistence in vivo may improve clinical outcomes and prevent patient sensitization toward donor antigens.
MSCs were originally identified by Friedenstein in mouse bone marrow and were characterized according to their multilineage potential1–3. Caplan later referred to these cells as mesenchymal stem cells4, yet to date rigorous in vivo demonstration of their stem cell properties has not been established. As a result of their original identification in the bone marrow, many referred to them as “bone marrow stromal cells.” However, MSCs have since been shown to be derived from both pericytes and adventitial progenitor cells from nearly all tissues5,6. Thus it may be appropriate to refer to MSCs as “multipotent perivascular-derived cells.” Regardless, the issue of MSC nomenclature remains contentious. As of December 17, 2013, there were 18,284 references in PubMed to “mesenchymal stem cell” or “mesenchymal stem cells,” 14,586 to “mesenchymal stromal cell” or “mesenchymal stromal cells,” 4,254 to “bone marrow stromal cell” or “bone marrow stromal cells,” and 183 to “multipotent stromal cell” or “multipotent stromal cells.”
Irrespective of the nomenclature, it is still unclear if the MSC phenotype exists in vivo. Although lineage-restricted, self-renewing skeletal progenitors (bone, cartilage, stromal progenitor)7 and osteolineage-restricted Mx1+ stromal cell populations8 were recently identified in mice, their relationship to MSCs is not clear. Indeed, assays to identify and characterize MSCs are mostly based upon in vitro work. Although pericytes and MSCs share properties, and it is possible that when pericytes become activated and leave vessels they differentiate into MSCs, this has not been conclusively demonstrated. In 2006 the International Society for Cellular Therapy established minimal criteria for designating a cell an MSC9; these include tri-lineage differentiation potential (osteogenic, adipogenic and chondrogenic), cell-surface expression of CD90, CD105 and CD73, and lack of cell surface CD45, CD34, CD14, CD79 and HLA-DR. However, culture-expanded MSCs consist of a heterogeneous population of cells exhibiting a spectrum of phenotypes and functional properties, and the extent of these properties is dependent on the tissue, donor and species of origin, isolation technique, culturing protocols and media used, and passage number. That said, heterogeneity is not unique to MSCs, as clones of hematopoietic stem cells, for example, can exhibit considerable functional heterogeneity after transplantation10,11.
In addition, the clinical value of MSCs thus far seems primarily derived from their non-stem/progenitor cell properties. Namely, MSCs produce extracellular vesicles, including exosomes, and a multitude of cytokines and growth factors that suppress immune responses by inhibiting B- and T-cell proliferation and monocyte maturation and by promoting generation of regulatory T cells and M2 macrophages12–15. Therefore, although some argue that MSCs should be defined based on in vivo differentiation potential or ability to support hematopoiesis16,17, others advocate for a broader definition that places less emphasis on the ‘stem’ properties of the cell and more on the trophic and immunomodulatory properties that render them potentially useful in treating numerous diseases18–22. As the trophic and immunomodulatory properties of MSCs are largely responsible for the rapid rise in the therapeutic exploration of major histocompatibility (MHC)-unmatched allogeneic MSCs, a broader definition of MSCs that includes these properties is more applicable to this Perspective. It is also important to consider that MSCs can easily be manipulated in culture to obtain phenotypes that more effectively treat one disease over another; these modified cells may still be considered MSCs in the broad sense without necessarily meeting all of the minimal criteria defined by the 2006 definition. Given the general lack of rigorous MSC phenotype assessment in the published literature, adopting a narrower definition of MSCs would preclude us from writing this Perspective. Therefore, here we consider MSC to be cells that are generally defined by the 2006 minimal criteria.
Positive data from preclinical models and elucidation of the immunomodulatory properties of MSCs have prompted a sharp rise in the number of clinical trials that use MSCs to treat diseases including myocardial infarction, stroke, graft versus host disease (GvHD), lupus, arthritis, Crohn’s disease, acute lung injury, chronic obstructive pulmonary disease (COPD), cirrhosis, multiple sclerosis, amyotrophic lateral sclerosis (ALS) and diabetes23. Notably, most patients receive allogeneic MSCs23; in this scenario there is no MHC matching before treatment. The assumption that allogeneic MSC preparations represent a one-size-fits-all, off-the-shelf, cell-based therapy originated in the assumption that MSCs are immune privileged. However, recent data indicate that allogeneic MSCs can provoke an immune response resulting in rejection. In this Perspective we attempt to understand the origin of the immune-privileged hypothesis by revisiting the early discoveries of the immunomodulatory potential of MSCs. We review the results of MSC clinical trials and consider ways in which MSC immune privilege or lack thereof may affect the efficacy of MSC therapy. Lastly, we suggest ways in which insights about MSC immunogenicity may be used to design improved MSC-based therapies.
The rise of allogeneic MSC therapy
From 1998 to 2000, researchers at Osiris Therapeutics presented a series of abstracts at the American Society of Hematology meetings suggesting that interactions between MSCs and hematopoietic cells did more than support hematopoiesis24,25; they proposed that MSCs act as immune regulators26–28. Specifically, human MSCs (hMSCs) suppressed proliferation of activated T cells28,29 and mixed lymphocyte reactions in a genetically unrestricted, allogeneic manner27,30. As even third-party, MHC-mismatched hMSCs were capable of suppressing an ongoing mixed lymphocyte reaction, Klyushnenkova et al. proposed the concept of generating a large supply of culture-expanded allogeneic (allo)-MSCs from a single, universal donor that could then be used to treat all patients27,30.
In 2002, Bartholomew et al. showed that the addition of baboon MSCs to cultures of stimulated allogeneic peripheral blood leukocytes resulted in suppression of leukocyte proliferation in an MSC dose–dependent manner31. This suppression occurred regardless of the MSC donor origin (autogeneic (auto)-, allo- and third-party MSCs). In addition, allo-MSCs inhibited T-cell proliferation induced by the potent mitogens, PHA, Con A and SpA32,33. Interestingly, MSC T-cell suppression was transient, as T cells respond to allo-antigen upon removal from MSC co-cultures34. Moreover, suppression was partially reversed by the addition of interleukin (IL)-2, suggesting MSCs did not induce T-cell anergy31. To test the immunosuppressive potential of MSCs in vivo, skin grafts from MHC-mismatched baboons were performed immediately before intravenous injection of donor-matched or third-party MSCs. Administration of donor or third-party MSCs extended the survival of the skin graft from 7 days (control without MSCs) to 11.3 and 11.8 days, respectively31. In parallel, Liechty et al. reported the finding that human MSCs could persist as long as 13 months after intraperitoneal injection into pre-immune (immune system has not yet developed) and immune-competent fetal sheep; in contrast, xenogenic or allogeneic hematopoietic stem cells (HSCs) are rejected in this model35. Others subsequently reported that donor-derived MSCs promote tolerance to other transplanted tissues, including pancreatic islets36 and heart allografts37; however, specific tolerance toward unmatched MSCs or repeat doses of MSCs has not been convincingly demonstrated. Together, these and other findings indicate that MSC immunomodulatory potential is MHC-unrestricted.
MSC-mediated immune suppression has been shown to depend on a myriad of factors including cell dose, proximity to immune cells, activation of Toll-like receptors and stimulation by inflammatory cytokines38,39. Over a decade ago, MSCs were postulated to act either through cell contact–dependent signaling or through secretion of cytokines and growth factors, leading to a series of mechanistic studies32,33. As culture-expanded hMSCs typically express low levels of MHC class I, and no MHC class II or co-stimulatory molecules (e.g., B7-1, B7-2, or CD40), cell contact may not be the primary mechanism of MSC immunomodulatory action29,32,40. This was verified by studies concluding that MSC-mediated immune suppression was not dependent on cell contact, but was augmented by close proximity to the target cell. Specifically, whereas hMSC maximally suppressed T cells in mixed co-cultures (which enable direct cell-cell contact), suppression was also observed in transwell assays33. Tse et al. examined the effect of soluble factors in a series of inhibitor studies. Inhibition of either prostaglandin E2 (PGE2) or indoleamine-2,3-dioxygenase (IDO) resulted in only a partial reduction of hMSC-mediated suppression29. These and other studies suggest that MSC suppressive potential cannot be pinned to a single factor or mechanism, but rather to a cocktail of soluble factors, many of which are induced in inflammatory environments15,39.
The occurrence of inflammation-responsive MSC-mediated suppression is supported by data showing enhanced T-cell suppression when MSCs are preconditioned by incubation with interferon (IFN)-γ for 48 h (ref. 40) and by data showing a limited suppressive effect in transwell33,34 and conditioned media34 experiments in which MSC exposure to IFN-γ is minimized or avoided. Furthermore, MSCs can be polarized to pro- or anti-inflammatory phenotypes by preconditioning with cytokines, including IFN-γ and TNF-α14,41, and by signaling through Toll-like receptors12,38,42,43. Reviews by Ren et al.44, Ranganath et al.39 and Prockop15 provide a thorough discussion of the MSC secretome and its immunosuppressive potential.
MSC clinical trials
Once the immunomodulatory potential of MSCs was established in vitro and in early preclinical models, MSCs were rapidly brought into the clinic. In 2004, Le Blanc et al. were among the first to clinically administer allo-MSCs45. Third-party haplo-identical MSCs were harvested from the mother of a 9-year-old boy suffering from treatment-resistant, grade IV GvHD. The boy received MSCs 73 and 170 days after bone marrow transplantation, with rapid recovery after each MSC infusion and survival beyond 1 year. In contrast, the 24 patients at the same treatment facility with acute grade IV GvHD who did not receive MSC therapy died an average of 2 months after bone marrow transplantation. This landmark case study provided an early glimpse of MSCs’ therapeutic potential. Just 8 months after the publication of Le Blanc et al.’s Lancet article, Osiris Therapeutics began recruiting patients for the first large-scale clinical trials of allo-MSCs for the treatment of acute GvHD and acute myocardial infarction.
Since then, MSC use in clinical trials soared. Today, numerous MSC cell preparations from academic and corporate institutions are being investigated in nearly 350 clinical trials (>80% of which are phase 1 or 2, Fig. 1a; 131 have reached their scheduled completion, and 37 have been reported to be placebo controlled). Clinical trials examining the safety and efficacy of MSCs have used both allogeneic (190) and auto-logous (150) cells (Fig. 1b). MSCs are typically manipulated by means of culture expansion, as they exist in limited quantities in situ; thus they require clinical trials to gain US Food and Drug Administration approval and have only recently begun to reach the market.
Figure 1.

The rise of MSC therapy. (a) The number of clinical trials in each phase using allogeneic or autologous MSCs. Trials in the registry that did not report a ‘phase’ are listed as “not reported.” (b) The cumulative total number of clinical trials that use allogeneic or autologous MSCs, plotted according to the year they were initiated. (c) Cumulative citations from early publications that support the ‘Universal Donor’ hypothesis (purple) and from work that highlights MSC immunogenicity (green), plotted from 2000–2012. Shades represent contributions of individual papers (references denoted on right). Contributions of papers are stacked upon one another with the most influential papers, as measured by citations, on the bottom and the least influential papers on the top. (d) Timeline of milestones that have marked the progress of MSC therapy. Data in a and b were collected from clinicaltrials.gov registry on December 15, 2013. Searches for “Mesenchymal Stem Cells,” “Mesenchymal Stromal Cells,” “Multipotent stromal cells,” “bone marrow stromal cells,” “Stem cells for Spinal Fusion,” “Prochymal” and “connective tissue progenitor” returned 347 unique MSC trials. Nine trials did not indicate the source (auto vs. allo) of the MSCs, and two reported using both allogeneic and autologous MSCs. Citation data for c was collected from Web of Knowledge (searched December 15, 2013).
Clinical trials exploring MSC therapy have been driven predominately by companies with proprietary allogeneic MSC (allo-MSC) preparations such as Osiris’ (now Mesoblast’s) Prochymal, Mesoblast’s Revascor, Athersys’ MultiStem, Stemedica’s Stemdyne-MSC, Allocure’s AC607, Cellerix’s Cx601, Stempeutic’s Stempeucel and Orthofix’s Trinity Evolution. Many of these preparations derive their product from a small number of donors, and subject cells to extensive culture expansion to generate therapeutic doses to treat entire cohorts of patients. Smaller academic-investigator–driven trials have also been conducted in medical centers and academic institutions, often under hospital exemption in Europe, although these studies typically use MSCs from much earlier passages46. Importantly, allo-MSC therapy has consistently been shown to be safe, enabling future trials to be conducted with improved trial design and using refined MSC-based approaches23,47.
Several recent industry-sponsored phase 2 clinical trials and academic-investigator–driven trials generated preliminary results that have been considered positive by some. It should be noted that industry-sponsored clinical trial data, specifically negative data, are often voluntarily unreported, and much of the data reported here have been gathered from non-peer-reviewed investor reports released from the sponsoring corporations: URL links to these references are included parenthetically. For example, Tigenix’s Cx611 culture-expanded adipose-derived allo-MSCs reduced joint swelling by 20% or more in 1/5 of patients at a 6-month follow-up, whereas placebo-treated patients showed no improvement (http://www.tigenix.com/public/uploads/pdf/en/f517459e904cc86.32694073_TiGenix%20Cx611%20Phase%20IIa%20results%20Final.pdf). Additionally, Mesoblast reported improved heart muscle function and a 78% reduction in major adverse cardiac events compared to placebo in congestive heart failure patients at an 18-month follow-up in a placebo-controlled phase 2 trial (http://www.prnewswire.com/news-releases-test/positive-results-from-phase-2-trial-of-mesoblasts-adult-stem-cell-therapy-presented-at-the-american-heart-association-annual-meeting-133835958.html). In late 2011 Athersys reported 13.5% and 10.9% increases in ejection fraction in acute myocardial infarction patients receiving 50 and 100 million MSCs, respectively, by means of intracoronary adventitial injections compared to historical controls (phase 1 trial)48. Osiris reported reduced stress-induced arrhythmias and hypertrophy in acute myocardial infarction patients receiving Prochymal compared to placebo in an ongoing phase 2 trial, although data on the primary endpoints have yet to be reported (http://investor.osiris.com/releasedetail.cfm?releaseid=688302).
In 2011, FBC-Pharmicell’s autologous MSC preparation, Hearticellgram-AMI, gained approval in South Korea, becoming the first culture-expanded MSC therapy to receive regulatory approval49. To date, one allogeneic culture-expanded MSC product has received regulatory approval: Osiris’ (now Mesoblasts’) Prochymal was approved in Canada in May of 2012 and shortly after in New Zealand for the treatment of steroid-refractory GvHD in children50. Although the original trial failed to demonstrate a significant advantage of Prochymal over placebo, retrospective subset analysis, which has been highly criticized in the field, revealed improvement in children and ultimately led to its approval in Canada (conditional upon post-market confirmatory studies to validate the efficacy of the product). Fortunately, results from a recent follow-up study are encouraging, showing a significant increase in 100-day survival (78% versus 30%) in patients that respond to Prochymal in the first 28 days of treatment (complete resolution of symptoms, n = 46) compared to patients who do not respond (no improvement in symptoms, n = 29)51. Prochymal is now available for adults and children in eight other countries including the United States for steroid refractory grade III and IV GvHD under an Expanded Access Program. Academic-investigator–driven studies without placebo controls have also shown a benefit with unmatched-allogeneic and haplo-identical MSCs in the prevention and treatment of acute GvHD52,53. A retrospective analysis of children treated with multiple infusions of MSCs revealed a significant increase in survival at 3 years (65% versus 0%) in children that had a complete response to the therapy (complete resolution of symptoms, n = 24) compared to those that had a partial or no response to therapy (minimal changes in symptoms following therapy, n = 13)54. Prophylactic treatment with MSCs at the time of bone marrow transplantation resulted in 0% incidence of grade III or IV GvHD (compared to 26% as shown in historical data; study approved by local Institutional Review Board)53. In another phase 2 study, 30/55 patients with severe GvHD showed a complete response to MSC therapy (resolution of all symptoms) and had significantly improved 1-year survival, 52% versus 16%, compared to patients that showed little or no improvement after MSC therapy52.
Whereas case studies45,55 and clinical studies of small groups of patients46,52 have suggested MSCs have significant clinical utility, demonstration of a beneficial effect from MSCs in a large placebo-controlled trial has remained elusive. Several placebo-controlled studies have yielded disappointing results, showing marginal or no benefit over placebo; these include Prochymal trials targeting steroid-resistant GvHD, first-line GvHD, COPD and type 1 diabetes (GvHD: http://investor.osiris.com/releasedetail.cfm?ReleaseID=407404; COPD: http://investor.osiris.com/releasedetail.cfm?ReleaseID=391580; diabetes: http://investor.osiris.com/releasedetail.cfm?ReleaseID=636520). Of note, MSC products in general have not been optimized to maximize therapeutic potential and it is believed that certain allo-MSC products may be overpassaged leading to reduced potency. Furthermore, cell therapy trials often suffer from a large placebo effect, making it difficult to show efficacy.
Allo-MSCs are not immune privileged
A potential limitation of MSC therapy is that MSCs do not persist following infusion. Using bioluminescence imaging, intravital micros copy, donor DNA analysis and donor RNA analysis, the persistence of human MSCs (in mice with severe combined immunodeficiency), mouse MSCs (mMSCs; in syngeneic mice) and rat MSCs (in allo-geneic rats) was limited, with the majority of cells dying within 48 h after systemic infusion56–58. The trend of early MSC death after infusion was recently confirmed through analysis of tissues at autopsy of patients who received allo-MSC infusions within a year before their death59. Tissues from 18 patients who received MHC-mismatched or haplo-identical MSCs were analyzed; no ectopic tissue was observed, and only one patient showed high (>1/1,000 cells) levels of donor DNA in multiple tissues. However, this patient was not representative of the average patient, as he was severely immunocompromised, septic and received the MSC infusion just 7 days before his death59. Although allo- and auto-MSCs alike may not persist following systemic infusion simply owing to stresses encountered during transplantation (nutrient and/or growth factor deprivation due to transport limitations of nutrients and oxygen60, shear stress, lack of attachment), it is likely that a more active immunological process is also responsible for the limited persistence of allo-MSCs. In fact, several preclinical and clinical observations have led multiple groups to question the immune-privileged status of MSCs, and subsequently the notion of using a universal donor for MSC therapy. Although the majority of in vitro studies have highlighted the immunosuppressive properties of MSCs, several studies have provided evidence that mismatched MSCs are immunogenic. For example, whereas culture-expanded MSCs express low levels of MHC class I and are negative for MHC class II, MSCs exposed to IFN-γ or differentiated into mature cell types can express significantly more MHC class I and MHC class II (ref. 40).
In one of the earliest reports of rejection of allo-MSC, Eliopoulos et al. examined the persistence of mMSCs transfected with erythropoietin (EPO-MSCs) in syngeneic and allogeneic unmatched hosts61. Specifically, C57Bl/6 EPO-MSCs were seeded in a collagen scaffold and injected subcutaneously into syngeneic or allogeneic (BALB/c) hosts. As a surrogate for MSC survival, the rise in hematocrit in response to EPO production was measured for over 140 days. Mice receiving syngeneic EPO-MSCs had a sustained increase in hematocrit, whereas those receiving allogeneic EPO-MSCs had a spike in hematocrit followed by a return to baseline. Analysis of collagen scaffolds removed 15 days after implantation revealed significant infiltration by CD8+ T cells and natural killer (NK) cells only in the allogeneic EPO-MSC grafts. In addition, administration of a second dose of allogeneic EPO-MSCs resulted in a second but diminished spike in hematocrit, suggesting the initial challenge may have sensitized the animals to allo-antigen61. In a separate study, injection of allogeneic EPO-MSCs into immune-competent hosts resulted in sensitization to the EPO antigen and an anti-EPO immune response62. Thus care must be taken with allogeneic products not to break tolerance to self-antigens, especially in the context of therapies engineered to overexpress proteins62.
Therefore, whereas allo-MSCs are not as immunogenic as unmatched fibroblasts or HSCs, which elicit rapid rejection in immunocompetent hosts, it is not appropriate to consider MSCs to be immune privileged because they do elicit a humoral and cellular immune response in vivo. Further supporting this conclusion, Zangi et al. injected luciferase-expressing mMSCs or fibroblasts into syngeneic and allogeneic hosts and compared their persistence63. The majority of syngeneic mMSCs and fibroblasts were detectable for the duration of the experiment (40 days), but in the allogeneic setting, fibroblasts died by day 10 and mMSCs by day 20. In addition, mice previously injected with allo-MSCs showed accelerated rejection of fibroblasts from the same donor (by day 2 compared to day 14 in naive mice). In accordance with this observation, allo-MSC–treated mice harbored more CD4+, CD122+, CD44+ and CD62Llow T cells, suggesting the formation of T-cell memory63. Others have since reported that infusion of allo-MSCs can induce immune memory. For example, mice inoculated with unmatched allo-MSCs exhibit rapid rejection of donor-derived splenocytes within 24 h of the second transplant64, and mice receiving intraperitoneal injections of allo-MSCs produced elevated titers of allo-reactive antibodies and rejected subsequent allogeneic skin grafts65. Despite subtle interspecies differences in the expression of surface markers and immunosuppressive factors, similar evidence of immune detection and lack of long-term engraftment of allo-MSCs has been observed in a variety of species66 including rat67,68, baboon69, rhesus macaquea70 and pig71.
Allo-MSC also seem to stimulate innate immune responses. For example, human MSCs promote macrophage and neutrophil infiltration to the injection site in rats and mice72,73. And late, but not early, passage MSCs elicit an instant blood-mediated inflammatory reaction in a donor- and dose-dependent manner74. Furthermore, hMSCs engage complement on their surface75, although the effect of complement on MSC function is currently debated and complement-mediated lysis of MSCs is likely to be allo-antibody dependent76,77.
In sum, although MSCs cannot be considered truly immune privileged, rejection of allo-MSCs occurs more slowly than rejection of other allogeneic cell types. The timing and severity of MSC rejection appears to be strongly dependent on context and dictated by a balance between MSC expression of immunogenic and immunosuppressive factors (Fig. 2). For example, failure to activate their immunosuppressive program leaves MSCs functioning much like antigen-presenting cells and able to promote inflammation in vivo78–80. In contrast, in in vitro assays (such as mixed lymphocyte reactions or T-cell suppression assays), where the concentration of MSCs is high enough to strongly influence the microenvironment within the cell culture well, the immunosuppressive properties of MSCs dominate. Illustrating the importance of context, unmatched allo- and xenogeneic-MSCs can preferentially persist within in vivo environments that are immune suppressed such as tumors, yet fail to persist in other tissues within the same animal58,81. This suggests that local immune suppression is required to mask MSC immunogenicity81. Detailed in vivo studies on MSC immunogenicity are needed using human MSCs or non-human primate MSCs that more closely mirror human biology, as murine and human MSCs exhibit differences in both host factors as well as MSC expression of immunomodulatory factors12,82. For example, hMSC-mediated immune modulation is often achieved in part through IDO, whereas mMSCs secrete virtually no IDO but high quantities of inducible nitric oxide synthase82.
Figure 2.

Immune suppression enables immune evasion. (a) MSC immunosuppressive potential and immunogenicity are influenced by levels of systemic or local inflammatory cytokines (secreted by T cells and other cell types). High immunosuppressive potential permits MSCs to suppress inflammation and delay or evade allo-rejection through suppression of T-cell activation and inhibition of antigen-presenting cell (APC) maturation. However, MSCs that do not tip the balance toward immunosuppression are prone to immune detection and destruction through multiple modes of rejection, as debris from dead MSCs are processed by APCs in the context of danger signals. (b) The rate of immune detection and elimination of allogeneic MSCs is dictated by the balance between a given cell’s relative expression of immunogenic and immunosuppressive factors. IFN-γ, interferon gamma; TNF-α, tumor necrosis factor alpha; MHC, major histocompatibility complex; TCR, T-cell receptor; IDO, indoleamine 2;3 dioxygenase; TGF-β, transforming growth factor beta; TSG-6; TNF-stimulated gene 6 protein; sHLA-G5, soluble human leukocyte antigen-g5; PGE2, prostaglandin E2.
Strategies to prolong MSC persistence
Importantly, it has yet to be shown clinically that improved MSC persistence or immune tolerance to MSCs leads to enhanced efficacy of MSC-based treatments. Many believe that the observed therapeutic effect of MSCs is due to a so-called hit-and-run mechanism mediated by the production of exosomes or secretion of trophic and immunomodulatory factors during the initial days following MSC injection15,59. Others believe that the main therapeutic benefit of MSCs may be achieved through reprogramming of the immune system through apoptotic bodies83. Nevertheless, conventional wisdom suggests that the beneficial effects of MSC therapy could be boosted by extending their persistence after injection39.
Strategies to boost MSC persistence in vivo may be gained from the field of transplantation biology, where overcoming MHC-barriers is very difficult. For example, recent progress indicates that solid organ transplantation can be helped by induction of mixed hematopoietic chimerism through donor HSC transplantation84. In addition, allogeneic cell products like Viacyte’s PEC-01 use immune isolation membrane-based devices (Encaptra) and tissue-engineered products like Organogenesis’ Apligraf and Shire’s SRM003 (formerly VASCUGEL) to temporarily prevent rejection by encasing differentiated allogeneic cells in collagen or gelatin gels. One recent study showed that subcutaneously injected mMSCs that were encapsulated in alginate persisted longer, and this approach significantly improved the survival of mice in a model of GvHD compared to systemically infused MSCs and locally injected MSCs without the alginate85.
Strategies to prolong MSC persistence by overcoming rejection of allo-MSCs can be divided into two primary categories: modification of the host and modification of MSCs. Several groups have experimented with combining MSCs and anti-rejection drugs classically used in organ transplantation. Interestingly, the strongest clinical evidence of an MSC therapeutic effect, as described above, was observed in patients with steroid-resistant acute GvHD, who received MSC therapy in the context of a standard regimen of immunosuppressant drugs. Although this observation is currently correlative, work is under way to investigate synergistic effects of immunosuppressant drugs and MSCs. Adding cyclosporine A to co-cultures of MSCs in an in vitro mixed lymphocyte reaction assay resulted in significantly enhanced suppression of cell lysis over cyclosporine A or MSCs alone86. However, dexamethasone, mycophenolate acid, rapamycin, cyclosporine A and tacrolimus individually synergized or antagonized MSC-mediated suppression of mixed lymphocyte reaction, depending on the dose of the drug and the responder/MSC ratio87. Unfortunately, the mechanism by which immunosuppressive drugs augmented or interfered with the immunosuppressive properties of MSCs was not evaluated, and detailed mechanistic studies are needed. In one of the few studies of immunosuppressive drug interactions with MSCs in vivo, Ge et al. investigated the effect of low-dose rapamycin and MSCs in a cardiac allograft mouse model88. Combining a 14-day course of low-dose rapamycin with unmatched allo-MSC therapy resulted in long-term persistence of GFP-labeled allo-MSCs and tolerance (100 days) of a heart graft of MSC donor origin88. The mice did not accept third-party allografts, however, indicating that any tolerance caused by the MSC treatment was specific to the donor antigens. They also contained more tolerogenic dendritic and T cells, but no allogeneic antibodies88. More research is needed to characterize the impact of immunosuppressive drugs on MSC phenotype and in vivo persistence. For example, short-course immunosuppression, which has shown promise in preventing GvHD89 and prolonging liver allograft survival90,91, may be sufficient to extend persistence of MSCs and augment their therapeutic effect. In addition, antibody-mediated depletion of immune cells such as NK or cytotoxic lymphocytes may prolong MSC persistence; however, such strategies could also increase the risk of infection in vulnerable patient populations92.
MSCs can also be directly modified to reduce their immunogenicity (Fig. 3). For example, de la Garza-Rodea et al. looked to viruses for inspiration to enable MSCs to evade immune detection. To permanently suppress MHC class I surface expression, they transfected hMSCs with viral immunoevasins from bovine herpes virus type 1, Epstein-Barr virus and human cytomegalovirus (HCMV)93. Of these, only the US11 protein from HCMV strongly suppressed hMSC expression of MHC class I. Moreover, US11-MSCs locally injected into the ear of immunocompetent mice persisted for a period similar to that of hMSC injected into immunodeficient mice. However MSC immune evasion was achieved only after NK cell depletion, as the lack of MHC class I expression made hMSCs susceptible to ‘missing self ’ NK cell-mediated lysis93. Soland et al. also infected hMSCs with immunoevasins from HCMV94 (US2, US3, US6 or US11). Expression of either US6 or US11 resulted in significant suppression of MHC class I surface expression, and facilitated hMSC evasion of recognition by cytotoxic lymphocytes. Interestingly, in contrast to de la Garza-Rodea’s report93, expression of US11 also resulted in protection from lysis in co-cultures with NK-92MI cells. However, the mechanism of hMSC evasion from NK cells was not fully elucidated. These engineered hMSCs, after injection into the liver of pre-immune fetal sheep, were less susceptible to NK cell–mediated lysis and had a 1.8-fold increase in engraftment (in terms of percent of donor cells found in the liver 66 days after injection) compared to unmodified hMSCs94. Whether these virus-inspired strategies will prevent immune memory in a fully immune-competent host or enhance the therapeutic effect of MSCs in disease models remains to be determined.
Figure 3.
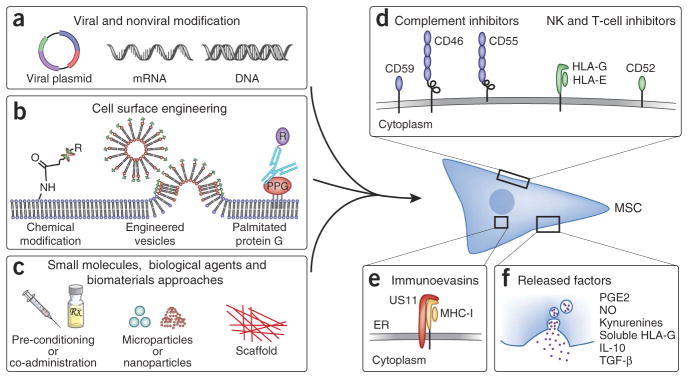
Strategies to facilitate MSC immune evasion. (a–c) A variety of engineering strategies can be applied to induce MSC expression of immunosuppressive and immunoevasive factors including forced expression through viral and nonviral modification (a), direct cell surface modification (b), and treatment with small molecules and/or biological agents directly or through controlled release biomaterials approaches (c). (c) The timing of MSC expression can potentially be extended by use of agent-doped, cell-internalized degradable microparticles99 or nanoparticles, or by use of agent-doped scaffolds that cells are seeded before in vivo implantation. (d) Decoy or inhibitory receptors can be directly engineered onto the cell surface through several techniques including chemical modification via covalent conjugation chemistry97,110, engineered vesicles98, or through insertion of antibody fusion proteins into the cell membrane via palmitated protein G (PPG) (b). (e,f) Increased persistence can also be achieved through reducing immunogenicity by the use of immunoevasins (e) or sustained release of immunosuppressive factors (f).
In addition to forced expression of viral immunoevasins (Fig. 3a,e), MSCs, as well as other therapeutic allogeneic cell types, could be engineered to overexpress molecules that suppress complement activation (CD46, CD55, CD59) or NK cell activation (HLA-E, HLA-G), and to secrete immunosuppressive factors (Fig. 3a,d,f). We have recently shown that MSCs can be transiently engineered to simultaneously overexpress IL-10 and homing receptors by using an mRNA transfection technique95. Cells can also be decorated with immune evasive moieties through chemical cell surface modification approaches96–98 (Fig. 3b,d). Alternatively, MSCs can be preconditioned or loaded99,100 with small-molecule drugs or biologics that increase the expression of surface receptors or the production of immunosuppressive factors (e.g., PGE2, IDO, HLA-G, IL-10) (Fig. 3c,f).
New vision for allogeneic MSC therapy
Although a comprehensive understanding of MSC allo-rejection is still under development and questions remain, it is clear that MSCs are not immune privileged, at least not to the extent that has been claimed in the literature; rather, MSCs could be considered ‘immune evasive’. Nevertheless, their use in clinical trials has continued to escalate (Fig. 1), and the community appears reluctant to abandon the immune-privileged paradigm as evidenced by the higher numbers of citations made to articles that support the ‘Universal Donor’ hypothesis, compared to articles that highlight MSC immunogenicity (Fig. 1c). Immunogenicity needs to be recognized as a characteristic of MSCs and its impact on MSC therapy needs to be examined. It is critical to consider that although infused MSCs may not express MHC class II, this will likely always be activated and/or expressed in vivo at sites of inflammation.
Allo-MSC therapy faces significant challenges, but auto-MSC therapy is not without drawbacks. Generating a therapeutic dose of auto-MSCs generally requires several weeks after cell harvest for ex vivo expansion101. Furthermore, significant variations have been observed in the secretome and immunomodulatory potency of MSCs from different donors14,102. Thus, auto-MSC therapy may not be suitable for the treatment of acute conditions, and variability between patient-derived MSCs is likely to lead to highly variable outcomes. Furthermore, auto-MSC therapy’s source of MSCs is limited, whereas allo-MSC therapy can harvest MSCs from healthy donors and select lots based on potency assays103.
Although both auto-MSCs and allo-MSCs are being used in clinical trials, direct comparisons between the two cell sources are scarce. In the recent POSEIDON trial, the safety and efficacy of auto- and allo-MSCs were compared after administration into the remodeled cardiac scar of patients with chronic cardiac ischemia104. Unfortunately, at 30 days, clinical improvement was limited in both groups. Auto-MSCs were associated with marginal improvements in a 6-min walk test and as recorded in the Minnesota Living with Heart Failure Questionnaire, whereas allo-MSCs were associated with fewer, but not statistically significant, arrhythmias. Administration of either auto- or allo-MSCs resulted in a small improvement in ejection fraction (~2%). Without a strong positive effect, a placebo group or analysis of donor cell persistence, it is difficult to use this trial to effectively compare the efficacy of allo- versus auto-MSCs. Although both cell sources were shown to be safe, comparisons of auto- and allo-MSCs are warranted in future studies.
Interestingly, although no major adverse events have been linked to administration of allo-MSCs, an anti-donor response has been observed. Both the POSEIDON trial and a recent phase 2 mesoblast trial reported generation of anti-donor antibodies in 13% of patients treated with allo-MSCs. However, the therapy still appears safe as Osiris and others frequently administer repeat doses without complications47. Moreover, it remains to be seen if allo-antibody production influences the efficacy of MSC therapy. For example, a phase 2 trial investigating the ability of MSCs to treat GvHD found no difference in efficacy between third-party and haplo-identical or human leukocyte antigen (HLA)-identical MSCs52. However, the degree of immune suppression in these patients may have masked differences between the third-party and HLA-identical MSCs. Measurements of anti-donor antibodies in clinical trials, which to date have been rare, should become routine as it will enable the field to understand the relationship between degree of the HLA mismatch, MSC rejection and treatment efficacy in specific conditions.
A better understanding of the particular MSC mechanisms that contribute to the therapeutic effect in each disease condition may also help clarify whether allo-MSCs or auto-MSCs are more appropriate. For example, if allo-MSCs are found to exhibit a more potent hit-and-run mechanism of action, they may be most appropriate where persistence is not required for a therapeutic effect.
Ultimately, the hypothesis that extended MSC persistence will translate to a sustained therapeutic effect and improved clinical outcomes has yet to be tested clinically. Nevertheless, the approach to allo-MSC therapy needs revision. Patients must be prevented from becoming immunized, which could blunt or inhibit the activity of future MSC therapies. In addition to the immune evasion strategies discussed above, banking MSCs, derived from patient tissues or induced pluripotent stem cells105, from a diverse donor population may help avoid patient sensitization. Due to the ease of MSC expansion, development of an MSC banking system, much like that for blood, is not difficult to imagine. MSCs from a representative subset of the population could be harvested, typed and expanded for clinical use. With time, healthy donors would populate the cell bank with a sufficient variety of HLA antigens. This should improve MSC persistence and, potentially, therapeutic outcomes; it should also enable serial injection of MSCs without rejection or co-administration of immunosuppressive drugs.
In addition, as MSC persistence correlates with the relative expression of immunogenic and immunosuppressive factors (Fig. 2), development of next-generation MSC therapies should focus on boosting expression of these factors. Specifically, MSC potency assays must be established based on MSC expression of immunogenic and immunosuppressive factors at baseline and after stimulation with molecules that mimic the physiological conditions to which the MSCs will be exposed in the patient (e.g., TNF-α, IFN-γ and IL-1β might simulate MSC exposure to inflammatory signals); these assays should be standardized to ensure that patients receive functional and comparable doses103,106. The 2006 International Society for Cellular Therapy minimal criteria that is often used in research cannot predict immunomodulatory function or immunogenicity of a batch of MSCs, and considerable variability in immunomodulatory potency between donors, tissue sources and culture conditions has been well documented14,102,107,108. MSC potency assays must also be validated to ensure they are predictive of patient outcomes. For example, in a recent study the degree of MSC suppression of in vitro mixed lymphocyte cultures did not correlate with clinical response in GvHD patients46. In addition to MSC potency assays, biomarkers that predict whether or not a patient is likely to respond to MSC therapy should also be explored12. There is also potential to develop in vivo potency assays where, for example, cell surface sensors could be harnessed to image the MSC secretome using intravital microscopy in animal models109. Once potency assays have been developed and validated, perhaps the definition of MSCs will once again be updated to include criteria that describe the MSC functional phenotype.
To realize the potential of MSC therapy in the next decade, we must be willing to let go of the old one-size-fits all strategy, accept that MSCs are immune evasive and not immune privileged, and consider approaches to avoid generation of allo-reactive antibodies. That said, allo-MSCs appear to be cleared only marginally faster than auto-MSCs. This implies that combination approaches to avoid allo-rejection and to mitigate transplantation shock would be most useful to extend MSC persistence. To maximize patient benefit and minimize patient risk, next-generation MSC therapies should be built on a foundation of thorough characterization and fine-tuning of MSC immunogenicity, survival, potency and disease-specific mechanisms of action.
Acknowledgments
This work was supported by National Institutes of Health grant HL095722, Department of Defense grant no. W81XWH-13-1-0305 and by a Movember–Prostate Cancer Foundation Challenge Award to J.M.K. J.A.A. was supported by the Hugh Hampton Young Memorial Fellowship.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Friedenstein AJ, Piatetzky-Shapiro II, Petrakova KV. Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol. 1966;16:381–390. [PubMed] [Google Scholar]
- 2.Friedenstein A, Gorskaja J, Kulagina N. Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp Hematol. 1976;4:267–274. [PubMed] [Google Scholar]
- 3.Friedenstein AJ, Chailakhyan RK, Gerasimov UV. Bone marrow osteogenic stem cells: in vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet. 1987;20:263–272. doi: 10.1111/j.1365-2184.1987.tb01309.x. [DOI] [PubMed] [Google Scholar]
- 4.Caplan AI. Mesenchymal stem cells. J Orthop Res. 1991;9:641–650. doi: 10.1002/jor.1100090504. [DOI] [PubMed] [Google Scholar]
- 5.James AW, et al. An abundant perivascular source of stem cells for bone tissue engineering. Stem Cells Transl Med. 2012;1:673–684. doi: 10.5966/sctm.2012-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crisan M, Corselli M, Chen WCW, Péault B. Perivascular cells for regenerative medicine. J Cell Mol Med. 2012;16:2851–2860. doi: 10.1111/j.1582-4934.2012.01617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan CKF, et al. Clonal precursor of bone, cartilage, and hematopoietic niche stromal cells. Proc Natl Acad Sci USA. 2013;110:12643–12648. doi: 10.1073/pnas.1310212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park D, et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell. 2012;10:259–272. doi: 10.1016/j.stem.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominici M, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 10.Copley MR, Beer PA, Eaves CJ. Hematopoietic stem cell heterogeneity takes center stage. Cell Stem Cell. 2012;10:690–697. doi: 10.1016/j.stem.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 11.Verovskaya E, et al. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood. 2013;122:523–532. doi: 10.1182/blood-2013-01-481135. [DOI] [PubMed] [Google Scholar]
- 12.Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13:392–402. doi: 10.1016/j.stem.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 13.Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110:3499–3506. doi: 10.1182/blood-2007-02-069716. [DOI] [PubMed] [Google Scholar]
- 14.François M, Romieu-Mourez R, Li M, Galipeau J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther. 2012;20:187–195. doi: 10.1038/mt.2011.189. [DOI] [PubMed] [Google Scholar]
- 15.Prockop DJ. Concise review: two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells. 2013;31:2042–2046. doi: 10.1002/stem.1400. [DOI] [PubMed] [Google Scholar]
- 16.Bianco P, et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013;19:35–42. doi: 10.1038/nm.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen H. Stricter standards sought to curb stem-cell confusion. Nature. 2013;499:389. doi: 10.1038/499389a. [DOI] [PubMed] [Google Scholar]
- 18.Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell. 2012;10:709–716. doi: 10.1016/j.stem.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 19.Caplan AI, Correa D. The MSC: an injury drugstore. Cell Stem Cell. 2011;9:11–15. doi: 10.1016/j.stem.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phinney DG, et al. MSCs: science and trials. Nat Med. 2013;19:812. doi: 10.1038/nm.3220. [DOI] [PubMed] [Google Scholar]
- 21.Fibbe WE, Dazzi F, LeBlanc K. MSCs: science and trials. Nat Med. 2013;19:812–813. doi: 10.1038/nm.3222. [DOI] [PubMed] [Google Scholar]
- 22.Pittenger MF. MSCs: science and trials. Nat Med. 2013;19:811. doi: 10.1038/nm.3219. [DOI] [PubMed] [Google Scholar]
- 23.Ankrum J, Karp J. Mesenchymal stem cell therapy: two steps forward, one step back. Trends Mol Med. 2010;16:203–209. doi: 10.1016/j.molmed.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haynesworth SE, Baber MA, Caplan AI. Cytokine expression by human marrow-derived mesenchymal progenitor cells in vitro: effects of dexamethasone and IL-1 alpha. J Cell Physiol. 1996;166:585–592. doi: 10.1002/(SICI)1097-4652(199603)166:3<585::AID-JCP13>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 25.Devine SM, Hoffman R. Role of mesenchymal stem cells in hematopoietic stem cell transplantation. Curr Opin Hematol. 2000;7:358–363. doi: 10.1097/00062752-200011000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Lazarus H, Curtin P, Devine S, McCarthy P, Holland K. Role of mesenchymal stem cells (MSC) in allogeneic transplantation: early phase I clinical results. Blood. 2000;96:392a. [Google Scholar]
- 27.Klyushnenkova E, Mosca JD, McIntosh KR, Thiede MA. Human mesenchymal stem cells suppress allogeneic T cell responses in vitro: implications for allogeneic transplantation. Blood. 1998;92:2652. [Google Scholar]
- 28.Tse WT, Beyer W, Pendleton JD, D’Andrea A. Bone marrow derived mesenchymal stem cells suppress T cell activation without inducing allogeneic anergy. Blood. 2000;96:1034a. [Google Scholar]
- 29.Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. 2003;75:389–397. doi: 10.1097/01.TP.0000045055.63901.A9. [DOI] [PubMed] [Google Scholar]
- 30.Klyushnenkova E, et al. T cell responses to allogeneic human mesenchymal stem cells: immunogenicity, tolerance, and suppression. J Biomed Sci. 2005;12:47–57. doi: 10.1007/s11373-004-8183-7. [DOI] [PubMed] [Google Scholar]
- 31.Bartholomew A, et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002;30:42–48. doi: 10.1016/s0301-472x(01)00769-x. [DOI] [PubMed] [Google Scholar]
- 32.Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringden O. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand J Immunol. 2003;57:11–20. doi: 10.1046/j.1365-3083.2003.01176.x. [DOI] [PubMed] [Google Scholar]
- 33.Di Nicola M, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838–3843. doi: 10.1182/blood.v99.10.3838. [DOI] [PubMed] [Google Scholar]
- 34.Krampera M, et al. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003;101:3722–3729. doi: 10.1182/blood-2002-07-2104. [DOI] [PubMed] [Google Scholar]
- 35.Liechty KW, et al. Human mesenchymal stem cells engraft and demonstrate site-specific differentiation after in utero transplantation in sheep. Nat Med. 2000;6:1282–1286. doi: 10.1038/81395. [DOI] [PubMed] [Google Scholar]
- 36.Ito T, et al. Mesenchymal stem cell and islet co-transplantation promotes graft revascularization and function. Transplantation. 2010;89:1438–1445. doi: 10.1097/tp.0b013e3181db09c4. [DOI] [PubMed] [Google Scholar]
- 37.Casiraghi F, et al. Pretransplant infusion of mesenchymal stem cells prolongs the survival of a semiallogeneic heart transplant through the generation of regulatory T cells. J Immunol. 2008;181:3933–3946. doi: 10.4049/jimmunol.181.6.3933. [DOI] [PubMed] [Google Scholar]
- 38.Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an immunosuppressive MSC2 phenotype. PLoS ONE. 2010;5:e10088. doi: 10.1371/journal.pone.0010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ranganath SH, Levy O, Inamdar MS, Karp JM. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cells. 2012;10:244–258. doi: 10.1016/j.stem.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringdén O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31:890–896. doi: 10.1016/s0301-472x(03)00110-3. [DOI] [PubMed] [Google Scholar]
- 41.Hemeda H, et al. Interferon-γ and tumor necrosis factor-α differentially affect cytokine expression and migration properties of mesenchymal stem cells. Stem Cells Dev. 2010;19:693–706. doi: 10.1089/scd.2009.0365. [DOI] [PubMed] [Google Scholar]
- 42.Mastri M, et al. Activation of Toll-like receptor 3 (TLR3) amplifies mesenchymal stem cell trophic factors and enhances therapeutic potency. Am J Physiol Cell Physiol. 2012;303:C1021–C1033. doi: 10.1152/ajpcell.00191.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delarosa O, Dalemans W, Lombardo E. Toll-like receptors as modulators of mesenchymal stem cells. Front Immunol. 2012;3:182. doi: 10.3389/fimmu.2012.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi Y, et al. How mesenchymal stem cells interact with tissue immune responses. Trends Immunol. 2012;33:136–143. doi: 10.1016/j.it.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le Blanc K, et al. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;363:1439–1441. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 46.von Bahr L, et al. Long-term complications, immunologic effects, and role of passage for outcome in mesenchymal stromal cell therapy. Biol Blood Marrow Transplant. 2012;18:557–564. doi: 10.1016/j.bbmt.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 47.Lalu MM, et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): a systematic review and meta-analysis of clinical trials. PLoS ONE. 2012;7:e47559. doi: 10.1371/journal.pone.0047559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Penn MS, et al. Adventitial delivery of an allogeneic bone marrow-derived adherent stem cell in acute myocardial infarction: phase I clinical study. Circ Res. 2012;110:304–311. doi: 10.1161/CIRCRESAHA.111.253427. [DOI] [PubMed] [Google Scholar]
- 49.Yang H. South Korea’s stem cell approval. Nat Biotechnol. 2011;29:857. [Google Scholar]
- 50.Cyranoski D. Canada approves stem cell product. Nat Biotechnol. 2012;30:571. [Google Scholar]
- 51.Kurtzberg J, et al. Allogeneic human mesenchymal stem cell therapy (Remestemcel-L, Prochymal) as a rescue agent for severe refractory acute graft-versus-host disease in pediatric patients. Biol Blood Marrow Transplant. 2013 doi: 10.1016/j.bbmt.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 52.Le Blanc K, et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371:1579–1586. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 53.Bernardo ME, et al. Co-infusion of ex vivo-expanded, parental MSCs prevents life-threatening acute GVHD, but does not reduce the risk of graft failure in pediatric patients undergoing allogeneic umbilical cord blood transplantation. Bone Marrow Transplant. 2011;46:200–207. doi: 10.1038/bmt.2010.87. [DOI] [PubMed] [Google Scholar]
- 54.Ball LM, et al. Multiple infusions of mesenchymal stromal cells induce sustained remission in children with steroid-refractory, grade III–IV acute graft-versus-host disease. Br J Haematol. 2013;163:501–509. doi: 10.1111/bjh.12545. [DOI] [PubMed] [Google Scholar]
- 55.Riordan NH, et al. Non-expanded adipose stromal vascular fraction cell therapy for multiple sclerosis. J Transl Med. 2009;7:29. doi: 10.1186/1479-5876-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toma C, Wagner WR, Bowry S, Schwartz A, Villanueva F. Fate of culture-expanded mesenchymal stem cells in the microvasculature: in vivo observations of cell kinetics. Circ Res. 2009;104:398–402. doi: 10.1161/CIRCRESAHA.108.187724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee RH, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5:54–63. doi: 10.1016/j.stem.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kidd S, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27:2614–2623. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.von Bahr L, et al. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long-term engraftment and no ectopic tissue formation. Stem Cells. 2012;30:1575–1578. doi: 10.1002/stem.1118. [DOI] [PubMed] [Google Scholar]
- 60.Muschler GF, Nakamoto C, Griffith LG. Engineering principles of clinical cell-based tissue engineering. J Bone Joint Surg Am. 2004;86-A:1541–1558. doi: 10.2106/00004623-200407000-00029. [DOI] [PubMed] [Google Scholar]
- 61.Eliopoulos N, Stagg J, Lejeune L, Pommey S, Galipeau J. Allogeneic marrow stromal cells are immune rejected by MHC class I- and class II-mismatched recipient mice. Blood. 2005;106:4057–4065. doi: 10.1182/blood-2005-03-1004. [DOI] [PubMed] [Google Scholar]
- 62.Campeau PM, et al. Mesenchymal stromal cells engineered to express erythropoietin induce anti-erythropoietin antibodies and anemia in allorecipients. Mol Ther. 2009;17:369–372. doi: 10.1038/mt.2008.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zangi L, et al. Direct imaging of immune rejection and memory induction by allogeneic mesenchymal stromal cells. Stem Cells. 2009;27:2865–2874. doi: 10.1002/stem.217. [DOI] [PubMed] [Google Scholar]
- 64.Nauta AJ, et al. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood. 2006;108:2114–2120. doi: 10.1182/blood-2005-11-011650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Badillo AT, Beggs KJ, Javazon EH, Tebbets JC, Flake AW. Murine bone marrow stromal progenitor cells elicit an in vivo cellular and humoral alloimmune response. Biol Blood Marrow Transplant. 2007;13:412–422. doi: 10.1016/j.bbmt.2006.12.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Griffin MD, et al. Anti-donor immune responses elicited by allogeneic mesenchymal stem cells: what have we learned so far? Immunol Cell Biol. 2013;91:40–51. doi: 10.1038/icb.2012.67. [DOI] [PubMed] [Google Scholar]
- 67.Camp DM, Loeffler DA, Farrah DM, Borneman JN, LeWitt PA. Cellular immune response to intrastriatally implanted allogeneic bone marrow stromal cells in a rat model of Parkinson’s disease. J Neuroinflammation. 2009;6:17. doi: 10.1186/1742-2094-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schu S, et al. Immunogenicity of allogeneic mesenchymal stem cells. J Cell Mol Med. 2012;16:2094–2103. doi: 10.1111/j.1582-4934.2011.01509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beggs KJ, et al. Immunologic consequences of multiple, high-dose administration of allogeneic mesenchymal stem cells to baboons. Cell Transplant. 2006;15:711–721. doi: 10.3727/000000006783981503. [DOI] [PubMed] [Google Scholar]
- 70.Isakova IA, Dufour J, Lanclos C, Bruhn J, Phinney DG. Cell-dose-dependent increases in circulating levels of immune effector cells in rhesus macaques following intracranial injection of allogeneic MSCs. Exp Hematol. 2010;38:957–967. doi: 10.1016/j.exphem.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Poncelet AJ, Vercruysse J, Saliez A, Gianello P. Although pig allogeneic mesenchymal stem cells are not immunogenic in vitro, intracardiac injection elicits an immune response in vivo. Transplantation. 2007;83:783–790. doi: 10.1097/01.tp.0000258649.23081.a3. [DOI] [PubMed] [Google Scholar]
- 72.Grinnemo KH, et al. Xenoreactivity and engraftment of human mesenchymal stem cells transplanted into infarcted rat myocardium. J Thorac Cardiovasc Surg. 2004;127:1293–1300. doi: 10.1016/j.jtcvs.2003.07.037. [DOI] [PubMed] [Google Scholar]
- 73.Xia Z, et al. Macrophagic response to human mesenchymal stem cell and poly(epsilon-caprolactone) implantation in nonobese diabetic/severe combined immunodeficient mice. J Biomed Mater Res A. 2004;71:538–548. doi: 10.1002/jbm.a.30185. [DOI] [PubMed] [Google Scholar]
- 74.Moll G, et al. Are therapeutic human mesenchymal stromal cells compatible with human blood? Stem Cells. 2012;30:1565–1574. doi: 10.1002/stem.1111. [DOI] [PubMed] [Google Scholar]
- 75.Moll G, et al. Mesenchymal stromal cells engage complement and complement receptor bearing innate effector cells to modulate immune responses. PLoS ONE. 2011;6:e21703. doi: 10.1371/journal.pone.0021703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Y, Lin F. Mesenchymal stem cells are injured by complement after their contact with serum. Blood. 2012;120:3436–3443. doi: 10.1182/blood-2012-03-420612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol. 2012;12:383–396. doi: 10.1038/nri3209. [DOI] [PubMed] [Google Scholar]
- 78.Chan JL, et al. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-gamma. Blood. 2006;107:4817–4824. doi: 10.1182/blood-2006-01-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stagg J, Pommey S, Eliopoulos N, Galipeau J. Interferon-gamma-stimulated marrow stromal cells: a new type of nonhematopoietic antigen-presenting cell. Blood. 2006;107:2570–2577. doi: 10.1182/blood-2005-07-2793. [DOI] [PubMed] [Google Scholar]
- 80.François M, et al. Mesenchymal stromal cells cross-present soluble exogenous antigens as part of their antigen-presenting cell properties. Blood. 2009;114:2632–2638. doi: 10.1182/blood-2009-02-207795. [DOI] [PubMed] [Google Scholar]
- 81.Dembinski JL, et al. Tumor stroma engraftment of gene-modified mesenchymal stem cells as anti-tumor therapy against ovarian cancer. Cytotherapy. 2013;15:20–32. doi: 10.1016/j.jcyt.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ren G, et al. Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem Cells. 2009;27:1954–1962. doi: 10.1002/stem.118. [DOI] [PubMed] [Google Scholar]
- 83.Voll RE, et al. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 84.Leonard DA, Cetrulo CL, Jr, McGrouther DA, Sachs DH. Induction of tolerance of vascularized composite allografts. Transplantation. 2013;95:403–409. doi: 10.1097/TP.0b013e31826d886d. [DOI] [PubMed] [Google Scholar]
- 85.Zanotti L, et al. Encapsulated mesenchymal stem cells for in vivo immunomodulation. Leukemia. 2013;27:500–503. doi: 10.1038/leu.2012.202. [DOI] [PubMed] [Google Scholar]
- 86.Maccario R, et al. Human mesenchymal stem cells and cyclosporin a exert a synergistic suppressive effect on in vitro activation of alloantigen-specific cytotoxic lymphocytes. Biol Blood Marrow Transplant. 2005;11:1031–1032. doi: 10.1016/j.bbmt.2005.08.039. [DOI] [PubMed] [Google Scholar]
- 87.Buron F, et al. Human mesenchymal stem cells and immunosuppressive drug interactions in allogeneic responses: an in vitro study using human cells. Transplant Proc. 2009;41:3347–3352. doi: 10.1016/j.transproceed.2009.08.030. [DOI] [PubMed] [Google Scholar]
- 88.Ge W, et al. Infusion of mesenchymal stem cells and rapamycin synergize to attenuate alloimmune responses and promote cardiac allograft tolerance. Am J Transplant. 2009;9:1760–1772. doi: 10.1111/j.1600-6143.2009.02721.x. [DOI] [PubMed] [Google Scholar]
- 89.Luznik L, et al. High-dose cyclophosphamide as single-agent, short-course prophylaxis of graft-versus-host disease. Blood. 2010;115:3224–3230. doi: 10.1182/blood-2009-11-251595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang WH, Yan Y, De Boer B, Bishop GA, House AK. A short course of cyclosporine immunosuppression inhibits rejection but not tolerance of rat liver allografts. Transplantation. 2003;75:368–374. doi: 10.1097/01.TP.0000044360.20396.AB. [DOI] [PubMed] [Google Scholar]
- 91.Huang WH, et al. A short course of mycophenolate immunosuppression inhibits rejection, but not tolerance, of rat liver allografts in association with inhibition of interleukin-4 and alloantibody responses. Transplantation. 2003;76:1159–1165. doi: 10.1097/01.TP.0000092304.18324.42. [DOI] [PubMed] [Google Scholar]
- 92.Forslöw U, et al. Treatment with mesenchymal stromal cells is a risk factor for pneumonia-related death after allogeneic hematopoietic stem cell transplantation. Eur J Haematol. 2012;89:220–227. doi: 10.1111/j.1600-0609.2012.01824.x. [DOI] [PubMed] [Google Scholar]
- 93.de la Garza-Rodea AS, et al. Exploitation of herpesvirus immune evasion strategies to modify the immunogenicity of human mesenchymal stem cell transplants. PLoS ONE. 2011;6:e14493. doi: 10.1371/journal.pone.0014493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Soland MA, et al. Modulation of human mesenchymal stem cell immunogenicity through forced expression of human cytomegalovirus US proteins. PLoS ONE. 2012;7:e36163. doi: 10.1371/journal.pone.0036163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Levy O, et al. mRNA-engineered mesenchymal stem cells for targeted delivery of interleukin-10 to sites of inflammation. Blood. 2013;122:e23–e32. doi: 10.1182/blood-2013-04-495119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ko IK, Kean TJ, Dennis JE. Targeting mesenchymal stem cells to activated endothelial cells. Biomaterials. 2009;30:3702–3710. doi: 10.1016/j.biomaterials.2009.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sarkar D, et al. Chemical engineering of mesenchymal stem cells to induce a cell rolling response. Bioconjug Chem. 2008;19:2105–2109. doi: 10.1021/bc800345q. [DOI] [PubMed] [Google Scholar]
- 98.Sarkar D, et al. Engineered mesenchymal stem cells with self-assembled vesicles for systemic cell targeting. Biomaterials. 2010;31:5266–5274. doi: 10.1016/j.biomaterials.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sarkar D, Ankrum J, Teo GSL, Carman CV, Karp JM. Cellular and extracellular programming of cell fate through engineered intracrine-, paracrine-, and endocrine-like mechanisms. Biomaterials. 2011;32:3053–3061. doi: 10.1016/j.biomaterials.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ankrum J, et al. Engineering cells with intracellular agent-loaded microparticles to control cell phenotype. Nat Protoc. 2014;9:233–245. doi: 10.1038/nprot.2014.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brandenberger R, et al. Cell therapy bioprocessing. Bioprocess Int. 2011;9:30–37. [Google Scholar]
- 102.Melief SM, Zwaginga JJ, Fibbe WE, Roelofs H. Adipose tissue-derived multipotent stromal cells have a higher immunomodulatory capacity than their bone marrow-derived counterparts. Stem Cells Transl Med. 2013;2:455–463. doi: 10.5966/sctm.2012-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bravery CA, et al. Potency assay development for cellular therapy products: an ISCT review of the requirements and experiences in the industry. Cytotherapy. 2013;15:9–19. doi: 10.1016/j.jcyt.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 104.Hare JM, et al. Comparison of allogeneic vs autologous bone marrow–derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: the POSEIDON randomized trial. J Am Med Assoc. 2012;308:2369–2379. doi: 10.1001/jama.2012.25321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jung Y, Bauer G, Nolta JA. Concise review: induced pluripotent stem cell-derived mesenchymal stem cells: progress toward safe clinical products. Stem Cells. 2012;30:42–47. doi: 10.1002/stem.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Krampera M, et al. Immunological characterization of multipotent mesenchymal stromal cells-The International Society for Cellular Therapy (ISCT) working proposal. Cytotherapy. 2013;15:1054–1061. doi: 10.1016/j.jcyt.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 107.Zhukareva V, Obrocka M, Houle JD, Fischer I, Neuhuber B. Secretion profile of human bone marrow stromal cells: donor variability and response to inflammatory stimuli. Cytokine. 2010;50:317–321. doi: 10.1016/j.cyto.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 108.Strioga M, Viswanathan S, Darinskas A, Slaby O, Michalek J. Same or not the same? Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells Dev. 2012;21:2724–2752. doi: 10.1089/scd.2011.0722. [DOI] [PubMed] [Google Scholar]
- 109.Zhao W, et al. Cell-surface sensors for real-time probing of cellular environments. Nat Nanotechnol. 2011;6:524–531. doi: 10.1038/nnano.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]