

Abstract
Objective
The objective of this study was to investigate whether cytosolic phospholipase A2 (cPLA2), an important isoform of PLA2 that mediates the release of arachidonic acid, plays a role in the pathogenesis of spinal cord injury (SCI).
Methods
A combination of molecular, histological, immunohistochemical, and behavioral assessments were used to test whether blocking cPLA2 activation pharmacologically or genetically reduced cell death, protected spinal cord tissue, and improved behavioral recovery after a contusive SCI performed at the 10th thoracic level in adult mice.
Results
SCI significantly increased cPLA2 expression and activation. Activated cPLA2 was localized mainly in neurons and oligodendrocytes. Notably, the SCI-induced cPLA2 activation was mediated by the extracellular signal-regulated kinase signaling pathway. In vitro, activation of cPLA2 by ceramide-1-phosphate or A23187 induced spinal neuronal death, which was substantially reversed by arachidonyl trifluoromethyl ketone, a cPLA2 inhibitor. Remarkably, blocking cPLA2 pharmacologically at 30 minutes postinjury or genetically deleting cPLA2 in mice ameliorated motor deficits, and reduced cell loss and tissue damage after SCI.
Interpretation
cPLA2 may play a key role in the pathogenesis of SCI, at least in the C57BL/6 mouse, and as such could be an attractive therapeutic target for ameliorating secondary tissue damage and promoting recovery of function after SCI.
Traumatic spinal cord injury (SCI) leads to neurological deficits and motor and sensory dysfunctions. In the United States alone, there were approximately 270,000 people living with SCI in 2012, and an additional 12,000 new SCI cases occur every year, most of them younger than 30 years (https://www.nscisc.uab.edu).1 To date, there is no effective pharmacological treatment for SCI.2 SCI is caused by mechanical damage that triggers cellular events culminating in the secondary injury phase, which provides an important therapeutic window for neuroprotective strategies to improve recovery of function after SCI. Previous studies indicate that multiple injury mechanisms, including inflammation, oxidative stress, and glutamate excitotoxicity,3–6 are involved in the secondary injury process after initial trauma, but exact mechanisms remain to be fully elucidated.
Several lines of evidence suggest that phospholipase A2 (PLA2) may play a key role in mediating multiple injury insults, as mentioned above, after SCI.7–11 PLA2 is a diverse family of enzymes that hydrolyze the acyl bond at the sn-2 position of glycerophospholipids to produce free fatty acids and lysophospholipids.8,12,13 These products are precursors of bioactive eicosanoids and platelet activating factor, which are well-known mediators of inflammation and tissue damage implicated in pathological states of several acute and chronic neurological disorders.8,12–14 Our previous study showed that PLA2 activity and expression increased after SCI in rats.15 Injections of exogenous PLA2 or melittin, a potent activator of endogenous PLA2, into the normal spinal cord resulted in inflammation and tissue damage.15 Administration of annexin A1, a nonselective inhibitor of PLA2, inhibited SCI-induced inflammation and reduced tissue damage after SCI.16 These findings suggest that PLA2 may be a potential therapeutic target for SCI.
PLA2 can be broadly classified into 3 major categories: secretory PLA2 (sPLA2), cytosolic PLA2 (cPLA2), and Ca2+-independent PLA2 (iPLA2).8 Among them, cPLA2 is considered to be the most important PLA2 isoform, because it has been implicated as an effector in receptor-mediated release of arachidonic acid (AA) and exhibits strong preference for deacylation of AA over other fatty acids.13,17 However, the role of cPLA2 in the pathogenesis of SCI has not yet been fully understood, and is even controversial.15,18 Here, we report that SCI significantly induced cPLA2 activation and expression. Blocking cPLA2 pharmacologically and genetically ameliorated motor deficits, and reduced cell loss and tissue damage after SCI in mice. Thus, cPLA2 may represent a therapeutic target for treatment after traumatic SCI.
Materials and Methods
All of the chemicals used in this study were from Sigma (St Louis, MO), except for those specifically indicated. Antibodies used in this study were from Cell Signaling Technology (Boston, MA), except for those specifically indicated.
Mice and Rats
Female C57BL/6 mice (12 weeks, 18–24g) were purchased from Jackson Laboratories (Bar Harbor, ME). Breeding pairs of male and female heterozygous (cPLA2+/−) mice were kindly provided by Dr J. Bonventre (Harvard Medical School). The breeding was carried out at Indiana University School of Medicine Laboratory Animal Resource Center. Female cPLA2−/− mice and wild-type (WT) littermates (12 weeks, 18–24g) generated from heterozygous breeding pairs were used in this study. The genotypes of the yielded litters were determined by polymerase chain reaction (PCR). All mice were on a C57/BL6 background.19,20 Female Sprague–Dawley rats (210–230g) were purchased from Harlan (Indianapolis, IN). Female animals are routinely used in SCI studies, because female animals allow for easier manual expression of bladders after SCI, less urinary tract infection, and less mortality.21–24 In addition, there are reports showing that no significant differences were detected in histological and behavioral outcomes between male and female animals after SCI.25,26 The animals were maintained on a 12/12-hour light/dark cycle with food and water freely available. All surgical interventions, treatments, and postoperative animal care were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council) and the Guidelines of the Institutional Animal Care and Use Committee of the Indiana University School of Medicine.
Contusive Spinal Cord Injury and Treatment
A contusive SCI in mice was performed at the T10 level using an Infinite Horizon Impactor (Infinite Horizons, Lexington, KY) at an impact force of 60 kdyne as described.27 The rats underwent a T10 contusive spinal cord injury using a New York University Impactor (10g, 12.5mm) as was described previously.16,28 After injury, the muscles and skin were closed in layers, and animals were placed in a temperature- and humidity-controlled chamber overnight. Manual bladder expression was carried out at least 3× daily until reflex bladder emptying was established. Control animals received laminectomy only. At 30 minutes after contusion injury, mice were treated with arachidonyl trifluoromethyl ketone (AACOCF3; Cayman Chemicals, Ann Arbor, MI), delivered intravenously (50μl of 4mM), followed by intraperitoneal injections of the compound (200μl of 4mM) every other day up to 2 weeks postinjury. The dose and treatment regimen were selected based on our pilot study and a previous published report.29 Another group of SCI animals received vehicle injections.
Western Blotting
Western blot analysis was performed as described previously with minor modification.16,28 For cPLA2 expression, primary antibodies included mouse monoclonal anti-cPLA2 antibody (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), polyclonal rabbit anti–phospho(p)-cPLA2 antibody (1:500), and mouse anti–β-tubulin antibody (1:1,000; Sigma). For active caspase-3 and poly(adenosine diphosphate ribose) polymerase (PARP) expression, primary antibodies included rabbit anti–caspase-3 antibody (cleaved, 1:1,000), rabbit anti–PARP-1 (cleaved, 1:500), and mouse anti–β-tubulin antibody (1:1,000; Sigma). For extracellular signal-regulated kinase (ERK) expression, primary antibodies included polyclonal rabbit anti-ERK1/2 antibody (1:1,000) and monoclonal mouse anti–p-ERK1/2 antibody (1:2,000). Secondary Alexa Fluor 680 goat antimouse (1:10,000; Invitrogen, Grand Island, NY) and IRDye 800 goat antirabbit (1:5,000; Rockland, Gilbertsville, PA) antibodies were used. The Western blot was imaged and quantified using a Li-Cor Odyssey Infrared Imaging system (LI-COR Biosciences, Lincoln, NE) according to the manufacturer's instruction.
Immunohistochemistry
Immunohistochemistry followed procedures described previously.15 One set of the sections was incubated with primary polyclonal rabbit anti–p-cPLA2 antibody (1:100) overnight at 4°C. On the second day, the sections were incubated with secondary biotinylated goat antirabbit immunoglobulin G antibody (1:400; Vector Laboratories, Burlingame, CA) for 1 hour at room temperature. Primary antibody omission controls were used to further confirm the specificity of the immunohistochemical labeling.
Immunofluorescence Double Labeling
This method has been described in our previous publication.15 Briefly, a mixture of rabbit polyclonal anti–phospho(p)-cPLA2 (1:100) and mouse anti-NeuN (1:100; Chemicon, Temecula, CA), anti–SMI-31 (1:2,000; Sigma), and anti-CC1 (APC-7, 1;100; Calbiochem, San Diego, CA) antibodies were used to examine colocalization of p-cPLA2 in neurons, axons, and oligodendrocytes, respectively. For colocalization of p-cPLA2 and p-ERK1/2, polyclonal anti–p-cPLA2 antibody (1:100) and monoclonal mouse anti–p-ERK1/2 antibody (1:400) were used. On the following day, the sections were incubated with fluorescein-conjugated goat antirabbit (1:100; ICN Biochemicals, Aurora, OH) and rhodamine-conjugated goat antimouse (1:100; ICN Biochemicals) antibodies. Primary antibody omission controls and cPLA2 knockout (KO) spinal cord sections were used to further confirm the specificity of the immunofluorescence double labeling, and secondary antibody omission controls were used to determine the degree of autofluorescence. Single-labeling controls were also used to assess any bleed-through. Images were acquired using an FluoView 500 Confocal Laser Scanning Microscope (Olympus America, Melville, NY) with a sequential scanning mode to minimize crosstalk among channels in multicolor images.
Intraspinal Injection of U0126
The bilateral microinjections (2 injections/side, 1μl/injection, total = 4μl) of vehicle (20% dimethylsulfoxide) or U0126 (0.5μg/μl) from Phoenix Pharmaceuticals (Burlingame, CA) were made into the spinal cord at 0.6mm from the midline and at a depth of 1.5mm from the dorsal cord surface on both sides using a glass micropipette attached to a pneumatic picopump (World Precision Instruments, Sarasota, FL). There was a 2mm distance between the 2 injections on each side.
Reverse Transcription PCR
Reverse transcription PCR (RT-PCR) was performed with the Access RT-PCR system (Promega, Madison, WI) according to the manufacturer's instructions. Sense primer 5′-AAG GCC AAG TGA CAC CAG CC-3′ and antisense primer 5′-GAA ACA GAG CAA CGA GAT GGG-3′ were used to yield a 452–base pair cPLA2 product. Primers for cyclophilin were used for control.
Spinal Cord Neuronal Culture, Cell Treatment, and Viability Assessment
Cells were obtained from embryonic day 14 rat spinal cords by gentle trituration according to our previously described protocol.15,16 Under this culture condition, a purity of >85% spinal cord neuronal population was obtained at the seventh day in vitro. Cultures were then treated with the designated concentration of ceramide-1-phosphate (C-1-P), A23187, and/or AACOCF3 for the designated time. The cultures were maintained for an additional 24 hours, and the culture medium of each well was removed for lactate dehydrogenase release assay using a CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega). In a subset of cultures, spinal cord neurons were treated using terminal deoxynucleotide transferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL) and immunofluorescent double labeling as well as Western blot.
TUNEL Assay
Apoptotic spinal cord neurons were detected by TUNEL and the immunostaining of the neuronal marker neurofilament protein (NFP) using an in situ cell death detection kit (TMR red; Roche Applied Science, Mannheim, Germany), according to the manufacturer's instructions. Primary rabbit polyclonal NFP antibody (1:400; Sigma) and secondary fluorescein-conjugated goat antirabbit antibody (1:100; ICN Biochemicals) were used.
Behavioral Assessments
All behavioral tests were blindly performed. The Basso Mouse Scale (BMS) locomotor test was performed weekly up to 6 weeks post-SCI by 2 observers lacking knowledge of the experimental groups according to a method published previously.30 Briefly, mice were placed in an open field (diameter = 42in) and observed for 4 minutes by 2 trained observers. The scores were on a scale of 0 to 9 (9 = normal locomotion; 0 = complete hind limb paralysis), which is based on hind limb movements made in an open field including hind limb joint movement, weight support, plantar stepping, coordination, paw position, and trunk and tail control.
Footprint analysis was used to examine the stepping patterns of the mice. The animals' hind paws were inked with blue dye, and the animals were required to traverse a narrow runway (100 × 4 × 4cm) lined with white paper. Only mice with frequently or consistently plantar stepping were tested (BMS score ≥ 5 for both hind limbs). Three separate traverses of the track (trials) were recorded per testing session. A minimum of 5 consecutive footprints were assessed to determine values for the trial, and the 3 trials were averaged to obtain the values for each parameter assessed per session. Six parameters including toe spread, paw length, paw rotation, stride length, stride width, and intermediary toes were analyzed.
Beam walking31 was evaluated in mice that showed frequent or consistent plantar stepping using a graded series of rough metal beams (24cm long) of various widths: 0.4, 0.8, 1.2, 1.6, and 2.0cm. The narrowest beam each mouse could traverse was recorded, along with the number of errors across 4 trials. Hind paw and whole body falls were both counted as errors. If an animal could not maintain placement of its hind paws on the beam, or if the animal was dragging its hindquarters across the beam, this was considered failing the task, and no score was recorded for the animal. The scoring was based on beam size and the number of errors.31 Data for this test were obtained by taking the average of 4 trials per beam per animal.
Histological Assessments
Spinal cord segments containing the epicenter were isolated from each animal, embedded, and cut into 25μm-thick serial sections (250μm apart and spanning the entire rostrocaudal extent of the lesion). One set of the sections was stained for myelin with Luxol fast blue, and the other was counterstained with cresyl violet–eosin. The lesion and spared white matter area of the injured cord were visualized, outlined, and quantified using an Olympus BX60 microscope equipped with a Neurolucida system (MicroBrightField, Colchester, VT). An unbiased estimation of the percentage of spared tissue and lesion volume were calculated using the Cavalieri method.16
Measurement of Na+, K+-Adenosine Triphosphatase Activity
Membrane-bound Na+, K+-adenosine triphosphatase (ATPase) was isolated as described previously with minor modification.32,33 Spinal cord tissue was homogenized in ice-cold isolation buffer (30mM histidine, 0.32M sucrose, and 1mM ethylenediaminetetraacetic acid [EDTA], pH 7.4). The homogenates were centrifuged at 1,000 × g for 20 minutes at 4°C. The pellet was discarded, and the supernatant was centrifuged at 14,000 × g for 60 minutes at 4°C. The pellet was resuspended in isolation buffer and stored at −80°C. The enzyme activity was assayed by the method previously described.34 Briefly, the Na+, K+-ATPase activity was assayed in an incubation medium consisting of 30mM histidine, 130mM NaCl, 20mM KCl, 5mM MgCl2, and 6.5μg membrane protein with or without 1mM ouabain. Inorganic phosphate was measured by the method of Fiske and Subbarow.34
Tissue of Prostaglandin E2 Determination
Tissue levels of prostaglandin E2 (PGE2) in the spinal cord were assayed using enzyme immunoassay (EIA; Prostaglandin E2 EIA kit; Cayman Chemical). Spinal cord segments containing the epicenter were removed and were homogenized in ice-cold lysis buffer (0.1M phosphate, pH 7.4, 1mM EDTA, 10μM indomethacin; Cayman Chemical) using a tube pestle. Acetone was added (2 × sample volume), and samples were centrifuged at 1,500 × g for 10 minutes. The supernatants were then stored at −80°C, and assay followed the manufacturer's instructions.
Myeloperoxidase Activity Assay
Myeloperoxidase (MPO) activity, an indicator of polymorphonuclear leukocyte accumulation, was performed as previously described.16 Briefly, the injured spinal cord segment (10mm) was removed and homogenized. The supernatant, after centrifugation at 14,000 × g for 25 minutes, was assayed for MPO activity.
PLA2 Activity Assay
A 10mm spinal cord segment containing the injury epicenter was dissected after intracardial perfusion of the mice with 10ml of saline under anesthesia. The cord segment was homogenized in 0.4ml of 50mM hydroxyethylpiperazine ethanesulfonic acid (HEPES), pH 7.4, containing 1mM EDTA and centrifuged at 10,000 × g for 15 minutes at 4°C. Supernatant was removed, and PLA2 activity was measured in the presence and absence of calcium according to the protocol in the PLA2 Assay Kits with minor modification (Cayman Chemical Company). Briefly, Total PLA2 activity was measured by incubating the samples with a substrate, arachidonoyl thio-PC, for 1 hour at room temperature in the assay buffer. The reactions were stopped by dithiobis nitrobenzoic acid (DTNB)/ethyleneglycoltetraacetic acid (EGTA) for 5 minutes, and the absorbances were determined at 405nm using a VICTOR3 V 1420 Multilabel Counter (PerkinElmer Wallac Oy, Turku, Finland). To detect the activity of iPLA2, the assay buffer was modified to Ca2+-free buffer (4mM EGTA, 160mM HEPES, pH 7.4, 300mM NaCl, 8mM Triton X-100, 60% glycerol, 2mg/ml of bovine serum albumin) as described previously.35 The iPLA2 activity was assayed by incubating the samples with the substrate, arachidonoyl thio-PC, for 1 hour at room temperature in the modified Ca2+-free buffer. The reaction was stopped by addition of DTNB/EGTA for 5 minutes, and the absorbance was determined at 405nm using the PerkinElmer VICTOR3 V 1420 Multilabel Counter. Activity of Ca+2-dependent PLA2 = total PLA2 activity − iPLA2 activity. Because C57BL/6 mice have a naturally occurring null mutation of the major form of sPLA2,36 Ca+2-dependent PLA2 activity reflects cPLA2 activity.
Statistical Analysis
All statistical analyses were performed using Prism software (version 6.00; GraphPad, La Jolla, CA) except for number of neurons. All data are presented as mean ± standard error of the mean values, and were analyzed by Student t tests or analysis of variance (ANOVA; 1-way, 2-way, or repeated measures as appropriate) followed by post hoc Dunnett or Tukey multiple comparison test. For the number of neurons, nonparametric repeated measures ANOVA was performed using SAS software (SAS Institute, Cary, NC), where the ranks of the outcome variable were used as the dependent variable, with group (WT and cPLA2 KO) and location as independent variables, including interaction. Potential correlation was adjusted for measurements obtained from the same animals. Bonferroni adjustment was used in post hoc analysis comparing WT to cPLA2 KO within each specific location. A p value of <0.05 was considered statistically significant.
Results
cPLA2 Activation in the Injured Spinal Cord following SCI
Mouse SCI models are being increasingly used because transgenic and KO mice are available for the study of cellular mechanisms. We found that cPLA2 expression in mice significantly increased after SCI, peaked at 7 days post-SCI, and remained highly expressed at 14 days (Fig 1). This profile of cPLA2 expression is consistent with our previous observation in rats.15 Because cPLA2 activation requires phosphorylation of cPLA2 by MAPK,17 we also examined p-cPLA2 as an indicator of cPLA2 activation. The p-cPLA2 expression was also significantly increased after SCI, with a similar expression profile to that of cPLA2. The expression of specific p-cPLA2 (ie, ratio of p-cPLA2/cPLA2) was also significantly increased at as early as 1 day and reached the highest at 14 days after SCI. Immunohistochemistry further revealed that the expression of p-cPLA2 increased as early as 8 hours post-SCI. Extensive p-cPLA2 expression was found in axons, particularly in those that underwent degeneration (see Fig 1E, right column, white matter) and in neurons and glial cells (see Fig 1E, left column, gray matter) between 1 and 7 days post-SCI. The p-cPLA2 expression was found not only in regions close to the injury (1.5mm) but also in areas distant from it (5mm). Immunofluorescence double labeling further confirmed that p-cPLA2 was expressed in neurons, swollen axons, and oligodendrocytes (Fig 2). The c-PLA2 expression and activation in mice was also confirmed in rats by immunohistochemistry (data not shown), protein,15 and mRNA (Fig 3) analyses.
Figure 1.
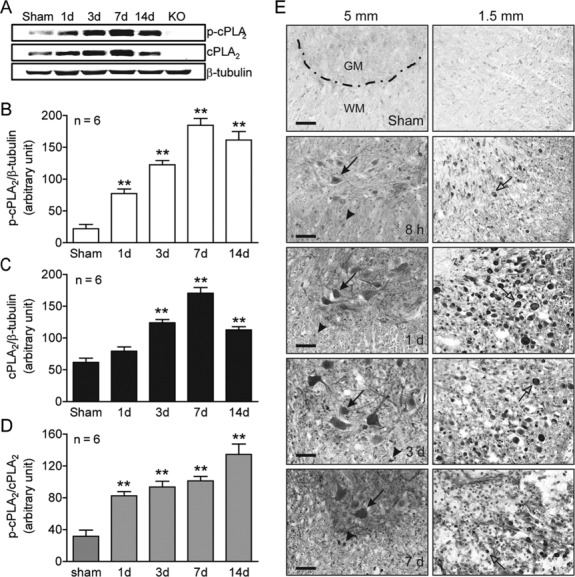
Cytosolic phospholipase A2 (cPLA2) activation following spinal cord injury. (A) Representative time courses of phosphorylated cPLA2 (p-cPLA2), cPLA2, and β-tubulin expression. (B) Compiled results in a bar graph for the ratio of p-cPLA2/β-tubulin expression. (C) Compiled results in a bar graph for the ratio of cPLA2/β-tubulin expression. (D) Compiled results in a bar graph for the ratio of p-cPLA2/cPLA2 expression. **p < 0.01 versus sham (1-way analysis of variance, Dunnett post hoc test, n = 6 mice/group). Error bars represent mean ± standard error of the mean. (E) Immunohistochemistry of mouse spinal cord sections at 1.5 and 5mm caudal to the injury epicenter shows increased p-cPLA2 expression in neurons (arrows), axons (open arrows), and glial cells (arrowheads). GM = gray matter; KO = knockout; WM = white matter. Bars = 40μm.
Figure 2.
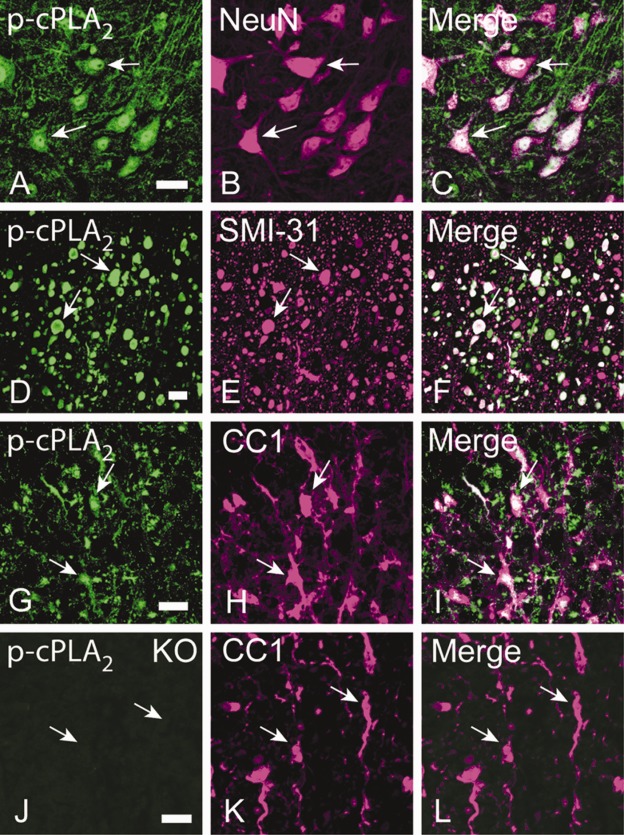
Cellular localization of phosphorylated cytosolic phospholipase A2 (p-cPLA2) expression in the mouse spinal cord after spinal cord injury (SCI). (A–C) p-cPLA2 immunoreactivity (IR) was localized in neurons indicated by NeuN IR (arrows). (D–F) p-cPLA2 IR was localized in a subpopulation of axons that show morphologically degenerating changes such as swelling (arrows). Axons were SMI-31 immunoreactive. (G–I) p-cPLA2 IR was localized in oligodendrocytes indicated by CC1 IR (arrows). (J–L) In spinal cord sections of cPLA2 knockout (KO) mice after SCI, p-cPLA2 IR was not observed, confirming the specificity of p-cPLA2 antibody (J). Bars = 20μm.
Figure 3.
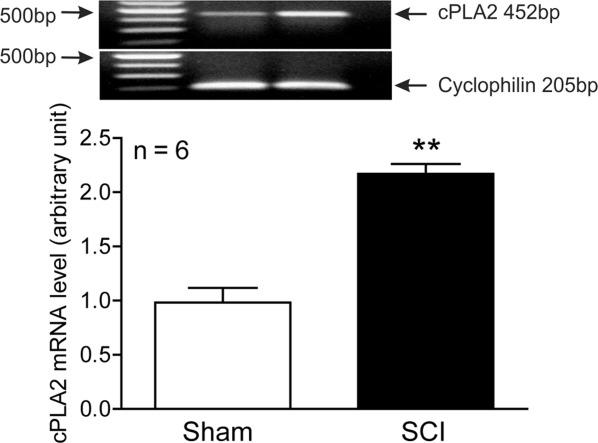
Expression of cytosolic phospholipase A2 (cPLA2) mRNA in the rat injured spinal cord at 7 days after spinal cord injury (SCI). Reverse transcription polymerase chain reaction was performed using specific primers designed for rat cPLA2 and cyclophilin. Bar graph indicates the mean ± standard error of the mean; n = 6 mice/group, **p < 0.01 versus sham-operated group (Student t test).
ERK1/2 Signaling Pathway Mediates SCI-Induced cPLA2 Phosphorylation In Vivo
To assess the mechanism of cPLA2 activation, we asked whether ERK1/2 signaling pathway plays a role in mediating cPLA2 phosphorylation. Our results showed colocalization of p-cPLA2 and p-ERK1/2 in neurons, degenerated axons, and glial cells at 24 hours after SCI (Fig 4). In sham-operated controls, p-ERK1/2 immunoreactivity (IR) at a very low level was observed in morphologically characteristic neurons; however, no p-cPLA2 IR was detected in these neurons or any other cells. Two-way ANOVA analysis showed that there were statistically significant effects of treatment group (F2,18 = 137.6, p < 0.0001), ERK1/2 (F1,18 = 83.38, p < 0.0001), and the interaction of treatment group and ERK1/2 (F2,18 = 11.86, p = 0.0005). Western blot analysis revealed that expressions of p-p44 (p-ERK1) and p-p42 (p-ERK2) were increased by 581.11% and 514.92%, respectively (p < 0.01) at 24 hours after SCI. Importantly, the increased expression of p-p44 and p-p42 were reversed by 59.82% (p < 0.01) and 41.61% (p < 0.05), respectively, by U0126, an ERK1/2 inhibitor. In the same animal model, SCI increased p-cPLA2 expression by 298.85% (p < 0.01). Interestingly, administration of the ERK1/2 inhibitor U0126 inhibited p-cPLA2 expression by 42.84% (p < 0.01).
Figure 4.
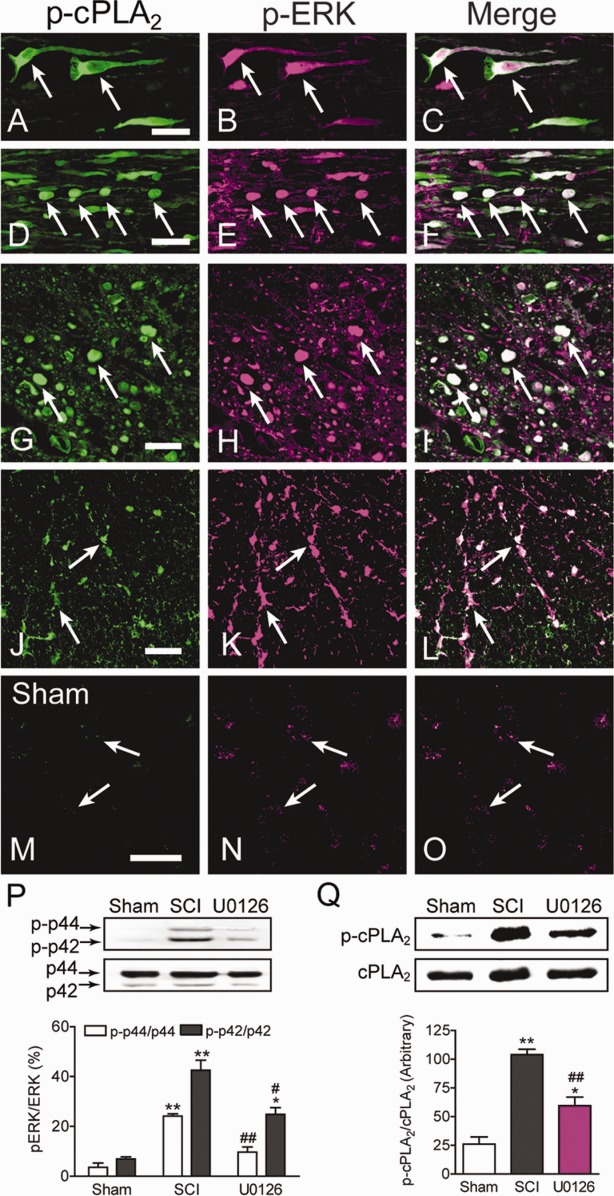
Extracellular signal-regulated kinase (ERK) 1/2 signaling pathway mediates cytosolic phospholipase A2 (cPLA2) phosphorylation induced by spinal cord injury (SCI). (A–L) Colocalization of phosphorylated cPLA2 (p-cPLA2) and phospho-ERK1/2 (p-ERK1/2) at 24 hours after SCI. (A–C) In a longitudinal section of the gray matter, neurons were positive for p-cPLA2 (A, arrows) and p-ERK1/2 (B, arrows). Coexistence of p-cPLA2 and p-ERK1/2 were in the same cells (C, arrows). (D–F) In a longitudinal section of the white matter, p-cPLA2 was colocalized with p-ERK1/2 in degenerated axons that showed beaded morphology (arrows). (G–L) In a cross section of the spinal cord, colocalization of p-cPLA2 and p-ERK1/2 was found mainly in axons undergoing degeneration (G–I, arrows) and in glial cells morphologically characteristic of oligodendrocytes (J–L, arrows). (M–O) In sham-operated controls, p-ERK1/2 immunoreactivity (IR) was observed at a very low level (N, arrows); however, no p-cPLA2 IR was detected in these cells (M, arrows). Bars = 40μm. (P) Representative photos and statistical comparison after normalization to ERK1/2. SCI induced p-ERK1/2 expression, which was reversed by U0126, a selective ERK1/2 inhibitor. *p < 0.05, **p < 0.01 versus sham; #p < 0.05, ##p < 0.01 versus vehicle-treated SCI (2-way analysis of variance, Tukey post hoc test); n = 4 mice/group; error bars represent mean ± standard error of the mean (SEM). (Q) Representative photos and statistical comparison after normalization to cPLA2. SCI induced p-cPLA2 expression, which was reversed by U0126, an ERK1/2 inhibitor. *p < 0.05, **p < 0.01 versus sham; ##p < 0.01 versus vehicle-treated SCI (1-way ANOVA, Tukey post hoc test); n = 4 mice/group; error bars represent mean ± SEM.
Activation of cPLA2 Induces Spinal Cord Neuronal Death In Vitro
Because we observed that cPLA2 activation was induced following SCI, the next question would be: could cPLA2 activation induce spinal cord neuronal death? To address this issue, we first examined the effects of C-1-P and A23187 on spinal cord neuronal death in vitro. C-1-P is a direct activator of cPLA2 through interaction with the CaLB/C2 domain.37 The calcium ionophore A23187 is an indirect activator of cPLA2 through elevations of intracellular free calcium.38 Our results showed that both C-1-P and A23187 induced cultured spinal neuronal death in a dose-dependent manner (Fig 5). Importantly, such C-1-P– or A23187-induced neuronal death could be significantly reversed by AACOCF3, a cPLA2 inhibitor. TUNEL staining revealed that C-1-P–induced spinal cord neuronal death took the form of apoptosis. Western blot analysis further confirmed that C-1-P–induced death of spinal cord neurons expressed apoptotic markers active caspase-3 (p < 0.05) and active PARP-1 (p < 0.01).
Figure 5.
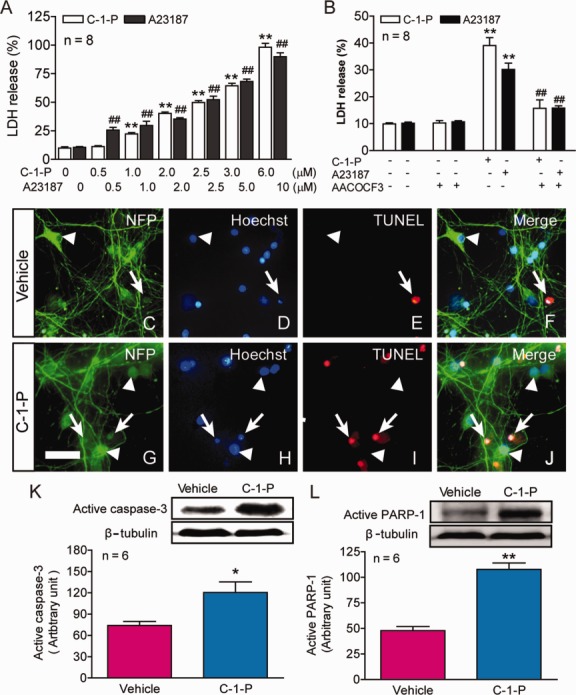
Effects of cytosolic phospholipase A2 (cPLA2) activation on spinal cord neuronal death in vitro. (A) Cultured spinal cord neurons were treated with the designated concentrations of ceramide-1-phosphate (C-1-P) or A23187 for 24 hours. Lactate dehydrogenase (LDH) release assay revealed that both cPLA2 activators, C-1-P and A23187, induced cultured spinal cord neuronal death in a dose-dependent manner. **p < 0.01, ##p < 0.01 versus the vehicle control, 1-way analysis of variance (ANOVA), Tukey post hoc test; n = 8. Data are shown as the mean ± standard error of the mean (SEM) from 3 independent experiments. (B) Importantly, neuronal death induced by C-1-P (2μM) or A23187 (5μM) was significantly reversed by AACOCF3 (15μM), a cPLA2 inhibitor. AACOCF3 was added 30 minutes before C-1-P or A23187, and the culture medium of each well was removed at 24 hours after the activator treatment for LDH release assay. **p < 0.01 versus the vehicle control, ##p < 0.01 versus the C-1-P or A23187 group, 2-way ANOVA, Tukey post hoc test, n = 8. Data are shown as the mean ± SEM from 3 independent experiments. (C–J) Administration of C-1-P (2.5μM) for 2.5 hours in vitro induced massive neuronal apoptosis (G–J) as compared to the vehicle controls (C–F). Apoptotic cells were identified by both Hoechst and triphosphate nick-end labeling (TUNEL) staining (arrows) and normal neurons were identified by staining for neurofilament protein (NFP) and Hoechst (arrowheads). Bar = 30μm. (K, L) C-1-P (2.5μM) for 2.5 hours also induced significant increase in the expression of active caspase-3 (K) and poly(adenosine diphosphate ribose) polymerase 1 (PARP-1) (L), two important components in the apoptotic pathway. *p < 0.05, **p < 0.01 versus the vehicle control (Student t test); n = 6. Data are shown as the mean ± SEM from 3 independent experiments.
Inhibition of cPLA2 Reduces SCI-Induced Tissue Damage and Improves Behavioral Recovery
To further assess whether activation of cPLA2 is sufficient to mediate the secondary SCI, we tested whether blocking cPLA2 activation with the cPLA2 inhibitor AACOCF3 would reduce injury-induced increases in eicosanoids (downstream metabolites of cPLA2), inflammation, and tissue damage and in turn enhance recovery after a contusive SCI in mice. AACOCF3 is a potent and selective inhibitor of cPLA2. This inhibitor shows slow tight binding to cPLA2 in the presence of Ca2+ and forms a covalent bond with a serine residue in the active site of the enzyme.39,40 This inhibitor is about 500-fold more potent at inhibiting cPLA2 than sPLA239 and may also be a weak inhibitor of iPLA2.41–43 AACOCF3 was delivered intravenously (50μl of 4mM) at 30 minutes in C57BL/6 mice after the injury followed by intraperitoneal injections of the compound (200μl of 4mM) every other day up to 2 weeks postinjury. Certain mouse strains, such as C57BL/6, 129/Sv, and B10.rIII, have a naturally occurring null mutation of the major form of sPLA2.36 Therefore, C57BL/6 mice in this study were deficient in sPLA2. To confirm cPLA2 inhibition in AACOCF3-treated mice, we measured PLA2 activity and its metabolite PGE2 at 24 hours after SCI. Our results showed that AACOCF3 treatment significantly reduced cPLA2 activity and PGE2 production by 42.9% (p < 0.01) and 35.1% (p < 0.01) after SCI (Fig 6). However, SCI-induced iPLA2 activation was not affected significantly by AACOCF3 (p > 0.05). AACOCF3 administration also restored Na+, K+-ATPase activity (a marker for membrane integrity or damage) by 38.1% (p < 0.05), and reduced SCI-induced MPO activity (a marker for neutrophil infiltration) by 67.1% (p < 0.01).
Figure 6.
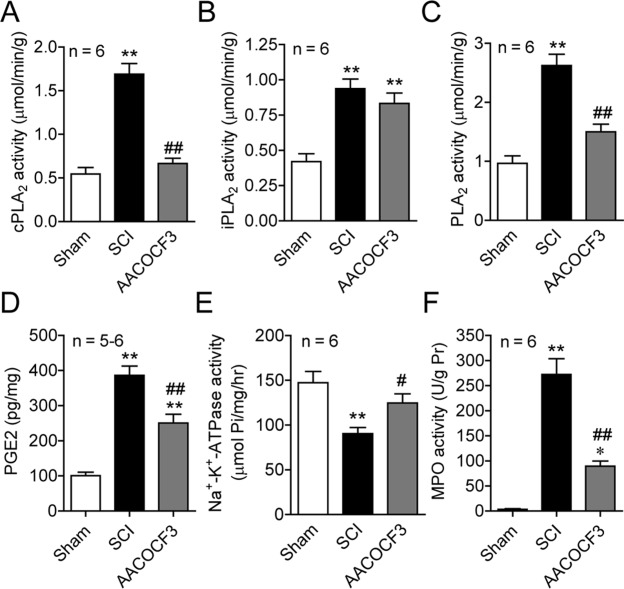
Pharmacological blockade of cytosolic phospholipase A2 (cPLA2) with AACOCF3 reduced cPLA2 activity, membrane injury, and inflammation induced by spinal cord injury (SCI). (A–C) AACOCF3 significantly inhibited an increase of cPLA2 (A) and total PLA2 (C) activities induced by SCI at 24 hours postinjury. However, AACOCF3 did not significantly affect SCI-induced Ca2+-independent PLA2 (iPLA2) activity (B). (D) AACOCF3 also inhibited an increase of prostaglandin E2 (PGE2), a downstream metabolite of PLA2, induced by SCI at 24 hours postinjury. (E) AACOCF3 also reversed the decrease of Na+, K+-adenosine triphosphatase (ATPase) activity at 24 hours after SCI. (F) AACOCF3 resulted in a decrease of myeloperoxidase (MPO) activity at 24 hours postinjury. *p < 0.05, **p < 0.01 versus sham; #p < 0.05, ##p < 0.01 versus vehicle-treated SCI (1-way analysis of variance, Tukey post hoc test); error bars represent mean ± standard error of the mean.
To determine whether inhibition of cPLA2 promotes functional recovery, an array of behavior tests were performed on consecutive days following SCI to evaluate motor and sensorimotor functions. There were statistically significant effects of treatment group (F2,21 = 40.17, p < 0.0001), test day (F7,147 = 53.32, p < 0.0001), and the interaction of treatment group and test day (F14,147 = 15.93, p < 0.0001; repeated measures ANOVA) for BMS for open field locomotion. AACOCF3 treatments significantly improved BMS scores for up to 6 weeks (Fig 7; p < 0.05–0.01). Repeated measures ANOVA also revealed that there were statistically significant effects of AACOCF3 treatment (F2,21 = 30.19, p < 0.0001) for beam walking scores at 4 and 6 weeks post-SCI. Footprint analysis showed that administration of AACOCF3 significantly improved the stride length (p < 0.05), paw rotation angle (p < 0.01), and intermediary toes (p < 0.01) at 5 weeks post-SCI.
Figure 7.
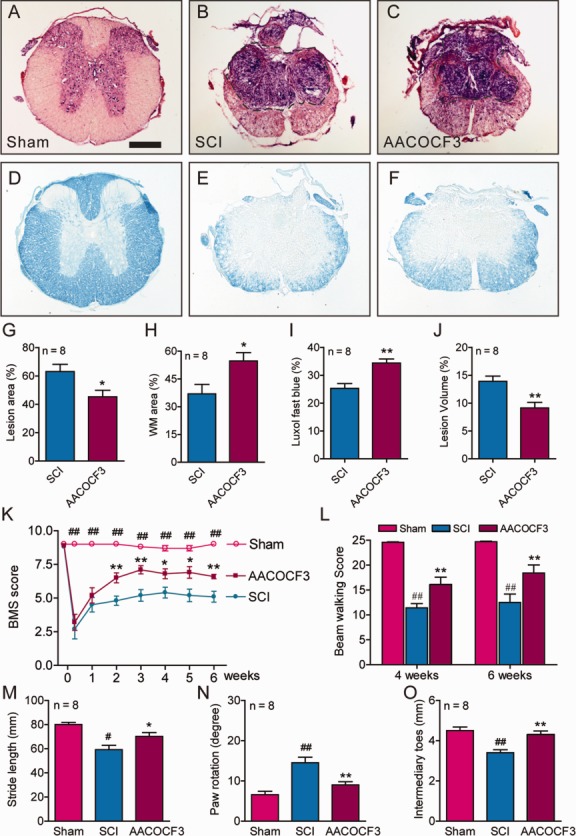
Pharmacological blockade of cytosolic phospholipase A2 (cPLA2) with AACOCF3 reduced tissue damage and improved behavioral recovery after spinal cord injury (SCI). (A–F) Representative sections show the lesion epicenter stained with cresyl violet and eosin (A–C) or with Luxol fast blue (D–F). AACOCF3 treatment significantly reduced lesion area by 28.1% (A–C, G, *p < 0.05), increased white matter (WM) sparing by 47.8% (A–C, H, *p < 0.05), enhanced myelin sparing by 35.5% (D–F, I, **p < 0.01), and reduced lesion volume by 34.4% (A–C, J, **p < 0.01). Bar = 300μm. (G–J) Student t test. (K) Administration of AACOCF3 significantly improved Basso Mouse Scale (BMS) locomotor scores up to 6 weeks post-SCI in C57BL/6 mice. *p < 0.05, **p < 0.01 versus vehicle-treated SCI; ##p < 0.01 versus vehicle- and AACOCF3-treated groups; repeated measures ANOVA, Tukey post hoc test. (L) AACOCF3 significantly increased beam walking score at 4 and 6 weeks postinjury. ##p < 0.01 versus sham; **p < 0.01 versus vehicle-treated SCI; repeated measures ANOVA, Tukey post hoc test). (M–O) AACOCF3 also significantly increased stride lengths (M), decreased paw rotation angles (N), and increased intermediary toes (O) in the foot print analysis at 5 weeks postinjury as compared to the vehicle-treated SCI. #p < 0.05, ##p < 0.01 versus sham; *p < 0.05, **p < 0.01 versus vehicle-treated SCI (1-way ANOVA, Tukey post hoc test). In G–O, n = 8 mice/group; error bars represent mean ± standard error of the mean.
Because we showed that administration of AACOCF3 significantly improved behavioral recovery after SCI, we next examined whether such a treatment also would result in tissue protection in vivo. To ensure that the entire rostrocaudal expansion of the lesion was examined, a 1.2cm-long cord segment was serially sectioned. Measurements of percentage total lesion volume, lesion area, and white matter sparing area were made from cresyl violet–stained and eosin-stained transverse sections spanning the entire lesion. Comparison of the lesion area at the injury epicenter demonstrated that AACOCF3 treatments resulted in a significant reduction of lesion area by 28.1% (see Fig 7; p < 0.05). Such reduction in lesion area was accompanied by a corresponding increase in the area of white matter sparing by 47.8% (p < 0.05) at 6 weeks post-SCI. In addition, Luxol fast blue staining showed that the AACOCF3 treatment resulted in a corresponding increase in myelin sparing by 35.5% (p < 0.01). Finally, stereological assessments of the lesion volume showed that AACOCF3 treatment resulted in a significant reduction in the percentage total lesion volume by 34.3% (p < 0.01).
Genetic Ablation of cPLA2 Reduces Cell Loss and Tissue Damage, and Improves Behavioral Recovery after SCI
To definitively determine the role of cPLA2 following SCI, we used cPLA2−/− mice and compared them with WT littermates (cPLA2+/+). All mice were on a C57/BL6 background and deficient in sPLA2.19 cPLA2−/− mice develop normally, gain weight at a rate equal to that of WT animals, and have a lifespan of >1 year.19,20 We found that at 24 hours after SCI, there was a marked loss of ventral horn neurons at and near the site of injury in the WT littermates (Fig 8). In contrast, in cPLA2 KO mice, the SCI-induced neuronal loss was significantly reduced at 24 hours postinjury. To test further whether cPLA2 ablation resulted in neuroprotection against cell death, particularly apoptosis, we examined the expression of active caspase-3, an enzyme known to be expressed following SCI and to be critically involved in the execution of the mammalian apoptotic cell death program.44 Western blot analysis revealed that SCI induced a significant increase of active caspase-3 expression (p < 0.01) in the WT littermates, which was significantly reduced by 67.1% (p < 0.01) in cPLA2 KO mice.
Figure 8.
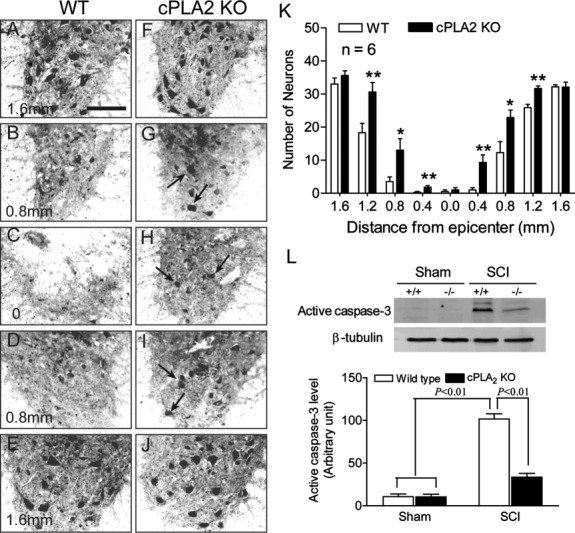
Cytosolic phospholipase A2 (cPLA2) ablation reduces neuronal loss induced by spinal cord injury (SCI) at 24 hours postinjury. (A–E) SCI induced a marked loss of ventral horn neurons (NeuN immunoreactivity) at and near the site of injury in a wild-type (WT) control mouse. (F-J) In contrast, numerous neurons survived the same injury at these regions such as at 0.8mm rostral (G) and caudal (I) to the injury epicenter (arrows). (K) Statistical analysis showed that significantly more neurons survived in the ventral gray matter of the cPLA2 knockout (KO) mice at distances 0.4 to 1.2mm rostral and caudal to the injury as compared to their WT littermates; n = 6 mice/group. *p < 0.05, **p < 0.01 versus WT (nonparametric repeated measures analysis of variance, Bonferroni post hoc test). (L) Expression of active caspase-3 expression at 24 hours after SCI. In cPLA2 KO mice, SCI-induced active caspase-3 expression was significantly reduced (p < 0.01), as compared to the WT group. Upper panel shows representative photograph of active caspase-3 expression; lower panel shows compilation of results in a bar graph; n = 6 mice/group. Data are shown as the mean ± standard error of the mean.
We also used an array of behavior tests to assess outcomes in cPLA2 KO mice and their WT littermates after SCI. The BMS locomotor scores were significantly improved in the cPLA2 KO mice (p < 0.05–0.01) for up to 6 weeks as compared to their WT littermates (Fig 9). The beam walking scores were also significantly improved in the cPLA2 KO mice (p < 0.05–0.01) at 4 and 6 weeks post-SCI as compared to their WT littermates. Footprint analysis showed that the toe spread (p < 0.05), stride width (p < 0.05), and base of support (p < 0.05) were all significantly improved in the cPLA2 KO mice at 5 weeks post-SCI as compared to their WT littermates.
Figure 9.
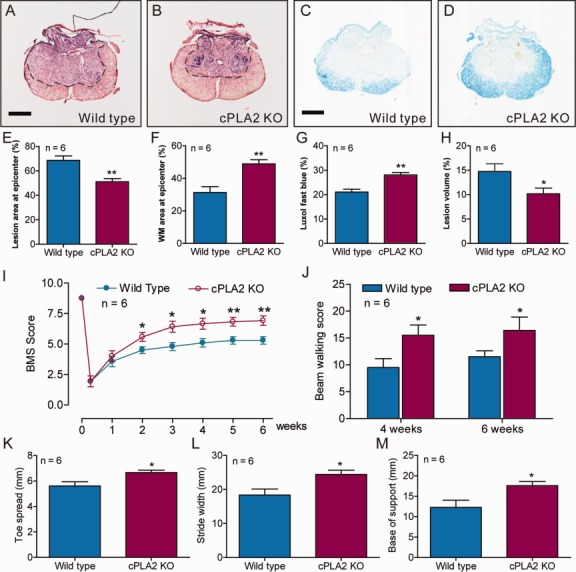
Cytosolic phospholipase A2 (cPLA2) ablation protects against tissue damage induced by spinal cord injury (SCI) and improves behavioral recovery in vivo. (A, B) Representative sections show the lesion epicenter stained with cresyl violet and eosin. (C, D) Representative sections show the lesion epicenter stained with Luxol fast blue. Bars = 300μm. (E–H) Bar graphs show that cPLA2 ablation reduced tissue damage (E), enhanced white matter (WM) sparing (F), increased myelin sparing (G), and reduced lesion volume (H) as compared to the wild-type controls. *p < 0.05, **p < 0.01 versus wild-type group, Student t test, n = 6 mice/group. Data are shown as the mean ± standard error of the mean (SEM). (I–M) Behavioral outcomes in wild-type and cPLA2 knockout (KO) mice after SCI. (I) Basso Mouse Scale (BMS) locomotor scores were significantly improved up to 6 weeks post-SCI in cPLA2 KO mice as compared to the wild-type controls (*p < 0.05, **p < 0.01, Student t test). (J) Beam walking scores were significantly increased at 4 and 6 weeks postinjury in cPLA2 KO mice as compared to the wild-type controls (*p < 0.05). (K–M) Toe spread (K), stride width (L), and base of support (M) in the foot print analysis were also significantly increased at 5 weeks postinjury as compared to the wild-type controls. *p < 0.05 versus the wild-type group (Student t test); n = 6 mice/group. Data are shown as the mean ± SEM.
Histological analysis further revealed that there was a significant reduction of lesion area by 25.6% (p < 0.01) in the cPLA2 KO mice compared to their WT littermates (see Fig 9). Such reduction in lesion area was accompanied by a corresponding increase in the area of white matter sparing by 56.2% (p < 0.01) in the cPLA2 KO mice. Luxol fast blue staining also showed that there was a corresponding increase in myelin sparing by 33.2% (p < 0.01) in the cPLA2 KO mice. Stereological assessments showed that there was a significant reduction in the percentage total lesion volume by 31.1% (p < 0.01) in the cPLA2 KO mice. Thus, genetic ablation of cPLA2 not only confirmed the previous observation of pharmacological inhibition of cPLA2 by AACOCF3, but also clearly indicated that cPLA2 could be an attractive target for intervention following SCI.
Discussion
The goal of this study was to determine whether targeting cPLA2 could be an effective strategy for functional repair after SCI. Our study showed that SCI induced an elevation of cPLA2 expression and activation. The elevated cPLA2 was localized mainly in neurons and oligodendrocytes. We also showed that the SCI-induced cPLA2 activation is mediated, at least in part, by ERK signal, revealing a molecular mechanism of cPLA2 activation. In vitro studies demonstrated that cPLA2 activation induced cultured spinal cord neuronal death. Most importantly, both pharmacological blockade and genetic deletion of cPLA2 significantly reduced inflammation, cell death, and tissue damage, as well as improved behavioral recovery after SCI. These findings collectively suggest that modulation of cPLA2 could represent a new therapeutic strategy for treatment of SCI.
SCI significantly induced cPLA2 activation, which was observed as early as 8 hours postinjury and peaked at 7 days postinjury. The activated cPLA2 was mainly localized in neurons and oligodendrocytes. The expression of cPLA2 mRNA was increased in the injured cord, which correlated well with increased expression of cPLA2 protein. Several earlier investigators found that AA and eicosanoids, metabolites of cPLA2, increased within 30 minutes after SCI.45,46 Others reported that increased eicosanoids were persistent at least for 3 days (the longest time point studied) after SCI.47 Furthermore, Demediuk et al reported that induced concentrations of free fatty acid quickly increased after SCI, peaked at 3 days, and remained significantly high at 7 days after SCI.48 The induction profiles of these PLA2 metabolites are similar to that of cPLA2 after SCI in the present study. These results indicate that a prolonged effect of cPLA2 exists after SCI, which suggests that there may be a unique therapeutic window for intervention. The finding that cPLA2 was mainly localized in neurons and oligodendrocytes is particularly interesting, because these 2 cell types not only play important roles in normal central nervous system (CNS) function but also are the most vulnerable cell types to injuries as compared to other CNS cell types such as astrocytes and microglia.49
Although cPLA2 activation and expression were significantly increased after SCI, the mechanism(s) by which they were increased remains to be determined. Our in vivo experiments revealed that ERK1/2 signaling pathway mediated SCI-induced cPLA2 activation. Our previous in vitro experiments7 also showed that ERK1/2 signaling pathway mediated cPLA2 phosphorylation, induced by glutamate and H2O2, two important injury mediators of secondary SCI5. We and others have reported that cPLA2 is induced by several toxic factors that are generated in the injured cord, including inflammatory cytokines,8,17 free radicals,7–9,11 and excitatory amino acids.7,8,10 Therefore, cPLA2 may serve as a central or convergence molecule that mediates multiple key mechanisms of secondary injury, making it an attractive therapeutic target to improve tissue protection and function recovery.
Our results clearly demonstrated that cPLA2 activation induced spinal cord neuronal death. This was in agreement with our previous finding that cPLA2 activation mediated cultured spinal cord neuronal death induced by glutamate and H2O2.7 Apoptosis has been considered a key mechanism of cell death following SCI.49,50 Caspase-3 plays a central role in the execution phase of apoptosis and is responsible for the cleavage of proteins such as the nuclear enzyme PARP. Our results showed that cPLA2 activation induced the expression of active caspase-3 and PARP-1. TUNEL staining further confirmed that cPLA2 activation induced neuronal apoptosis, which was supported by cPLA2-mediated neural apoptosis induced by Aβ.51 These results suggest that cPLA2 activation induced neuronal death through apoptosis, at least in part.
A significant finding of this study is that pharmacological blockade of cPLA2 with AACOCF3 inhibited inflammation and membrane injury, reduced tissue damage, and improved behavioral recovery in C57BL/6 mice after SCI. Notably, the cPLA2 inhibitor was administered after trauma. Our results showed a long beneficial effect of targeting cPLA2 on anatomical and functional recoveries. In agreement with our observation, several studies have reported a detrimental effect of cPLA2 in other CNS diseases such as ischemia,20,52 experimental autoimmune encephalomyelitis,29,53 and Alzheimer disease.54 In contrast, there is a recent report showing that activation of cPLA2 is beneficial. In that study, both BALB/c mice treated with AX059, a cPLA2 inhibitor, and cPLA2-null BALB/c mice displayed greater neuronal and myelin loss after SCI.18 The contrary results between this mouse strain and others remain unclear, and they may be related to different mouse backgrounds (C57BL/6 vs BALB/c) and inhibitors (AACOCF3 vs AX059) that were used. It has been reported that different strains of mice display distinctly different responses to trauma injury, including inflammation, histology, and behavioral recovery.30,55–57 For example, post-traumatic inflammation was markedly reduced in BALB/c mice compared with C57BL/6 mice.55 After SCI, a densely packed cellular matrix fills necrotic cavities. The magnitude of this response was greatest for C57BL/6 mice and least for BALB/c mice.55 Kipnis and colleagues also showed that BALB/c mice exhibited a T-cell–dependent neuroprotective response, whereas trauma- or glutamate-mediated neuronal cell loss was enhanced in C57BL/6 mice.57 These results suggest that genetic differences may confer different responses to traumatic injury, which may modify the secondary injury processes after SCI. Thus, the contrary results from C57BL/6 and BALB/c mice may be related to genetic differences including sPLA2.
We previously demonstrated increased sPLA2 expression following SCI.9 Injection of sPLA2 into the normal spinal cord resulted in tissue damage, demyelination, and behavioral impairment in vivo.15 Importantly, administration of a sPLA2 inhibitor GK511 in BALB/c mice reduced tissue damage and improved behavioral recovery after SCI.18 In the current study, sPLA2 action was excluded in both sham and SCI groups, because C57BL/6 mice have a naturally occurring null mutation of sPLA2.36 iPLA2 is generally considered to be a housekeeping enzyme for the maintenance of membrane phospholipids.8 Lopez-Vales et al reported that iPLA2 was upregulated after SCI and was expressed mainly in oligodendrocytes.18 Using FKGK11, a potent and highly selective iPLA2 inhibitor in BALB/c mice with SCI, they further showed that iPLA2 appeared to play a minor detrimental role in SCI.18 Although AACOCF3 has been reported to be a weak inhibitor of iPLA2,41–43 our results showed that there was no significant difference of iPLA2 activity between the SCI and AACOCF3-treated groups. These results suggest that AACOCF3 exerts neuroprotection mainly via inhibition of cPLA2.
A definitive finding of the present study is that genetic deletion of cPLA2 resulted in neuroprotection and behavioral recovery following SCI. Genetic deletion of cPLA2 also inhibited the expression of active caspase-3 after SCI, suggesting that cPLA2 activation mediates neural apoptosis. Our observation is supported by several other reports that cPLA2−/− mice show protection in ischemic brain damage, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity, and neurodegeneration.20,54,58 cPLA2−/− mice also show significant reductions in AA release and eicosanoid production in response to a variety of stimuli.19,20 cPLA2 may contribute to injury by a direct effect on cell membranes and/or indirectly through generation of its metabolites, which are inflammatory and vasoconstrictive mediators.8,59 Our results suggest that cPLA2 contributes to the pathogenesis of SCI and that targeting cPLA2 could be an effective therapeutic strategy for intervention after SCI.
Acknowledgments
This work was supported by the NIH NINDS (1R01 NS059622, 1R01 NS050243, 1R01 NS073636; X.-M.X.), the Indiana Clinical and Translational Sciences Institute Collaboration in Biomedical/Translational Research Pilot Grant Program (RR025761; X.-M.X.), the Indiana Spinal Cord and Brain Injury Research Foundation, Mari Hulman George Endowment Funds (X.-M.X.), the Indiana State Department of Health (A70-2-079609, A70-9-079138; N.-K.L.), and the NIH National Institute of Diabetes and Digestive and Kidney Diseases (DK39773, DK 054741; J.V.B.).
We thank Dr S. Gao, a biostatistician, for statistical assistance; P. Raley, a medical editor, for critical reading of the manuscript; and Dr C. L. Hammer, a medical fellow, for some in vitro experiments.
Authorship
N.-K.L. designed experiments, performed experiments, analyzed data, and wrote the paper. L.-X.D. and J.-G.H. performed in vitro experiments. Y.P.Z., Q.-B.L., and X.-F.W. performed in vivo experiments. E.O. performed some behavioral assessments. J.V.B. provided cPLA2 knockout mice and edited the manuscript. C.B.S. designed and edited the manuscript. X.-M.X. designed experiments, reviewed the data, and wrote the manuscript.
Potential Conflicts of Interest
Nothing to report.
References
- 1.Spinal cord injury facts and figures at a glance. J Spinal Cord Med. 2012;35:480–481. doi: 10.1179/1079026812Z.000000000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rabchevsky AG, Patel SP, Springer JE. Pharmacological interventions for spinal cord injury: where do we stand? How might we step forward? Pharmacol Ther. 2011;132:15–29. doi: 10.1016/j.pharmthera.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Young W. Secondary injury mechanisms in acute spinal cord injury. J Emerg Med. 1993;11(suppl 1):13–22. [PubMed] [Google Scholar]
- 4.Hall ED, Braughler JM. Role of lipid peroxidation in post-traumatic spinal cord degeneration: a review. Cent Nerv Syst Trauma. 1986;3:281–294. doi: 10.1089/cns.1986.3.281. [DOI] [PubMed] [Google Scholar]
- 5.Park E, Velumian AA, Fehlings MG. The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma. 2004;21:754–774. doi: 10.1089/0897715041269641. [DOI] [PubMed] [Google Scholar]
- 6.Buki A, Okonkwo DO, Wang KK, Povlishock JT. Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci. 2000;20:2825–2834. doi: 10.1523/JNEUROSCI.20-08-02825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao Z, Liu N, Huang J. Inhibition of cPLA2 activation by Ginkgo biloba extract protects spinal cord neurons from glutamate excitotoxicity and oxidative stress-induced cell death. J Neurochem. 2011;116:1057–1065. doi: 10.1111/j.1471-4159.2010.07160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu NK, Xu XM. Phospholipase A2 and its molecular mechanism after spinal cord injury. Mol Neurobiol. 2010;41:197–205. doi: 10.1007/s12035-010-8101-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Titsworth WL, Cheng X, Ke Y. Differential expression of sPLA2 following spinal cord injury and a functional role for sPLA2-IIA in mediating oligodendrocyte death. Glia. 2009;57:1521–1537. doi: 10.1002/glia.20867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein J. Membrane breakdown in acute and chronic neurodegeneration: focus on choline-containing phospholipids. J Neural Transm. 2000;107:1027–1063. doi: 10.1007/s007020070051. [DOI] [PubMed] [Google Scholar]
- 11.van Rossum GS, Drummen GP, Verkleij AJ. Activation of cytosolic phospholipase A2 in Her14 fibroblasts by hydrogen peroxide: a p42/44(MAPK)-dependent and phosphorylation-independent mechanism. Biochim Biophys Acta. 2004;1636:183–195. doi: 10.1016/j.bbalip.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 12.Titsworth WL, Liu NK, Xu XM. Role of secretory phospholipase a(2) in CNS inflammation: implications in traumatic spinal cord injury. CNS Neurol Disord Drug Targets. 2008;7:254–269. doi: 10.2174/187152708784936671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farooqui AA, Yang HC, Rosenberger TA, Horrocks LA. Phospholipase A2 and its role in brain tissue. J Neurochem. 1997;69:889–901. doi: 10.1046/j.1471-4159.1997.69030889.x. [DOI] [PubMed] [Google Scholar]
- 14.Bonventre JV. Roles of phospholipases A2 in brain cell and tissue injury associated with ischemia and excitotoxicity. J Lipid Mediat Cell Signal. 1996;14:15–23. doi: 10.1016/0929-7855(96)00503-2. [DOI] [PubMed] [Google Scholar]
- 15.Liu NK, Zhang YP, Titsworth WL. A novel role of phospholipase A2 in mediating spinal cord secondary injury. Ann Neurol. 2006;59:606–619. doi: 10.1002/ana.20798. [DOI] [PubMed] [Google Scholar]
- 16.Liu NK, Zhang YP, Han S. Annexin A1 reduces inflammatory reaction and tissue damage through inhibition of phospholipase A2 activation in adult rats following spinal cord injury. J Neuropathol Exp Neurol. 2007;66:932–943. doi: 10.1097/nen.0b013e3181567d59. [DOI] [PubMed] [Google Scholar]
- 17.Clark JD, Schievella AR, Nalefski EA, Lin LL. Cytosolic phospholipase A2. J Lipid Mediat Cell Signal. 1995;12:83–117. doi: 10.1016/0929-7855(95)00012-f. [DOI] [PubMed] [Google Scholar]
- 18.Lopez-Vales R, Ghasemlou N, Redensek A. Phospholipase A2 superfamily members play divergent roles after spinal cord injury. FASEB J. 2011;25:4240–4252. doi: 10.1096/fj.11-183186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sapirstein A, Bonventre JV. Specific physiological roles of cytosolic phospholipase A(2) as defined by gene knockouts. Biochim Biophys Acta. 2000;1488:139–148. doi: 10.1016/s1388-1981(00)00116-5. [DOI] [PubMed] [Google Scholar]
- 20.Bonventre JV, Huang Z, Taheri MR. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 21.Khan T, Havey RM, Sayers ST. Animal models of spinal cord contusion injuries. Lab Anim Sci. 1999;49:161–172. [PubMed] [Google Scholar]
- 22.Widenfalk J, Lundstromer K, Jubran M. Neurotrophic factors and receptors in the immature and adult spinal cord after mechanical injury or kainic acid. J Neurosci. 2001;21:3457–3475. doi: 10.1523/JNEUROSCI.21-10-03457.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lasiene J, Shupe L, Perlmutter S, Horner P. No evidence for chronic demyelination in spared axons after spinal cord injury in a mouse. J Neurosci. 2008;28:3887–3896. doi: 10.1523/JNEUROSCI.4756-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker KA, Hagg T. An adult rat spinal cord contusion model of sensory axon degeneration: the estrus cycle or a preconditioning lesion do not affect outcome. J Neurotrauma. 2005;22:415–428. doi: 10.1089/neu.2005.22.415. [DOI] [PubMed] [Google Scholar]
- 25.Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp Neurol. 1996;139:244–256. doi: 10.1006/exnr.1996.0098. [DOI] [PubMed] [Google Scholar]
- 26.Luchetti S, Beck KD, Galvan MD. Comparison of immunopathology and locomotor recovery in C57BL/6, BUB/BnJ, and NOD-SCID mice after contusion spinal cord injury. J Neurotrauma. 2010;27:411–421. doi: 10.1089/neu.2009.0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engesser-Cesar C, Anderson AJ, Basso DM. Voluntary wheel running improves recovery from a moderate spinal cord injury. J Neurotrauma. 2005;22:157–171. doi: 10.1089/neu.2005.22.157. [DOI] [PubMed] [Google Scholar]
- 28.Liu N, Han S, Lu PH, Xu XM. Upregulation of annexins I, II, and V after traumatic spinal cord injury in adult rats. J Neurosci Res. 2004;77:391–401. doi: 10.1002/jnr.20167. [DOI] [PubMed] [Google Scholar]
- 29.Kalyvas A, David S. Cytosolic phospholipase A2 plays a key role in the pathogenesis of multiple sclerosis-like disease. Neuron. 2004;41:323–335. doi: 10.1016/s0896-6273(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 30.Basso DM, Fisher LC, Anderson AJ. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- 31.Hill RL, Zhang YP, Burke DA. Anatomical and functional outcomes following a precise, graded, dorsal laceration spinal cord injury in C57BL/6 mice. J Neurotrauma. 2009;26:1–15. doi: 10.1089/neu.2008.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skou JC, Esmann M. Preparation of membrane-bound and of solubilized (Na+ + K+)-ATPase from rectal glands of Squalus acanthias. The effect of preparative procedures on purity, specific and molar activity. Biochim Biophys Acta. 1979;567:436–444. doi: 10.1016/0005-2744(79)90129-3. [DOI] [PubMed] [Google Scholar]
- 33.Sweadner KJ. Purification from brain of an intrinsic membrane protein fraction enriched in (Na+ + K+)-ATPase. Biochim Biophys Acta. 1978;508:486–499. doi: 10.1016/0005-2736(78)90094-9. [DOI] [PubMed] [Google Scholar]
- 34.Esmann M. ATPase and phosphatase activity of Na+,K+-ATPase: molar and specific activity, protein determination. Methods Enzymol. 1988;156:105–115. doi: 10.1016/0076-6879(88)56013-5. [DOI] [PubMed] [Google Scholar]
- 35.Smani T, Zakharov SI, Leno E. Ca2+-independent phospholipase A2 is a novel determinant of store-operated Ca2+ entry. J Biol Chem. 2003;278:11909–11915. doi: 10.1074/jbc.M210878200. [DOI] [PubMed] [Google Scholar]
- 36.Kennedy BP, Payette P, Mudgett J. A natural disruption of the secretory group II phospholipase A2 gene in inbred mouse strains. J Biol Chem. 1995;270:22378–22385. doi: 10.1074/jbc.270.38.22378. [DOI] [PubMed] [Google Scholar]
- 37.Pettus BJ, Bielawska A, Subramanian P. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J Biol Chem. 2004;279:11320–11326. doi: 10.1074/jbc.M309262200. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura H, Wakita S, Suganami A. Modulation of the activity of cytosolic phospholipase A2alpha (cPLA2alpha) by cellular sphingolipids and inhibition of cPLA2alpha by sphingomyelin. J Lipid Res. 2010;51:720–728. doi: 10.1194/jlr.M002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Street IP, Lin HK, Laliberte F. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry. 1993;32:5935–5940. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- 40.Trimble LA, Street IP, Perrier H. NMR structural studies of the tight complex between a trifluoromethyl ketone inhibitor and the 85-kDa human phospholipase A2. Biochemistry. 1993;32:12560–12565. doi: 10.1021/bi00210a002. [DOI] [PubMed] [Google Scholar]
- 41.Riendeau D, Guay J, Weech PK. Arachidonyl trifluoromethyl ketone, a potent inhibitor of 85-kDa phospholipase A2, blocks production of arachidonate and 12-hydroxyeicosatetraenoic acid by calcium ionophore-challenged platelets. J Biol Chem. 1994;269:15619–15624. [PubMed] [Google Scholar]
- 42.Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca(2+)-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- 43.Ghomashchi F, Loo R, Balsinde J. Trifluoromethyl ketones and methyl fluorophosphonates as inhibitors of group IV and VI phospholipases A(2): structure-function studies with vesicle, micelle, and membrane assays. Biochim Biophys Acta. 1999;1420:45–56. doi: 10.1016/s0005-2736(99)00056-5. [DOI] [PubMed] [Google Scholar]
- 44.Springer JE, Azbill RD, Knapp PE. Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat Med. 1999;5:943–946. doi: 10.1038/11387. [DOI] [PubMed] [Google Scholar]
- 45.Demediuk P, Saunders RD, Anderson DK. Early membrane lipid changes in laminectomized and traumatized cat spinal cord. Neurochem Pathol. 1987;7:79–89. doi: 10.1007/BF02834293. [DOI] [PubMed] [Google Scholar]
- 46.Demediuk P, Saunders RD, Anderson DK. Membrane lipid changes in laminectomized and traumatized cat spinal cord. Proc Natl Acad Sci U S A. 1985;82:7071–7075. doi: 10.1073/pnas.82.20.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Resnick DK, Nguyen P, Cechvala CF. Regional and temporal changes in prostaglandin E2 and thromboxane B2 concentrations after spinal cord injury. Spine J. 2001;1:432–436. doi: 10.1016/s1529-9430(01)00130-9. [DOI] [PubMed] [Google Scholar]
- 48.Demediuk P, Daly MP, Faden AI. Changes in free fatty acids, phospholipids, and cholesterol following impact injury to the rat spinal cord. J Neurosci Res. 1989;23:95–106. doi: 10.1002/jnr.490230113. [DOI] [PubMed] [Google Scholar]
- 49.Liu XZ, Xu XM, Hu R. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crowe MJ, Bresnahan JC, Shuman SL. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- 51.Kriem B, Sponne I, Fifre A. Cytosolic phospholipase A2 mediates neuronal apoptosis induced by soluble oligomers of the amyloid-beta peptide. FASEB J. 2005;19:85–87. doi: 10.1096/fj.04-1807fje. [DOI] [PubMed] [Google Scholar]
- 52.Tabuchi S, Uozumi N, Ishii S. Mice deficient in cytosolic phospholipase A2 are less susceptible to cerebral ischemia/reperfusion injury. Acta Neurochir Suppl. 2003;86:169–172. doi: 10.1007/978-3-7091-0651-8_36. [DOI] [PubMed] [Google Scholar]
- 53.Marusic S, Leach MW, Pelker JW. Cytosolic phospholipase A2 alpha-deficient mice are resistant to experimental autoimmune encephalomyelitis. J Exp Med. 2005;202:841–851. doi: 10.1084/jem.20050665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanchez-Mejia RO, Newman JW, Toh S. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer's disease. Nat Neurosci. 2008;11:1311–1318. doi: 10.1038/nn.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol. 2006;494:578–594. doi: 10.1002/cne.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Inman D, Guth L, Steward O. Genetic influences on secondary degeneration and wound healing following spinal cord injury in various strains of mice. J Comp Neurol. 2002;451:225–235. doi: 10.1002/cne.10340. [DOI] [PubMed] [Google Scholar]
- 57.Kipnis J, Yoles E, Schori H. Neuronal survival after CNS insult is determined by a genetically encoded autoimmune response. J Neurosci. 2001;21:4564–4571. doi: 10.1523/JNEUROSCI.21-13-04564.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klivenyi P, Beal MF, Ferrante RJ. Mice deficient in group IV cytosolic phospholipase A2 are resistant to MPTP neurotoxicity. J Neurochem. 1998;71:2634–2637. doi: 10.1046/j.1471-4159.1998.71062634.x. [DOI] [PubMed] [Google Scholar]
- 59.Sapirstein A, Bonventre JV. Phospholipases A2 in ischemic and toxic brain injury. Neurochem Res. 2000;25:745–753. doi: 10.1023/a:1007583708713. [DOI] [PubMed] [Google Scholar]