

Abstract
Upon ingestion of contaminated food, Listeria monocytogenes can cause serious infections in humans that are normally treated with β-lactam antibiotics. These target Listeria's five high molecular weight penicillin-binding proteins (HMW PBPs), which are required for peptidoglycan biosynthesis. The two bi-functional class A HMW PBPs PBP A1 and PBP A2 have transglycosylase and transpeptidase domains catalyzing glycan chain polymerization and peptide cross-linking, respectively, whereas the three class B HMW PBPs B1, B2 and B3 are monofunctional transpeptidases. The precise roles of these PBPs in the cell cycle are unknown. Here we show that green fluorescent protein (GFP)-PBP fusions localized either at the septum, the lateral wall or both, suggesting distinct and overlapping functions. Genetic data confirmed this view: PBP A1 and PBP A2 could not be inactivated simultaneously, and a conditional double mutant strain is largely inducer dependent. PBP B1 is required for rod-shape and PBP B2 for cross-wall biosynthesis and viability, whereas PBP B3 is dispensable for growth and cell division. PBP B1 depletion dramatically increased β-lactam susceptibilities and stimulated spontaneous autolysis but had no effect on peptidoglycan cross-linkage. Our in vitro virulence assays indicated that the complete set of all HMW PBPs is required for maximal virulence.
Introduction
Listeria monocytogenes is an ubiquitously occurring Gram-positive rod belonging to the firmicutes. It is present in the soil, on plant surfaces or decaying plant material, where it lives as a saprophyte (Freitag et al., 2009). However, the bacterium is also able to cause infections in humans upon ingestion of contaminated food. Listeriosis involves self-limiting gastrointestinal symptoms in otherwise healthy individuals but can also develop into more systemic conditions, primarily affecting the brain as well as the fetus in pregnant women (Allerberger and Wagner, 2010). In such severe invasive cases, mortality rates of up to 30% have been reported despite antibiotic therapy (Swaminathan and Gerner-Smidt, 2007). L. monocytogenes can induce its uptake into the cytosol of nonphagocytic human host cells via a transient passage through a primary vacuole. The bacterium multiplies inside the host cell cytoplasm from where it can even spread into neighboring cells. This mechanism enables the bacterium to breach all main barriers of the human body (Cossart and Toledo-Arana, 2008). L. monocytogenes is sufficiently susceptible to a wide range of antibiotics in vitro, but its intracellular growth complicates the antimicrobial treatment of listeriosis due to poor accessibility of antibiotics (Hof, 2004). This is why listeriosis generally requires therapy with high doses of β-lactam antibiotics such as ampicillin or amoxicillin. This treatment can be combined with gentamycin in nonpregnancy cases to achieve synergistic killing effects, as the β-lactams and gentamycin alone are bacteriostatic, whereas the combination of both have bactericidal effects at least in vitro (Hof2003;2004; Allerberger and Wagner, 2010). However, the efficacy of a dual gentamicin-ampicillin therapy has been challenged by a recent epidemiological study, which could not confirm any beneficial effects of such a combination used in a retrospective study on more than hundred cases of invasive listeriosis (Munoz et al., 2012). As alternative drugs for anti-listerial treatment, meropenem and cotrimoxazole have also been used with some success (Grant et al., 2010; Matano et al., 2010; Munoz et al., 2012).
β-lactam antibiotics covalently bind to and block the active site serine in the transpeptidase domain of penicillin-binding proteins (PBPs) and can therefore be used for their detection using radioactively labeled penicillin. Early studies using this technique uncovered five PBPs in cellular extracts of L. monocytogenes, initially denoted PBP1-5 (Vicente et al., 1990). Sequence analysis of the L. monocytogenes EGD-e genome (Glaser et al., 2001) has later confirmed the existence of five high molecular weight (HMW) PBPs and five low molecular weight PBPs (Guinane et al., 2006; Bierne and Cossart, 2007; Korsak et al., 2010). The five HMW PBPs are also present in the genome of L. innocua strain CLIP 11262 and in the 45 L. monocytogenes strains for which completely assembled genomes are available at the NCBI server (http://blast.ncbi.nlm.nih.gov, as of 1 August 2014). Class A HMW PBPs are characterized by a short N-terminal cytoplasmic part, a single membrane-spanning region followed by the extracellular transglycosylase and transpeptidase domains. In class B PBPs, the transglycosylase domain is replaced by a noncatalytic domain. L. monocytogenes possesses two bi-functional class A HMW PBPs encoded by the lmo1892 (PBP A1) and the lmo2229 (PBP A2) genes, and three class B HMW PBPs, encoded by lmo1438 (PBP B1), lmo2039 (PBP B2) and lmo0441 (PBP B3) (Fig. 1A). Deletion and overexpression studies have assigned PBP3 to PBP B1 (Krawczyk-Balska et al., 2012), PBP4 to PBP A2 (Zawadzka-Skomial et al., 2006; Van de Velde et al., 2009) and PBP5 to the D-alanyl-D-alanine-carboxypeptidase PBP D1 (lmo2754) (Korsak et al., 2005) that cleaves off the terminal D-alanine from murein peptide stems. Inactivation of HMW PBPs of L. monocytogenes using gene disruption by integration of temperature sensitive plasmids demonstrated the essentiality of lmo2039 (encoding PBP B2), whereas the other HMW PBPs were shown to be dispensable for viability suggesting functional redundancies among the HMW PBPs (Guinane et al., 2006). Inactivation of PBP A1 or PBP B1 caused severe virulence defects in mice (Guinane et al., 2006), and inactivation of PBP A2 and PBP B3 resulted in increased susceptibilities against β-lactams (Guinane et al., 2006; Van de Velde et al., 2009). These phenotypes suggest that all HMW PBPs contribute to listerial peptidoglycan biosynthesis; however, it is likely that this is achieved via their distinct as well as overlapping physiological functions. Interestingly, PBP A1 genes of L. monocytogenes isolates J0161 (reference number: NC_017545.1), J1816 (NC_021829.1) and WSLC1042 (CP007210.1) have acquired premature stop codons leading to truncated proteins lacking up to 110 C-terminal amino acids (Klumpp et al., 2014). These truncations do, however, not extend into the transpeptidase domains and, hence, might be without functional consequences. In contrast, more severe truncations are observed in PBP B3 genes of L. monocytogenes isolates La111 (NC_020557.1) and N53-1 (NC_020558.1) (Holch et al., 2013). In these strains, pre-mature stop codons in the 5′ regions of the PBP B3 genes cause expression of 105 aa long PBP B3 fragments devoid of nearly their complete extracellular domains.
Figure 1.
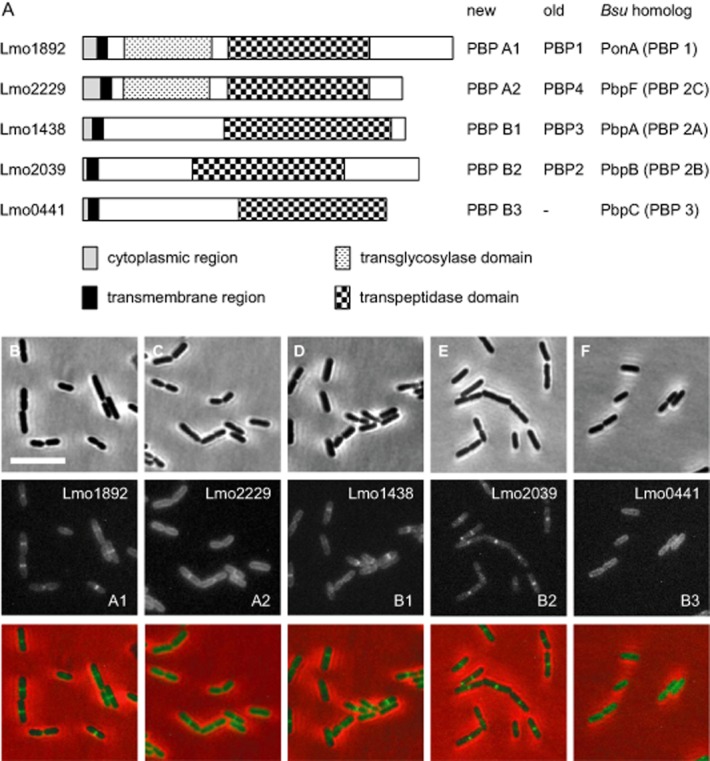
High molecular weight penicillin-binding proteins of L. monocytogenes and their subcellular localization.A. HMW PBPs of L. monocytogenes. Schematic illustration to show domain organization of the five HMW PBP genes that are present in the L. monocytogenes EGD-e genome. PBP names are according to the nomenclature proposed by others (Bierne and Cossart, 2007; Korsak et al., 2010). Old PBP names are according to the apparent molecular weights of the individual PBPs on SDS-PAGE gels stained with radiolabeled penicillin (Vicente et al., 1990). The corresponding B. subtilis homologs are indicated on the right side.B–F. Micrographs showing L. monocytogenes cells expressing GFP-PBP fusions. Cells of L. monocytogenes LMS44 (gfp-lmo1892, B), LMS38 (gfp-lmo2229, C), LMS46 (gfp-lmo1438, D), LMS45 (gfp-lmo2039, E) and LMS47 (gfp-lmo0441, F) were grown in BHI broth at 30°C up to mid-logarithmic growth phase and subjected to epifluorescence microscopy (middle panels). Mnemonics in the lower right corners indicate the type of the respective PBPs. Micrographs showing phase contrast images (upper panels) and merged images (lower panels) are included for better comparison. Scale bar is 5 μm.
Here we analyzed the contribution of all five HMW PBPs to growth and physiology of L. monocyctogenes. GFP fusions of the individual HMW PBPs showed distinct localization patterns suggesting discrete functions. We constructed a set of genetically stable, clean deletion mutants and analyzed their phenotypes. For the essential HMW PBP genes, we generated isopropyl-β-d-thiogalactopyranoside (IPTG)-dependent conditional mutant strains. Our data demonstrated the essentiality of PBP B2 and the synthetic lethality of the two bi-functional PBPs, PBP A1 and PBP A2. The individual HMW PBPs had specific functions in cell growth and/or cell division. Finally, antibiotic susceptibility assays demonstrated that PBP B1 contributes most significantly to the inherent resistance of L. monocytogenes against β-lactam antibiotics.
Results
Subcellular localization of HMW PBPs
For a better understanding of the role of each listerial HMW PBP, we first determined their subcellular localization. For this purpose, the gfp gene was fused to each HMW PBP gene allowing the ectopic expression of GFP-PBP fusion proteins in the wild-type. GFP-PBP A1, GFP-PBP A2 and GFP-PBP B1 were expressed as demonstrated by Western blotting. GFP-PBP B2 and GFP-PBP B3 are expressed at lower levels and showed signs of proteolytic degradation (Fig. S1A). The successful labeling with the fluorescent β-lactam bocillin-fl indicated correct folding of GFP-PBP A1, GFP-PBP A2, GFP-PBP B1 and GFP-PBP B2 (Fig. S1B). Only GFP-PBP B3 was invisible in the bocillin-fl stained gels, even though it was clearly detected in the Western blot. To our knowledge, PBP B3 has never been detected in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels using labeled penicillins, suggesting that GFP-PBP B3 cannot be stained.
Epifluorescence microscopy showed that GFP-PBP A1 (Fig. 1B), GFP-PBP A2 (Fig. 1C) and GFP-PBP B1 (Fig. 1D) localized to the cell periphery and the septum, albeit to different degrees and not in all cells of the population. In contrast, GFP-PBP B2 localized exclusively to septal sites (Fig. 1E), whereas GFP-PBP B3 produced only peripheral fluorescence signals (Fig. 1F). The distinct subcellular localization patterns possibly reflect functional differences in the listerial HMW PBPs.
Deletion of L. monocytogenes HMW PBP genes
A collection of insertional disruption mutants in HMW PBP genes of L. monocytogenes, which had been generated by the help of plasmids containing the temperature sensitive repAts allele, was described earlier (Guinane et al., 2006). Insertional mutants constructed by such Campbell-like plasmid integrations have, at least in our experience, the tendency to revert in the absence of selection, if the selection pressure for reversion is strong enough. In order to obtain a genetically stable set of mutants in HMW PBP genes of L. monocytogenes, we therefore decided to construct strains with marker-less clean deletions of the lmo1892, lmo2229, lmo1438, lmo2039 and lmo0441 genes. Derivatives of the pMAD plasmid, allowing scar-less removal of chromosomal sequences (Arnaud et al., 2004), were used to generate internal deletions of the lmo1892 and lmo2229 genes, encoding the two class A PBPs, PBP A1 and PBP A2 respectively. These deletions were designed in a way that the gene regions comprising the C-terminal part of the transglycosylase domains and N-terminal part of the transpeptidase domains were removed and replaced by premature stop codons (Fig. S2). Likewise, an internal region of lmo0441, encoding PBP B3, was removed and replaced by a stop codon (Fig. S2). For removal of the lmo1438 (PBP B1) and lmo2039 genes (PBP B2), pMAD derivatives were constructed, which would allow for the removal of the complete open reading frames. However, this strategy was not successful, most likely due to the essentiality of both these genes. In contrast, deletion of both genes was possible in strains LMJR5 and LMJR20, which contained IPTG-inducible, ectopic copies of lmo2039 and lmo1438 respectively (Fig. S2). This suggested that PBP A1, PBP A2 and PBP B3 are dispensable, whereas PBP B1 and PBP B2 are essential under standard laboratory conditions.
Detection of HMW PBPs in pbp mutant strains of L. monocytogenes
Labeling with the fluorescent penicillin bocillin-fl (Korsak et al., 2010) was used to verify the absence or IPTG-dependent expression of HMW PBPs in the constructed mutants. To this end, L. monocytogenes strains EGD-e (wild type), LMS57 (Δlmo1892), LMS64 (Δlmo2229), LMJR18 (Ilmo2039, I – inducible; this designation is used for IPTG-dependent conditional mutants), LMJR27 (Ilmo1438), as well as LMJR41 (Δlmo0441) were cultivated in brain heart infusion (BHI) broth with or without 1 mM IPTG. Membrane protein fractions were isolated, stained with bocillin-fl and analyzed by 8% SDS-PAGE. The membrane protein extract of the wild-type strain contained four fluorescent protein bands after bocillin-fl staining (Fig. 2). The most prominent band at approximately 100 kDa was absent in extracts from the Δlmo1892 mutant LMS57 and therefore corresponds to PBP A1. Extracts of strain LMS64 (Δlmo2229) lacked the second intense band at around 70 kDa, which had previously been assigned to PBP A2 (Zawadzka-Skomial et al., 2006; Van de Velde et al., 2009). Depletion of PBP B1 (Lmo1438) in strain LMJR27 caused the loss of the lower band of a ∼ 80 kDa-doublet, and depletion of PBP B2 (Lmo2039) in strain LMJR18 led to the loss of the upper band of the same doublet. As indicated in Fig. 2, these results are in good agreement with the theoretical molecular masses of these PBPs. Strain LMJR41 lacking PBP B3 (Lmo0441) showed the same PBP staining pattern as wild type, and the expression of a second ectopic copy of PBP B3 did not produce an additional band (data not shown). PBP B3 bands were also not detected with higher bocillin-fl concentrations (data not shown). This suggests that PBP B3 either generally escapes detection by bocillin-fl or that it is not stable enough to accumulate to a level above the detection limit.
Figure 2.
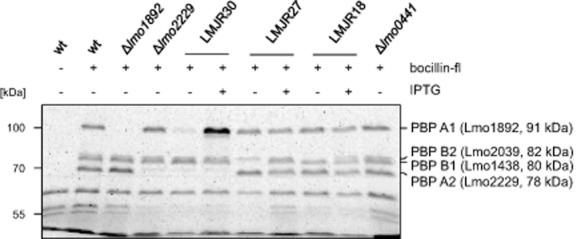
Detection of L. monocytogenes penicillin-binding proteins using bocillin-fl. Cells of L. monocytogenes EGD-e (wt), LMS57 (Δlmo1892), LMS64 (Δlmo2229), LMJR30 (Ilmo1892 Δlmo2229, I – IPTG-inducible), LMJR27 (Ilmo1438), LMJR18 (Ilmo2039) and LMJR41 (Δlmo0441) were grown in BHI broth (containing 1 mM IPTG where indicated) at 37°C to an OD600 of 1.0 and membrane fractions were isolated. Aliquots thereof were mixed with bocillin-fl and separated by SDS-PAGE (8% polyacrylamide). Fluorescently labeled PBPs were detected with a Fuji raytest FLA 2000 fluorescence scanner. An unlabeled wild-type control was included to identify nonspecific background fluorescence. Note that the PBP A1/PBP A2 double mutant strain LMJR30 is described in a separate results chapter.
Growth of L. monocytogenes HMW PBP mutants
In order to record possible growth phenotypes, all mutant strains were cultivated in BHI broth and the optical density was measured in hourly intervals. The absence of PBP A1 or PBP A2 did not alter growth under standard laboratory conditions (Fig. 3A) but caused a slight growth defect at 42°C (Fig. S3A). To test the effect of PBP B1 and PBP B2 depletion on growth of L. monocytogenes, overnight cultures of strains LMJR27 and LMJR18, respectively, were grown in BHI broth supplemented with 1 mM IPTG and used to inoculate depletion cultures without inducer the next morning. Cells were grown for 24 h at 37°C and used as starting material to inoculate cultures containing or not containing 1 mM IPTG. The depletion of PBP B1 led to a severe reduction of growth of strain LMJR27 (Fig. 3B) at 37°C and a complete growth arrest at 42°C (Fig. S3B). In contrast, the depletion of PBP B2 prevented any growth of strain LMJR18 (Fig. 3C). In order to record the effect of increased temperature on PBP B2 depleted cells, strain LMJR18 was pre-grown in overnight cultures containing IPTG. Cells from these cultures were washed and used to inoculate fresh cultures containing or not containing 1 mM IPTG, and growth was monitored at 37°C and at 42°C. Although there was no growth defect visible at 37°C (Fig. S4A), LMJR18 cells cultivated in the absence of IPTG stopped growth at 42°C, as soon as PBP B2 became depleted, and the cells started to lyse as it is indicated by the constant decline in optical density (Fig. S4B). Finally, strain LMJR41, lacking PBP B3, did not show any growth defects at 37°C or 42°C (Fig. 3D, Fig. S3C). Taken together, these results show that (i) PBP A1, PBP A2 and PBP B3 are not required during growth at 37°C, that (ii) PBP B1 depletion dramatically reduces growth rate, and that (iii) PBP B2 is essential under standard laboratory conditions. Furthermore, loss or depletion of most PBPs causes temperature sensitive phenotypes.
Figure 3.
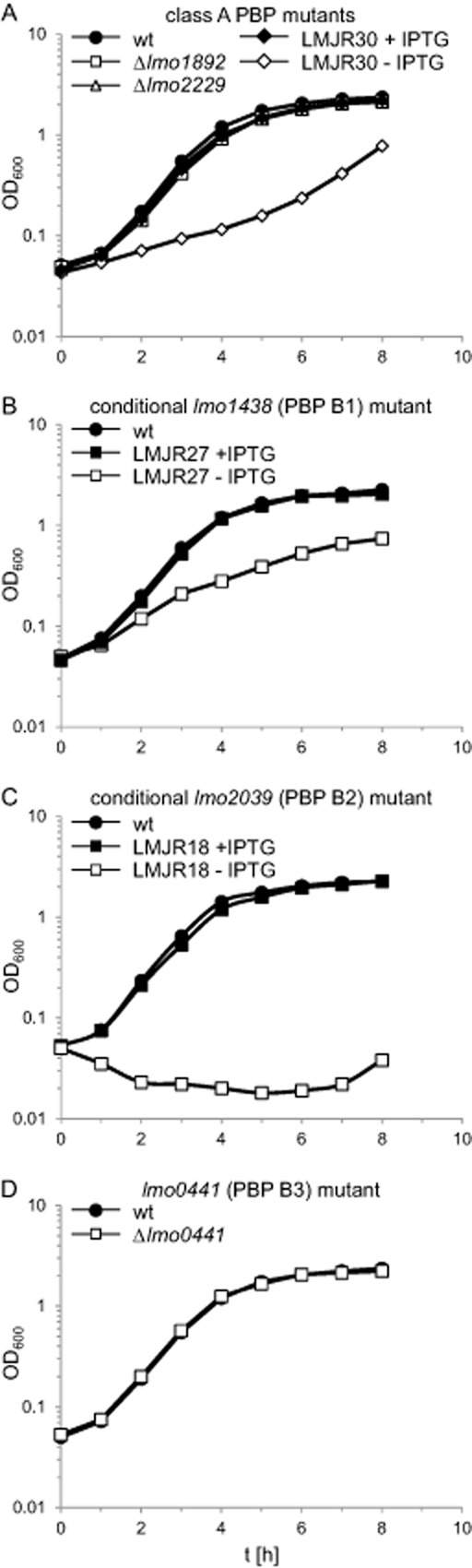
Growth of L. monocytogenes strains lacking penicillin-binding proteins.A. Growth of strains lacking class A high molecular weight penicillin-binding proteins PBP A1 (Lmo1892), PBP A2 (Lmo2229) or both. Strains LMS57 (Δlmo1892), LMS64 (Δlmo2229) and LMJR30 (Ilmo1892 Δlmo2229) where cultivated in BHI broth (containing IPTG where indicated) at 37°C and optical density was measured at hourly intervals.B–D. Growth of strains lacking or conditionally expressing class B high molecular weight proteins. Strains LMJR27 (Ilmo1438, inducible PBP B1), LMJR18 (Ilmo2039, inducible PBP B2), as well as LMJR41 (Δlmo0441, PBP B3) were cultivated in BHI broth (containing 1 mM IPTG where indicated) and growth was recorded based on optical density.
Cellular morphology of HMW PBP mutants
To better characterize the contribution of the listerial HMW PBPs to cell wall assembly, ultrathin sections of all five pbp mutant strains were subjected to transmission electron microscopy. Cells of LMS57 (Δlmo1892, Fig. 4C), LMS64 (Δlmo2229, Fig. 4E) and LMJR41 (Δlmo0441, Fig. 5E) produced normal lateral and septal walls. Septal cross-wall synthesis appeared to be slightly delayed in strain LMS57 (Δlmo1892, Fig. 4C). In sharp contrast, depletion of PBP B1 in strain LMJR27 caused malformed cross-walls, which regularly were found at off-center positions (Fig. 5A). Cells depleted for PBP B1 also seemed to be unable to hydrolyse cross-walls: Often, a thick triangular cell wall (peptidoglycan) mass occupied the transition zones from the lateral walls to the cross-walls; this material is normally degraded with ongoing constriction and cross-wall maturation (Fig. 5A). Depletion of PBP B2 resulted in the initiation of constriction and the formation of small invagination marks at potential division sites, but septation did not continue and cross-walls were not formed (Fig. 5C). Scanning electron microscopy (SEM) showed that lack of PBP A2 and PBP B3 did not cause any changes in cell shape (Figs. 4F and 5F). Lack of PBP A1 in strain LMS57 (Δlmo1892) resulted in slightly elongated and often bent cells (Fig. 4D), indicating a role for PBP A1 in cell division and synthesis of lateral peptidoglycan. Depletion of PBP B1 in cells of strain LMJR27 caused irregularly formed, partially swollen or almost coccoid cells (Fig. 5B), suggesting that PBP B1 primarily functions in lateral peptidoglycan biosynthesis. In good agreement with PBP B2's essential function in cross-wall formation, LMJR18 cells depleted of PBP B2 were extremely elongated (Fig. 5D).
Figure 4.
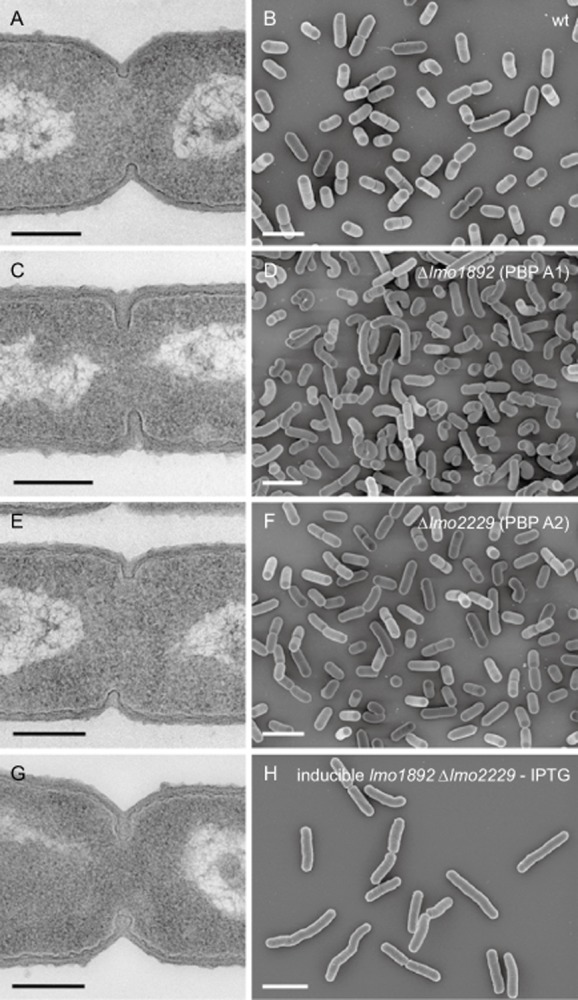
Effect of PBP A1 and PBP A2 inactivation on cell morphology. Transmission electron microscopy of ultrathin sections (A, C, E, G) and scanning electron microscopy (B, D, F, H) of fixed whole cells of L. monocytogenes strains devoid of class A high molecular weight penicillin-binding proteins. L. monocytogenes strains EGD-e (wt), LMS57 (Δlmo1892), LMS64 (Δlmo2229) and the inducible double mutant strain LMJR30 (Ilmo1892 Δlmo2229) were grown to mid-logarithmic growth phase in BHI at 37°C and subjected to chemical fixation and subsequent electron microscopy as described in the experimental procedures section. Scale bars: left column 200 nm, right column 2 μm.
Figure 5.
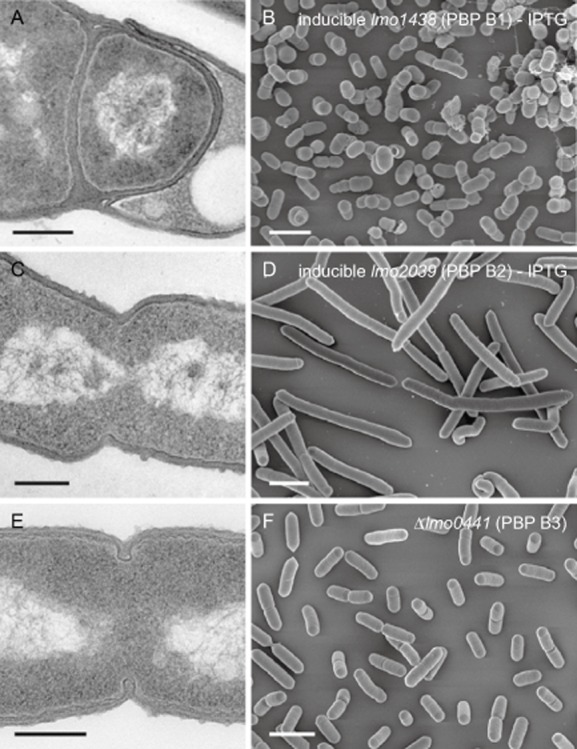
Effect of PBP B1, PBP B2 and PBP B3 inactivation on cell morphology. Transmission electron microscopy of ultrathin sections (A, C, E) and scanning electron microscopy (B, D, F) of fixed whole cells of L. monocytogenes strains devoid of class B high molecular weight penicillin-binding proteins. L. monocytogenes strains LMJR27 (Ilmo1438), LMJR18 (Ilmo2039) and LMJR41 (Δlmo0441) were grown to mid-logarithmic growth phase in BHI at 37°C and subjected to chemical fixation and subsequent electron microscopy as described in the experimental procedures section. Scale bars: left column 200 nm, right column 2 μm.
For a more precise view on the contribution of the individual HMW PBPs to cell division, we stained all pbp mutants with nile red and measured the lengths of 600 cells (Fig. 6A). Consistent with the SEM images, the deletion of PBP A1 caused an increase of the average cell length in strain LMS57 (1.44 ± 0.2 μm) as compared with wild type (1.16 ± 0.2 μm). In contrast, deletion of PBP A2 and PBP B3 in strains LMS64 (1.26 ± 0.2 μm) and LMJR41 (1.18 ± 0.3 μm) did not significantly affect cell length (Fig. 6B). Depletion of PBP B1 led to shorter cells (0.88 ± 0.2 μm, Fig. 6B), whereas PBP B2-depleted cells grew as 10.9 ± 5 μm long, nonseptated filaments (Fig. 6A), whereby the cell lengths were extremely heterogeneous and could reach values of up to 30 μm (Fig. 6C). Taken together, these data demonstrate a general role of PBP A1 in division and maintenance of cell shape. PBP B1 primarily functions in peptidoglycan biosynthesis at the lateral wall, whereas the function of PBP B2 is specific for cross-wall biosynthesis.
Figure 6.
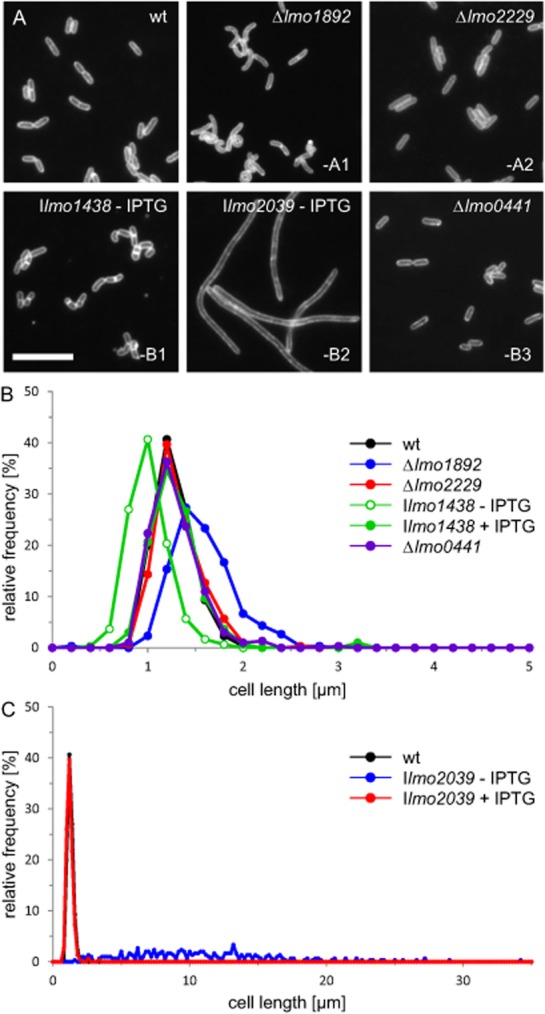
Division phenotypes of L. monocytogenes pbp mutants.A. Fluorescence micrographs of L. monocytogenes pbp mutant strains after membrane staining using nile red. L. monocytogenes strains EGD-e (wt), LMS57 (Δlmo1892), LMS64 (Δlmo2229), LMJR27 (Ilmo1438), LMJR18 (Ilmo2039) and LMJR41 (Δlmo0441) were cultivated in BHI broth to mid-logarithmic growth phase. For depletion of PBP B1 (Lmo1438) and PBP B2 (Lmo2039), strains LMJR27 and LMJR18, respectively, were pregrown overnight in the presence of IPTG and then used to start a depletion culture in the absence of IPTG. Mnemonics in the lower right corners indicate the type of the respective deleted or depleted PBPs. (B and C) Cell lengths of 600 cells per strain were measured and visualized as frequency plots. Cell length distribution of PBP B2-depleted cells is illustrated in a separate diagram due to their extreme filamentous growth in the absence of IPTG (C).
Lack of HMW PBPs weakens the cell wall without changing its composition
The contribution of HMW PBPs to resistance against clinically relevant antibiotics was analyzed by E-tests, using minimum inhibitory concentration (MIC) test strips with antibiotics targeting the cell wall (penicillin, ampicillin, amoxicillin, meropenem and vancomycin) and gentamycin as a control. Susceptibilities to gentamycin and vancomycin did not vary much among the different pbp mutant strains. In contrast, deletion or depletion of any of the HMW PBPs caused alterations in the susceptibility to β-lactams (Table 1). Deletion of PBP A1 or PBP A2 resulted in modest effects on β-lactam susceptibility, whereas strong effects were observed with PBP B1 and PBP B2 depleted cells. LMJR18 cells depleted for PBP B2 showed a minor increase in penicillin susceptibility but are two- to threefold more susceptible to meropenem and ampicillin. Depletion of PBP B1 in strain LMJR27 had the strongest effect and caused a three- to fivefold reduction in the MIC for penicillin, ampicillin and meropenem (Table 1). Interestingly, LMJR41 cells lacking PBP B3 also showed an effect as they were roughly twofold more susceptible to penicillin and ampicillin. This was the first indication, which appeared in the course of this study, that the lmo0441 gene is expressed and that PBP B3 is enzymatically active.
Generally, β-lactams do not have a bactericidal effect in L. monocytogenes, but PBP inactivation might induce bacteriolysis upon antibiotic treatment. Our quantitative autolysis assays demonstrated that individual deletion or depletion of most HMW PBPs did not induce autolysis in response to penicillin (Fig. 7A). The only exception was the depletion of PBP B1 that drastically enhanced bacteriolysis (Ilmo1438, Fig. 7A). Roughly two-thirds of this effect was due to spontaneous endogenous autolysis of PBP B1-depleted LMJR27 cells. However, the addition of penicillin further magnifies this effect (Fig. 7B). This result shows that an active PBP B1 is required for cellular integrity and intrinsic penicillin resistance of L. monocytogenes.
Figure 7.
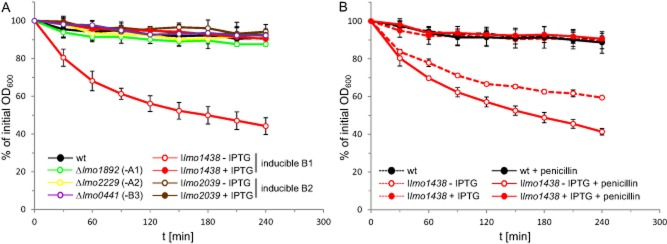
Effect of PBP inactivation on penicillin-induced autolysis.A. Autolysis assay of L. monocytogenes pbp mutants. L. monocytogenes strains EGD-e (wt), LMS57 (Δlmo1892), LMS64 (Δlmo2229), LMJR27 (Ilmo1438), LMJR18 (Ilmo2039) and LMJR41 (Δlmo0441) were pregrown in BHI broth (containing 1 mM IPTG for LMJR18 and LMJR27) at 37°C overnight and used to inoculate fresh BHI medium (containing 1 mM IPTG where indicated). Cultures were grown to mid-log phase (OD600∼0.8). Cells were harvested, washed and resuspended in 50 mM Tris-HCl pH 8.0 buffer. Penicillin was added at a concentration of 25 μg ml−1, and cells were incubated on a shaker at 37°C.B. Autolysis assay to record the contribution of endogenous autolysis to the effect of PBP B1 depletion on penicillin-induced autolysis. L. monocytogenes strains EGD-e (wt) and LMJR27 (Ilmo1438) were prepared as described in panel A, and the decrease in optical density (λ = 600 nm) over time was recorded photometrically in the presence or absence of penicillin. All values were expressed as relative values. Standard deviations were calculated from experiments performed in triplicate.
We have next analyzed the peptidoglycan composition of PBP-deleted/-depleted cells. The muramidase cellosyl released six major muropeptides that were separated and quantified by high-pressure liquid chromatography (HPLC) (Fig. 8A). Mass spectrometry confirmed the identity of these main fragments (Fig. 8B). Listerial peptidoglycan features an amidated meso-diaminopimelic acid residue at positon 3 of the stem peptide and has a high percentage of glucosamine residues originating from deacetylation of N-acetylglucosamine (Vollmer and Tomasz, 2000; Boneca et al., 2007). The different PBP depletion strains showed only minor differences in their muropeptide profile compared with wild type (Fig. 8). The cross-linkage was virtually unaffected, indicating that all combinations of the remaining four HMW PBPs are capable of producing a normally cross-linked peptidoglycan. Cells without PBP B1 had moderately enhanced levels of fully acetylated dimeric muropeptide, indicating a reduced activity of peptidoglycan deacetylase in this strain.
Figure 8.
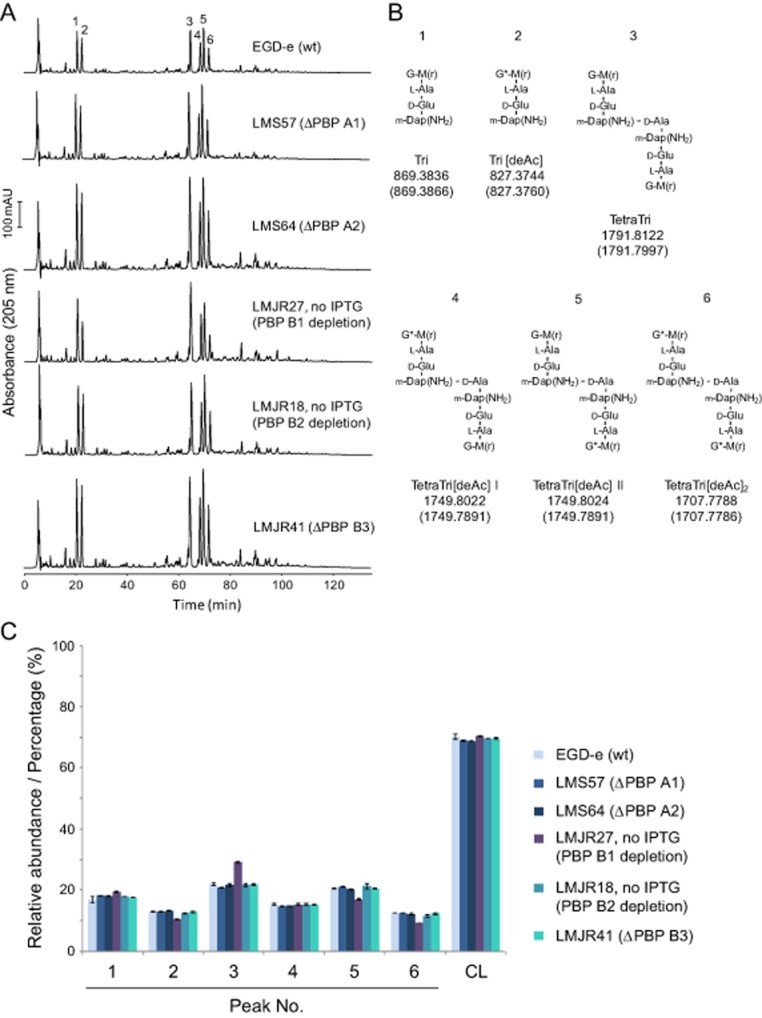
Peptidoglycan composition of pbp mutants.A. Muropeptide profiles obtained by HPLC analysis. The strain names and PBP affected are indicated. Major muropeptide peaks are numbered.B. Proposed muropeptide structures corresponding to the major peaks in (A). The names of the muropeptide and the calculated neutral m/z values are given below the structures (upper value, determined neutral m/z; lower value in brackets, theoretical value). G, N-acetylglucosamine; G*, glucosamine; M, N-acetylmuramic acid; m-Dap(NH2), amidated meso-diaminopimelic acid. TetraTri[deAc] exists in two isomers (I and II) in which the positions of the G and G* residues are not known.C. Quantification of the relative abundance of muropeptides 1–6 and of the percent peptides in cross-links (CL) in the peptidoglycan of strains indicated on the right side. The values are the average ± SD of two independent experiments.
Lack of both bi-functional HMW PBPs is lethal
The effects of individual PBP A1 and PBP A2 knock-outs on L. monocytogenes were rather mild, hence both genes might have overlapping functions and could possibly complement each other. In order to test this hypothesis, the temperature-sensitive plasmid pSH243 was introduced into the PBP A1 deletion strain LMS57 (Δlmo1892). pSH243 was designed for disruption of the lmo2229 gene encoding PBP A2 in a Campbell-type recombination event (Fig. S5C). The resulting strain (LMS143) could grow in the presence of erythromycin at the permissive temperature (30°C, Fig. S5A), when plasmid replication was unaffected. However, it did not form colonies under restrictive conditions (42°C), due to integration of pSH243 into the chromosome to confer erythromycin resistance. In contrast, strain LMS141 (wild-type background) formed colonies even at 42°C (Fig. S5B) and these colonies had pSH243 integrated in the lmo2229 open reading frame (data not shown). These results indicated that the simultaneous inactivation of both bi-functional HMW PBPs is not tolerated in L. monocytogenes.
In order to obtain a conditional PBP A1 PBP A2 double mutant, an internal deletion of lmo2229 was generated in strain LMJR21 (Δlmo1892 pIMK3-lmo1892). The resulting strain (LMJR30) was devoid of PBP A2, whereas the expression of PBP A1 was dependent on IPTG (Fig. 2). Growth of LMJR30 cells was retarded when no IPTG was added to the culture as compared with the presence of the inducer and dramatically lagged behind that of PBP A1 and PBP A2 single mutants (Fig. 3A). Background expression of PBP A1 (Fig. 2) presumably accounted for residual growth in the absence of IPTG. The growth defect of strain LMJR30 was even more pronounced at 42°C, where cells went through only one to two doublings during the 8 h interval, as long as no IPTG was present (Fig. S3A). Obvious ultrastructural changes other than that seen for the PBP A1 mutant LMS57 were not observed for the inducible double mutant (Fig. 4G). However, the slightly increased susceptibilities of the PBP A1 and PBP A2 single mutants to various β-lactam antibiotics appeared to be additive, resulting in a stronger reduction of resistance against penicillin, ampicillin, amoxicillin and meropenem in the conditional double mutant strain, whereas its susceptibilities to vancomycin and gentamycin were unaffected (Table 1). Taken together, depletion of PBP A1 in a ΔPBP A2 background severely impairs growth, heat sensitivity as well as resistance against β-lactams.
Infection experiments
In order to test whether inactivation of HMW PBPs would impair pathogenicity of L. monocytogenes, several in vitro infection assays were performed. First, the ability of the HMW PBP mutants to invade nonphagocytic eukaryotic host cells was analyzed in invasion experiments using HeLa cells. In our experimental setup, 1.5 ± 0.2% (n = 3) of the wild type cells from the initially applied inoculum invaded the HeLa cells, which is comparable with reported invasion rates (Gaillard et al., 1991). In contrast, invasion rates of the strains lacking PBP A1 and PBP A2 were reduced approximately four- and sixfold, respectively (Fig. 9A), indicating a special role for class A HMW PBPs during entry into eukaryotic cells. In order to measure the effect of PBP B1 and PBP B2 depletion, strains LMJR27 and LMJR18 were grown over night in BHI broth containing 1 mM IPTG and were used to inoculate fresh cultures with or without 1 mM IPTG the next morning. When the cultures had reached an OD600 of 0.4, the bacteria were harvested, processed and used for infection. Depletion of PBP B1 resulted in a roughly sevenfold reduction of invasion rate in strain LMJR27 when compared with the wild-type situation (13.4 ± 6.3% of wild-type level). Under conditions of PBP B1 expression, however, the invasion rate of LMJR27 cells reached 52.9 ± 5.8% of the wild-type level (Fig. 9B). Likewise, PBP B2 depletion in strain LMJR18 led to a 22-fold reduction of the invasion rate (4.5 ± 1.2% of wild-type level), whereas an invasion efficiency corresponding to 50.1 ± 8.1% of wild type level was observed for LMJR18 cells that had been cultivated in the presence of IPTG (Fig. 9B). In contrast, invasion rate of the PBP B3 deletion mutant LMJR41 slightly exceeded that of the wild type strain (130.1 ± 19.9% of the wild type) (Fig. 9B).
Figure 9.
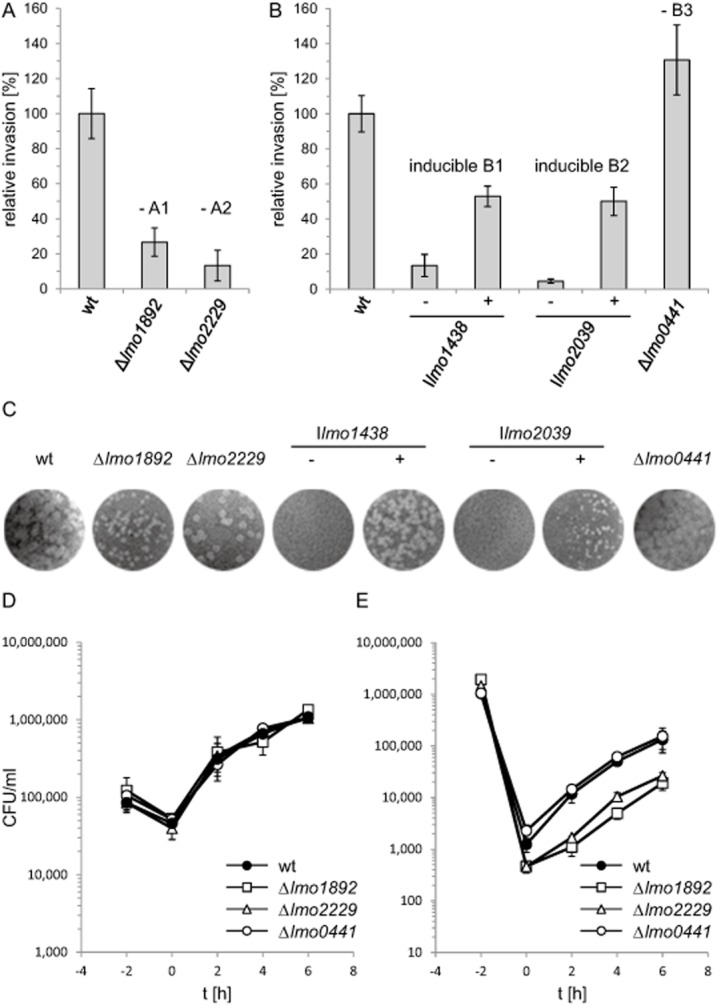
Attenuation of L. monocytogenes pbp mutants in in vitro infection models.A and B. Invasion of L. monocytogenes into HeLa cells. (A) L. monocytogenes strains lacking class A HMW PBPs, LMS57 (Δlmo1892) and LMS64 (Δlmo2229) were grown in BHI broth and used to infect HeLa cells. Noninvasive bacteria were washed away and killed using gentamycin. Invasion rates are the ratios of the number of intracellular bacteria versus the number of initially applied bacteria in the inoculum. Values are expressed relative to wild type levels (n = 3). (B) L. monocytogenes strains conditionally expressing or lacking class B HMW PBPs, LMJR18 (Ilmo2039), LMJR27 (Ilmo1438) and LMJR41 (Δlmo0441) were tested in the same way as described for panel A in a separate experiment. Strains LMJR18 and LMJR27 were cultivated in BHI broth either containing (+) or not containing IPTG (−). Bacterial samples recovered from the HeLa cells were plated on BHI agar plates containing 1 mM IPTG for CFU counting.C. Plaque formation assays of L. monocytogenes pbp mutant strains. Same set of strains as in panels A and B was used to infect 3T3 mouse embryo fibroblasts. IPTG-dependent strains (LMJR18 and LMJR27) were cultivated in the presence of IPTG prior to infection to ensure wild type-like invasion, but after infection the infected cell cultures were either incubated in the absence or presence of 1 mM IPTG. Zones of killed host cells resulted from spreading bacteria and appeared after 3 days post infection and counterstaining with neutral red.D. Intracellular replication of L. monocytogenes PBP A1, PBP A2 and PBP B3 deletion mutants in mouse macrophages. L. monocytogenes strains EGD-e (wt), LMS57 (Δlmo1892), LMS64 (Δlmo2229) and LMJR41 (Δlmo0441) were grown in BHI broth at 37°C and used to infect J774.A1 mouse ascites macrophages at a multiplicity of infection of 0.167. Sampling was performed right after infection (t = 0 h) and then in intervals of 2 h.E. Intracellular multiplication of the same set of strains in HeLa cells. Experiments were performed as described in the experimental procedures section. Average values and standard deviations were calculated from experiments performed in triplicate.
The capability of the HMW PBP mutants to spread from cell to cell was analyzed in a plaque formation assay using 3T3 mouse embryo fibroblasts. L. monocytogenes cells infect this cell type and spread circularly from the initial point of infection, forming zones of killed host cells that appear as bright plaques after neutral red staining (Fig. 9C). In the absence of PBP A1, the size but not the number of these plaques was considerably reduced. In contrast, absence of PBP B2 primarily reduced the plaque number but had no effect on the plaque size. This indicates that PBP A1 is required for cell-to-cell spread but does not affect the invasiveness, whereas deletion of PBP A2 has the opposite effect. The contribution of PBP B1 and PBP B2 to cell-to-cell spread was assessed by using LMJR27 and LMJR18 bacteria, which were precultivated in the presence of IPTG to allow wild type–like host cell invasion. After invasion, the infected cell cultures were either left without IPTG or cultivated in the presence of IPTG. In the absence of IPTG, both strains were unable to form plaques, whereas plaques were observed in the presence of the inducer, albeit with reduced size. In contrast, absence of PBP B3 did not affect plaque formation at all. These results demonstrate that PBP A1, PBP B1 and PBP B2 play crucial roles for cell-to-cell spread of L. monocytogenes, whereas PBP A2 and PBP B3 are of minor importance for spreading.
The multiplication of mutants lacking PBP A1, PBP A2 or PBP B3 inside eukaryotic cells was investigated using the J774 mouse ascites macrophage cell line. All strains were phagocytosed at a similar rate and grew like the wild-type strain inside these cells (Fig. 9D). When the same set of strain was used to infect HeLa cells, strains lacking PBP A1 and PBP A2 again showed the above-mentioned invasion defect, but all strains multiplied inside HeLa cells with a rate comparable with that of the wild-type strain EGD-e (Fig. 9E).
Cell wall retention of surface proteins
The cell wall is an anchor for surface proteins that covalently or noncovalently interact with the peptidoglycan (via LPXTG motifs or LysM domains, respectively) or with lipoteichoic acids (via GW modules) (Bierne and Cossart, 2007). In order to test, whether the surface attachment of such proteins is affected in the pbp mutant strains, we analyzed cell wall retention of all three protein classes by Western blotting. Internalin A (InlA) is a LPXTG protein (Lebrun et al., 1996). It was present in preparations of cell wall associated proteins of all pbp mutants and even slightly enriched upon depletion of PBP B1 and PBP B2 (Fig. S6A). Only background InlA levels were observed in culture supernatants of the pbp mutants indicating efficient cell wall retention. In contrast, InlA is released from the peptidoglycan and secreted into the growth medium in a ΔsrtA mutant (Fig. S6A). This strain lacks sortase A required for linking LPXTG proteins to the peptidoglycan (Bierne et al., 2002). Likewise, the internalin-like LPXTG protein Lmo0610 (Bierne and Cossart, 2007) is present in cell wall fractions of all pbp mutants, and its cell wall retention is lost in a ΔsrtA mutant (Fig. S6B). Lmo0880 is another LPXTG protein with a yet unknown function (Quereda et al., 2013) and also contains a LysM domain (Bierne and Cossart, 2007). Its cell wall retention was not affected in any of the pbp mutants, but SrtA-dependent like the other LPXTG proteins (Fig. S6C). Remarkably, the amount of secreted Lmo0880 was reduced in PBP B1 depleted cells (Fig. S6C). As with the LPXTG proteins, no clear differences in surface retention of the autolysins p60 and MurA (both containing LysM domains) or internalin B (GW protein) were observed in strains lacking PBP A1, PBP A2, PBP B2 or PBP B3 and, as expected, deletion of srtA had also no effect (Fig. S6D–F). Cells depleted for PBP B1 had reduced extracellular levels of p60 and MurA, which is consistent with the observed defects in cross-wall degradation (Fig. 5A). Reduction of extracellular p60, MurA and Lmo0880 levels might be explained by the massive lysis of PBP B1-depleted cells, as a result of which extracellular proteins could possibly be degraded by cytosolic proteases.
Discussion
The Achilles' heel of L. monocytogenes having the strongest clinical relevance for antibiotic therapy is the family of PBPs. More than 20 years ago, Vicente et al. suggested that the primary lethal target of β-lactam antibiotics of L. monocytogenes must be PBP3 (Vicente et al., 1990), which we here confirm to be PBP B1 by inducible expression of lmo1438 and subsequent bocillin-fl staining of membrane protein extracts. Depletion of PBP B1 had indeed the strongest effect on antibiotic susceptibility of L. monocytogenes dramatically reducing the MICs to all β-lactams tested. Similar effects on antibiotic resistance were not observed when any of the other HMW PBPs were inactivated. Depletion of PBP B1 furthermore predisposes L. monocytogenes to autolysis, and this effect could be further enhanced by addition of penicillin, whereas inactivation of any of the other HMW PBPs did neither stimulate spontaneous nor penicillin-induced autolysis. These results suggest that the other two transpeptidases, PBP B2 and PBP B3, must have rather specific (PBP B2) or only secondary (PBP B3, see below) functions in peptidoglycan cross linking, which are less important for maintenance of general cellular integrity. However, our data also show that transpeptidation of peptidoglycan strands by PBP B1 is required for the intrinsic resistance of L. monocytogenes against β-lactam antibiotics and thus supports the initial suggestion by Vicente (Vicente et al., 1990). Analysis of muropeptide composition showed normal peptidoglycan cross-linkage upon PBP B1 depletion but reduced levels of GlcNAc deacetylation. The latter could enhance the activity of endogenous peptidoglycan hydrolases like the muramidase MurA (NamA) or the glucosaminidase Auto (Carroll et al., 2003; Cabanes et al., 2004; Bublitz et al., 2009), leading to increased autolysis. That individual inactivation of none of the other PBPs had an effect on the muropeptide pattern is likely explained by the redundancy of the PBPs, which may compensate for each other.
A major problem in listeriosis treatment using β-lactams is that these antibiotics do not induce efficient bacteriolysis (Hof, 2004; Lemaire et al., 2005; Grayo et al., 2008). Remarkably, the β-lactams with the lowest MICs against L. monocytogenes, are the ones that exhibit the highest binding affinities to PBP B1, and these are penicillin G and imipenem (Vicente et al., 1990). This shows that anti-listerial intervention strategies with these antibiotics already target the PBP whose inactivation causes the strongest bacteriolytic effect. However, inactivation of PBP B1 by β-lactams can only prevent transpeptidation from the time of treatment onward. This is the difference to our depletion experiments and therefore resolves the obvious contradiction between the low bacteriolytic activities of β-lactams (Lemaire et al., 2005) and the strong induction of autolysis upon PBP B1 depletion. Compounds inhibiting PBP B1 by a different molecular mechanism or with a higher efficiency than the β-lactams could possibly improve antibiotic therapy of listeriosis by promoting bacteriolysis in situ.
In contrast to the pleiotropic PBP B1 phenotype, inactivation of PBP B2 activity through depletion of the lmo2039 gene product led to a more specific defect and was limited to septum formation and cross-wall biosynthesis. PBP B2 is homologous to PBP 2B (PbpB) of Bacillus subtilis, which has been described as one of the late division proteins that is recruited to the divisome after assembly of the FtsZ-ring (Daniel et al., 2000; Gamba et al., 2009). Depletion of B. subtilis PBP 2B causes a block in cell division leading to filament formation as is observed here with PBP B2-depleted L. monocytogenes cells. FtsZ-rings still assemble in PBP 2B-depleted cells of B. subtilis, but septal recruitment of late division proteins, such as DivIC and DivIB, is severely impaired, preventing completion of divisome assembly, constriction, cross-wall biosynthesis and cell division (Daniel et al., 2000). The function of L. monocytogenes PBP B2 seems to be identical to that of B. subtilis PBP 2B, and the concept of a divisome-specific role of PBP B2 is in good agreement with the observation that GFP-PBP B2 localized exclusively to the division septa (Fig. 1E). In contrast, inactivation of PBP B3 had no effect on growth, morphology or virulence, consistent with the finding that the lack of its homologue PbpC in B. subtilis causes no phenotypical effect (Murray et al., 1996). The only phenotype associated with lack of L. monocytogenes PBP B3 was a slight reduction in the susceptibility to several β-lactams and a slightly increased invasion efficiency (Table 1, Fig. 9B). This showed that PBP B3 is not a silent gene but rather must be an actively expressed gene. Possibly, its function is redundant and taken over by one of the other class B HMW PBPs in cells lacking PBP B3. It would be interesting to see, if deletion of PBP B3 in the inducible PBP B1 mutant would generate a strain that would not grow in the absence of IPTG at all.
Table 1.
Antibiotic susceptibilities of L. monocytogenes penicillin-binding protein mutants
| Strain | Genotype | affected PBP | Minimal Inhibitory Concentration (μg ml−1)a |
|||||
|---|---|---|---|---|---|---|---|---|
| Penicillin | Ampicillin | Amoxicillin | Meropenem | Vancomycin | Gentamycin | |||
| EGD-e | wt | – | 0.074 ± 0.017 | 0.053 ± 0.018 | 0.0183 ± 0.004 | 0.048 ± 0.016 | 1.5 ± 0.5 | 0.25 ± 0.00 |
| LMS57 | Δlmo1892 | A1 | 0.029 ± 0.005 | 0.053 ± 0.018 | 0.016 ± 0.00 | 0.052 ± 0.01 | 1.33 ± 0.288 | 0.253 ± 0.109 |
| LMS64 | Δlmo2229 | A2 | 0.029 ± 0.005 | 0.037 ± 0.008 | < 0.016 | 0.034 ± 0.012 | 1.5 ± 0.05 | 0.21 ± 0.035 |
| LMJR30 – IPTG | I1892 Δ2229 | A1, A2 | 0.016 ± 0.00 | 0.016 ± 0.00 | < 0.016 | 0.026 ± 0.005 | 1.67 ± 0.29 | 0.25 ± 0.11 |
| LMJR27 – IPTG | Ilmo1438 | B1 | < 0.016 | < 0.016 | < 0.016 | 0.011 ± 0.004 | 1.5 ± 0.00 | 0.31 ± 0.16 |
| LMJR18 – IPTGb | Ilmo2039 | B2 | 0.053 ± 0.01 | 0.016 ± 0.00 | < 0.016 | 0.023 ± 0.00 | 1.83 ± 0.29 | 0.21 ± 0.035 |
| LMJR41 | Δlmo0441 | B3 | 0.027 ± 0.009 | 0.027 ± 0.009 | < 0.016 | 0.042 ± 0.009 | 1.83 ± 0.29 | 0.27 ± 0.1 |
Antibiotic susceptibilities are expressed as average values of minimal inhibitory concentrations ± standard deviations, calculated from three independent repetitions.
BHI plates containing 0.025 mM IPTG were used to deplete PBP B2 (Lmo2039).
A synthetic lethal effect was observed with the two bi-functional class A HMW PBPs, PBP A1 and PBP A2, demonstrating that the presence of one of the two is essential for viability of L. monocytogenes. There are no other known transglycosylase genes present in the listerial genome, so the inactivation of PBP A1 and PBP A2 genes is expected to halt peptidoglycan biosynthesis. Depletion of PBP A1 in a ΔPBP A2 background resulted in a significant reduction of growth rate. Depleted cells still had background levels of PBP A1 (Fig. 2), explaining why growth was not completely abolished (Fig. 3A). Remarkably, the situation is different in B. subtilis: In this species the deletion of all four bi-functional HMW PBPs was tolerated and generated a viable strain, which still synthesized peptidoglycan presumably by an unknown transglycosylase (Popham and Setlow, 1996; McPherson and Popham, 2003). Our results suggest that this activity is not present or is present and cannot support growth of L. monocytogenes lacking PBP A2 and PBP A1. Perhaps a simultaneous deletion of both corresponding genes is possible under osmoprotective conditions, which would support stabilization of protoplasted cells, or in the cell wall deficient L-form mode of growth (Dell'Era et al., 2009).
Many surface proteins are covalently linked to the cell wall (LPXTG proteins) or associated to peptidoglycan components via noncovalent interactions (proteins containing GW modules or LysM domains). Among the LPXTG proteins are several internalins, including the major internalin InlA. The second major internalin InlB is associated with the cell wall via GW modules, and LysM domains for peptidoglycan binding are present in the two major autolysins p60 and MurA (Machata et al., 2005; Bierne and Cossart, 2007). All of these proteins are required for invasion into different cell types (Gaillard et al., 1991; Bergmann et al., 2002; Pilgrim et al., 2003; Halbedel et al., 2012), whereas p60 also contributes to cell-to-cell spread (Pilgrim et al., 2003). We only observe slight aberrations in cell wall retention of these proteins in some of the pbp mutants and the observed changes do not fit with their attenuation patterns in our virulence assays. Hence, distortions in net cell wall retention of any of these proteins do not account for the observed virulence defects. We cannot rule out that the distribution patterns of these proteins on the cell surface are altered or that cell wall retention of other surface proteins, critical for invasion and cell-to-cell spread is affected in the pbp mutants. Alternatively, changes in architecture, ultrastructure or physico-chemical properties of the peptidoglycan sacculus per se could contribute to the observed effects. Future experiments will have to clarify this question.
Experimental procedures
Bacterial strains and growth conditions
All bacterial strains used in this study are listed in Table 2. Cells of L. monocytogenes were routinely grown in BHI broth or on BHI agar plates at 37°C if not stated otherwise. If required, antibiotics and supplements were added to the growth media at the following concentrations: erythromycin (5 μg ml−1), kanamycin (50 μg ml−1), X-Gal (100 μg ml−1) and IPTG (1 mM). Escherichia coli TOP10 was used as the standard host for all cloning procedures (Sambrook et al., 1989).
Table 2.
Strains and plasmids used in this study
| Name | Relevant characteristics | Source/reference |
|---|---|---|
| Plasmids | ||
| pAUL-A | lacZα erm | (Chakraborty et al., 1992) |
| pIMK3 | Phelp-lacO lacI neo | (Monk et al., 2008) |
| pMAD | bla erm bgaB | (Arnaud et al., 2004) |
| pSH195 | Phelp-gfp neo | (Halbedel et al., 2012) |
| pUC19 | bla lacZα | Invitrogen |
| pJR4 | bla Δlmo1438 | This work |
| pJR5 | bla erm bgaB Δlmo1438 | This work |
| pJR7 | bla Δlmo2039 | This work |
| pJR8 | bla erm bgaB Δlmo2039 | This work |
| pJR10 | Phelp-lacO-lmo2039 lacI neo | This work |
| pJR17 | Phelp-lacO-lmo1438 lacI neo | This work |
| pJR18 | Phelp-lacO-lmo1892 lacI neo | This work |
| pJR20 | bla erm bgaB Δlmo2229 | This work |
| pJR24 | bla erm bgaB Δlmo0441 | This work |
| pSH239 | Phelp-gfp-lmo2229 neo | This work |
| pSH243 | erm ‘lmo2229’ | This work |
| pSH248 | Phelp-gfp-lmo1892 neo | This work |
| pSH249 | Phelp-gfp-lmo2039 neo | This work |
| pSH250 | Phelp-gfp-lmo1438 neo | This work |
| pSH251 | Phelp-gfp-lmo0441 neo | This work |
| pSH253 | bla erm bgaB 'gfp-lmo1892 | This work |
| pSH257 | bla erm bgaB Δlmo1892 | This work |
| pSH258 | Phelp-gfp-Δlmo2229 neo | This work |
| pSH277 | Phelp-gfp-Δlmo0441 neo | This work |
| pSH286 | Phelp-lacO-gfp-lmo2039 lacI neo | This work |
| L. monocytogenes strains | ||
| EGD-e | Wild type, serovar 1/2a strain | Lab collection |
| BUG1777 | ΔsrtA | (Bierne et al., 2002) |
| LMS22 | erm | (Halbedel et al., 2012) |
| LMS38 | attB::Phelp-gfp-lmo2229 neo | pSH239 → EGD-e |
| LMS44 | attB::Phelp-gfp-lmo1892 neo | pSH248 → EGD-e |
| LMS45 | attB::Phelp-gfp-lmo2039 neo | pSH249 → EGD-e |
| LMS46 | attB::Phelp-gfp-lmo1438 neo | pSH250 → EGD-e |
| LMS47 | attB::Phelp-gfp-lmo0441 neo | pSH251 → EGD-e |
| LMS57 | Δlmo1892 | pSH257 ↔ EGD-e |
| LMS64 | Δlmo2229 | pJR20 ↔ EGD-e |
| LMS141 | 'lmo2229' erm | pSH243 → EGD-e |
| LMS142 | Δlmo1892 erm | pAUL-A → LMS57 |
| LMS143 | Δlmo1892 'lmo2229' erm | pSH243 → LMS57 |
| LMJR5 | attB::Phelp-lacO-lmo2039 lacI neo | pJR10 → EGD-e |
| LMJR18 | Δlmo2039 attB::Phelp-lacO-lmo2039 lacI neo | pJR8 ↔ LMJR5 |
| LMJR20 | attB::Phelp-lacO-lmo1438 lacI neo | pJR17 → EGD-e |
| LMJR21 | Δlmo1892 attB::Phelp-lacO-lmo1892 lacI neo | pJR18 → LMS57 |
| LMJR27 | Δlmo1438 attB::Phelp-lacO-lmo1438 lacI neo | pJR5 ↔ LMJR20 |
| LMJR30 | Δlmo1892 Δlmo2229 attB::Phelp-lacO-lmo1892 lacI neo | pJR20 ↔ LMJR21 |
| LMJR41 | Δlmo0441 | pJR24 ↔ EGD-e |
The arrow (→) stands for a transformation event, and the double arrow (↔) indicates gene deletions obtained by chromosomal insertion and subsequent excision of pMAD plasmid derivatives (see Experimental procedures for details).
General methods, manipulation of DNA and oligonucleotide primers
Standard protocols were used for transformation of E. coli and isolation of plasmid DNA (Sambrook et al., 1989). Generation of electro-competent L. monocytogenes cells and transformation of plasmid DNA into L. monocytogenes were performed as described (Monk et al., 2008). Enzymatic modification of plasmid DNA was carried out as described by the instructions given by the manufacturers. Quickchange mutagenesis was employed for restriction free modification of plasmids (Zheng et al., 2004). DNA sequences of oligonucleotide primers are listed in Table 3.
Table 3.
Oligonucleotides used in this study
| Name | Sequence (5′→3′) |
|---|---|
| SHW164 | CTTAGGTACCTTTGTATAGTTCATCCATGCC |
| SHW165 | GACCGCTCGAGCCCAGCTTTTGTTCCCTTTAG |
| SHW166 | CTTAGGTACCGACAAATTCAAACAGCAACTTATTA |
| SHW167 | GACCGCTCGAGTTAATTACCTATCGAATCGATTAAG |
| SHW182 | AATGCAGTAGTTTCCATTGAAG |
| SHW183 | TTATTTGTATTTATCGCCTTCTGC |
| SHW184 | CTTAACTAGTTTTGTATAGTTCATCCATGCC |
| SHW185 | CTTAACTAGTGCAGATAAACCGCAGACAAG |
| SHW186 | GACCGCTCGAGTTAACTATCTGGAATTTTAACAGTGG |
| SHW187 | CTTAACTAGTAAACGGCGTATAGGTAACATG |
| SHW188 | GACCGCTCGAGTTAATTGAGCAAATCACCGATAC |
| SHW189 | CTTAACTAGTAAACTAAATTTTAGAAAAAAGAAAAAAG |
| SHW190 | GACCGCTCGAGTTAATTTTCGGTTTGTTCTGATTG |
| SHW191 | CTTAACTAGTGCTAGTTATGGTGGGAAAAAGAG |
| SHW192 | GACCGCTCGAGTTATTTATACATACTTTCAATTACAG |
| SHW215 | CACGCGTCGACTTAGTGAACACGGTCTGACATATAG |
| SHW216 | CACGCGTCGACGGAGCAAACAGTTCCAACCC |
| SHW217 | CACGCGTCGACTTACCACTCCCCATTACCAAAG |
| SHW218 | CACGCGTCGACTCCACGATGAAGCCACTCG |
| SHW271 | AAAGTGCGCATCTAAGGCTTGAAAAAATTTGATTTTGATAAG |
| SHW272 | TTTTTTCAAGCCTTAGATGCGCACTTTATCGGTTTTC |
| JR10 | CGGGATCCATGAGCAGAAAAATTGCAAGCATT |
| JR11 | ATATGTCGACTTTCACGCATAGTTACCTCACTTT |
| JR12 | ATATGTCGACAATTAATCAAAAACCACTTTCATTTAT |
| JR13 | GCATGCCATGGAGTCATTCTCTGTGAAAACGTCAA |
| JR20 | ATACGAGATCTGTCGTCGCGAAGTGAAACCAATTG |
| JR33 | TATTAGTCGACTTTCATTGACCATCTACCACTTTC |
| JR34 | TGAAAGTCGACTAATAATAGGAAGATAGAAGTATGTC |
| JR35 | CGTGGATCCGATCTTGAGACAAATTCATAAAC |
| JR48 | ACGCCGTTTATCCTTTTCCATGGGTTTCAC |
| JR49 | AAGGATAAACGGCGTATAGGTAACATGAG |
| JR53 | GCGCCCATGGCAGATAAACCGCAGACAAG |
| JR54 | GCGCGGATCCTTAACTATCTGGAATTTTAACAGTGG |
| JR57 | TCCCCCCGGGCAAACTAAATTTTAGAAAAAAGAAAAAAGATTC |
| JR58 | GCCCATCGATTTAATTTTCGGTTTGTTCTGATTGTGC |
Construction of strains expressing fluorescent fusion proteins
In order to obtain fluorescent variants of PBP proteins, GFP was fused to the N-terminus of all five HMW PBPs. The lmo2229 open reading frame was amplified in a polymerase chain reaction (PCR) using the primers SHW166/SHW167 and KpnI/XhoI-cloned into the backbone of plasmid pSH195 that had been linearized by PCR using the oligonucleotides SHW164/SHW165 and cut with the same enzymes. This resulted in plasmid pSH239. Likewise, the lmo1892 (SHW185/186), lmo2039 (SHW187/SHW188), lmo1438 (SHW189/SHW190) and lmo0441 (SHW191/SHW192) open reading frames were amplified and SpeI/XhoI-cloned into pSH195 which, however, had been linearized in a PCR using the primers SHW184/SHW165 and cut with SpeI/XhoI. The obtained plasmids were sequenced and named pSH248, pSH249, pSH250 and pSH251 respectively.
Plasmids pSH239 and pSH248-pSH251 were introduced into L. monocytogenes strains by electroporation, and kanamycin resistant clones were selected. Plasmid insertion at the attB site of the tRNAArg locus was verified by PCR.
Construction of pbp mutant strains
For deletion of an internal fragment of lmo1892 from the L. monocytogenes chromosome, plasmid pSH257 was constructed, which was obtained in two steps. First, the BamHI/XhoI 'gfp-lmo1892 fragment of plasmid pSH248 was cloned into pMAD, which had been linearized with BamHI/SalI. From the resulting plasmid (pSH253), a fragment corresponding to lmo1892 amino acid codons 206–485 was replaced by a TAA stop codon and a SalI site, using PCR and primers SHW215/SHW216. The resulting fragment was cut with SalI, self-ligated and yielded plasmid pSH257 upon transformation.
Plasmid pJR20 was constructed to facilitate deletion of an internal lmo2229 fragment. First, an internal fragment corresponding to amino acid residues 205–393 of the lmo2229 open reading frame of plasmid pSH239 was replaced by a stop codon followed by a SalI site in a PCR using the primer pair SHW217/SHW218. The obtained PCR fragment was cut with SalI, self-ligated and resulted in plasmid pSH258 after transformation. The EcoRI/BamHI 'gfp-Δlmo2229 fragment of plasmid pSH258 was then subcloned into EcoRI/BamHI cut pMAD to give plasmid pJR20. Plasmid pSH243, designed for insertional disruption of lmo2229, was constructed by blunt end cloning of an internal lmo2229 fragment, which was obtained using primers SHW182/SHW183, into SmaI cut pAUL-A. The reverse primer, SHW183, introduced a premature stop codon.
For deletion of the lmo1438 gene, plasmid pJR5 was constructed in two steps. First, up- and downstream regions of the lmo1438 gene were amplified using the primers JR10/JR11 and JR12/JR13 respectively. Both fragments were joined by ligation after their ends had been made compatible by SalI digestion and the joined fragment was amplified from the ligation mixture by PCR using JR10/JR13 as the primers. This fusion fragment was then blunt-end cloned into SmaI cut pUC19 and subcloned from the resulting plasmid (pJR4) into pMAD using NcoI restriction digestion.
Plasmid pJR8 was constructed for the removal of the lmo2039 gene and was generated in a similar way. Up- and downstream regions of lmo2039 were PCR amplified using primers JR20/JR33 and JR34/JR35, respectively, mixed and used as a template in a small overlapping extension PCR with the primer pair JR20/JR35. The fusion product was blunt-end cloned into SmaI cut pUC19 and subcloned from the resulting plasmid (pJR7) into pMAD using BamHI/BglII as the restriction enzymes.
To facilitate deletion of lmo0441 from the chromosome, plasmid pJR24 was generated. To this end, an internal lmo0441 fragment, corresponding to amino acids 168–501, was replaced by a premature stop codon from plasmid pSH251 in a PCR using the primer pair SHW271/SHW272. The BamHI/XhoI 'gfp-Δlmo0441 fragment of the resulting plasmid (pSH277) was then subcloned into BamHI/EcoRI cut pMAD.
For IPTG-inducible expression of lmo2039, the BamHI/XhoI gfp-lmo2039 fragment of plasmid pSH249 was first subcloned into BamHI/SalI cut pIMK3, resulting in plasmid pSH286. Later, the gfp part of the gfp-lmo2039 fusion of pSH286 was removed in a PCR using the primer pair JR48/JR49, and the resulting plasmid was named pJR10. For inducible expression of lmo1438, the lmo1438 open reading frame was amplified from chromosomal DNA with primers JR57/JR58, cut with ClaI/XmaI and ligated to the ClaI/XmaI cut backbone of pIMK3. This resulted in plasmid pJR17. Plasmid pJR18 was constructed to allow for IPTG-controllable expression of lmo1892. It was generated by amplification of the lmo1892 open reading frame, which had been obtained in a PCR using primers JR53/JR54, into pIMK3 after NcoI/BamHI digestion. Plasmids pJR10, pJR17 and pJR18 were inserted into the attB site of the tRNAArg locus as mentioned above. The chromosomal pbp genes were deleted in strain EGD-e or in strains containing ectopic, IPTG-controllable pbp genes using plasmids pSH257, pJR5, pJR8, pJR20, pJR24 and a plasmid integration/excision strategy described by others (Arnaud et al., 2004).
Isolation, separation and detection of proteins
Cells were cultivated in BHI broth at 37°C and harvested by centrifugation when the culture reached an optical density of 1.0. The cell pellet was washed once with ZAP buffer (10 mM Tris/HCl pH 7.5 and 200 mM NaCl), resuspended in 1 ml ZAP buffer also containing 1 mM phenylmethylsulfonyl fluoride (PMSF) and disrupted by sonication. Cell debris was removed by centrifugation (1 min, 13.000 rpm in a table top centrifuge), and membrane proteins were collected from the resulting supernatant by ultracentrifugation at 100.000 × g for 30 min at 4°C. The membrane fraction in the resulting pellet was resuspended in 100 μl ZAP buffer. In order to detect PBPs in SDS-PAGE gels, aliquots corresponding to 20 μg of membrane proteins were incubated with 3 μM bocillin-fl (Molecular Probes) for 20 min at 37°C. The binding reaction was stopped by addition of loading dye and incubation for 5 min at 65°C. Samples were separated by SDS-PAGE (8% acrylamide, 0.067% bis-acrylamide), and PBPs were detected using a Fuji raytest FLA 2000 fluorescence scanner.
For isolation of surface proteins, a previously published protocol was used with minor modifications (Jonquieres et al., 1999). Bacteria were grown in 50 ml BHI broth at 37°C until an OD600 of 2.0 and harvested by centrifugation. Proteins from the culture supernatant were precipitated using trichloroacetic acid (TCA) (Halbedel et al., 2014), and the cell pellet was washed once with phosphate buffered saline (PBS), once with TS buffer (10 mM Tris-HCl pH 6.9, 10 mM MgCl2, 0.5 M sucrose) and resuspended in 1.5 ml TS buffer containing 1 mM PMSF, 100 μg ml−1 lysozyme and 250 μg ml−1 RNase. After incubation for 1 h at 37°C with gentle shaking, cells were removed by centrifugation (13.000 rpm for 5 min in a table top centrifuge), and the released surface proteins were precipitated from the supernatant by addition of TCA (16% final concentration) over night at 4°C. Precipitated proteins were collected by centrifugation and dissolved in 100 μl 8 M urea.
In order to detect proteins by Western blotting, protein extracts were separated by standard SDS polyacrylamide gel electrophoresis and transferred onto positively charged polyvinylidene fluoride (PVDF) membranes using a semi-dry transfer unit. Proteins were immune-stained using polyclonal rabbit antisera recognizing GFP (lab stock), Lmo610 and Lmo0880 (Quereda et al., 2013) or monoclonal mouse antibodies against InlA, InlB (Lingnau et al., 1995) and p60 (MyBioSource, USA) as the primary and anti-rabbit or anti-mouse immunoglobulins G conjugated to horseradish peroxidase as the secondary antibody. The ECL(Tm) chemiluminescence detection system (Thermo Scientific) was used for detection of the peroxidase conjugates on the PVDF membrane. Zymography was performed as described earlier (Halbedel et al., 2014).
Determination of minimal inhibitory concentrations
Test strips with the following concentration gradients were used to determine the MICs of selected antibiotics: Amoxicillin (0.016–256 μg ml−1), ampicillin (0.016–256 μg ml−1), gentamicin (0.016–256 μg ml−1), meropenem (0.002–32 μg ml−1), penicillin G (0.016–256 μg ml−1), vancomycin (0.016–256 μg ml−1, all from Bestbiondx, Germany). L. monocytogenes strains were cultivated on BHI agar, containing 1 mM IPTG where necessary, at 37°C overnight, and several colonies were resuspended in 5 ml BHI broth. This resuspension was used to swab-inoculate BHI agar plates. MIC test strips were placed on top of the agar surface, and the plates were incubated at 37°C for 1 day.
Autolysis assay
Bacterial strains were cultivated in BHI broth (containing 1 mM IPTG where required) at 37°C up to an OD600 of around 0.8. Cells were collected by centrifugation (6000 × g, 5 min) and resuspended in 50 mM Tris/HCl pH 8.0 to an optical density of OD600 = 0.6. Penicillin was added to a final concentration of 25 μg ml−1, and the cells were shaken at 37°C. Autolysis was recorded by measuring the decline in optical density (λ = 600 nm) over time.
Peptidoglycan analysis
Cell wall from L. monocytogenes was isolated as described for S. pneumoniae (Bui et al., 2012). Wall teichoic acid was released by incubating the cell wall with 48% hydrofluoric acid at 4°C for 48 h, followed by washing of the resulting peptidoglycan as described (Bui et al., 2012). Peptidoglycan (2.5 mg ml−1) was incubated with 25 μg ml−1 of the muramidase cellosyl in 20 mM sodium phosphate pH 4.8 at 37°C for 24 h, followed by heating at 100°C for 10 min to inactivate the enzyme. The sample was centrifuged for 30 min, and the supernatant was reduced with sodium borohydride as described (Bui et al., 2012). The resulting reduced muropeptides were separated by HPLC (Glauner, 1988) at conditions optimized for Listeria muropeptides. A Prontosil 120-3-C18 AQ reversed phase column (250 × 4.6 mm, 3 μm) from Bischoff (Leonberg, Germany) was operated at 55°C at a flow rate of 0.5 ml min−1 with a 135-min gradient from 10 mM sodium phosphate, pH 6.0, 0.065% NaN3 to 10 mM sodium phosphate, pH 6.0, 15% MeOH, 0.033% NaN3. Muropeptides were detected by absorbtion at 205 nm. The chromatograms were analyzed using the LAURA software version 4.1.7.70 (LabLogic Systems Ltd). The main peaks were collected and analyzed by ESI-MS/MS as described (Bui et al., 2009). MS data obtained were consistent with previously published muropeptide structures (Boneca et al., 2007).
In vitro virulence assays
To record invasion of L. monocytogenes strains into HeLa cells, an infection protocol, which we had described elsewhere, was used (Halbedel et al., 2012). Briefly, 105 HeLa cells were seeded into the wells of a 24–multi-well plate and infected with 2 × 106 bacterial cells taken from logarithmically growing cultures, which were cultivated in BHI broth (supplemented with 1 mM IPTG where necessary) at 37°C. Sampling was performed by lysing the cells in 1 ml of ice-cold PBS containing 0.1% Triton X-100 right after infection. In order to determine the number of recovered bacteria, serial dilutions were plated on BHI agar plates (containing 1 mM IPTG where necessary) and incubated over night at 37°C.
Plaque formation assays to investigate cell-to-cell spread of L. monocytogenes strains and assays monitoring intracellular multiplication in HeLa cells and J774.A1 mouse ascites macrophages were essentially performed as described earlier (Halbedel et al., 2012; 2014).
Microscopy
Sample aliquots (0.4 μl) from logarithmically growing cultures were spotted onto microscope slides coated with a thin film of agarose (1.5% in distilled water), air-dried and covered with a coverslip. For staining of membranes, 1 μl of nile red (100 μg ml−1 in dimethyl sulfoxide [DMSO]) was added to 100 μl of culture, and the mixture was shaken for 20 min before the cells were prepared for microscopy. Images were taken with a Nikon Eclipse Ti microscope coupled to a Nikon DS-MBWc CCD camera and processed using the NIS elements AR software package (Nikon).
SEM
L. monocytogenes strains maintained in BHI broth were sedimented at 2000 g for 2 min. The pellets were collected in 1 ml of fixative (2 h at room temperature in 1% paraformaldehyde, 2.5% glutardialdehyde in 0.05 M HEPES buffer, pH 7.2) and stored in a refrigerator at 4°C until further processing. Suspensions were sedimented at 2000 g for 10 min and washed in double distilled water. The pellets were resuspended in 100 μl of water, and 30 μl of the suspension was dropped onto a coverslip that had been covered by a carbon layer and treated with alcian blue (1% in 1% acetic acid, Sigma Aldrich) to improve adhesion. After 30 min of adsorption in a wet chamber, the coverslips were transferred to multi-well plates (12-well, TPP, Switzerland) and covered with 2.5% glutardialdehyde (TAAB, Laboratories Equipment Ltd., UK) in HEPES buffer for further fixation (30 min at room temperature). The coverslips were washed five times in water, dehydrated in an ethanol series (30, 50, 70, 90 and 96% for 15 min each and absolute ethanol for 30 min) and critical point dried (CPD 030, Bal-Tec, Liechtenstein) using liquid carbon dioxide. Finally, the sample surface was coated with a gold/palladium layer (approx. 2 nm) in a sputter coater (E5100, Polaron, UK) and examined under high vacuum conditions in a field emission SEM (Leo 1530 Gemini, Carl Zeiss Microscopy, Germany) at 5 KV using the in-lens secondary electron detector.
Ultrathin section transmission electron microscopy
L. monocytogenes cells were fixed, sedimented and washed (0.05 M HEPES buffer, pH 7.2) as described above. Washed pellets were embedded in low melting point agarose (3%, mixed 1:1 with the sample; Sigma, Germany), trimmed into small pieces (about 1 × 1 mm) and fixed in 2.5% glutardialdehyde in 0.05 M HEPES buffer, pH 7.2. Samples were postfixed in osmium tetroxide (1% in water) for 60 min and 2 h in uranyl acetate (2% in water) at room temperature and dehydrated in an ethanol series (30, 50, 70, 90, 96% and absolute ethanol for 20 min). Samples were infiltrated with a mixture (1:1) of ethanol and resin (LR White hard grade; Science Services, Germany), embedded in pure resin and finally polymerized at 60°C overnight. Ultrathin sections of about 60–70 nm were cut with an ultramicrotome (Leica Microsystems, Germany), poststained with uranyl acetate followed by lead citrate. Sections were examined using a JEM-2100 transmission electron microscope (Jeol, Japan) at 200 KV. Images were recorded at full resolution of 2k × 2k with a Veleta side-mounted CCD camera (Olympus SIS, Germany).
Acknowledgments
This work was supported by the DFG (HA 6830/1-1, to SH), the Fonds der chemischen Industrie (FCI, to SH) and the BBSRC (BB/G015902/1, to WV). We are grateful to Pascale Cossart (Institut Pasteur, France) for the gift of the ΔsrtA mutant strain, to Francisco García-del Portillo (Spanish National Biotechnology Center, Spain) for kindly sharing antisera against LPXTG proteins and to Torsten Hain (University of Gießen, Germany) for sharing InlA and InlB antibodies. We thank Richard Daniel (Newcastle University, UK) for helpful discussions, Stephanie Großhennig for help with some experiments, and Birgit Hahn and Petra Kaiser for excellent technical assistance. Karan G. Kaval is acknowledged for critical comments on the manuscript.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
References
- Allerberger F. Wagner M. Listeriosis: a resurgent foodborne infection. Clin Microbiol Infect. 2010;16:16–23. doi: 10.1111/j.1469-0691.2009.03109.x. [DOI] [PubMed] [Google Scholar]
- Arnaud M, Chastanet A. Debarbouille M. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol. 2004;70:6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann B, Raffelsbauer D, Kuhn M, Goetz M, Hom S. Goebel W. InlA- but not InlB-mediated internalization of Listeria monocytogenes by non-phagocytic mammalian cells needs the support of other internalins. Mol Microbiol. 2002;43:557–570. doi: 10.1046/j.1365-2958.2002.02767.x. [DOI] [PubMed] [Google Scholar]
- Bierne H. Cossart P. Listeria monocytogenes surface proteins: from genome predictions to function. Microbiol Mol Biol Rev. 2007;71:377–397. doi: 10.1128/MMBR.00039-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H, Mazmanian SK, Trost M, Pucciarelli MG, Liu G, Dehoux P, et al. Inactivation of the srtA gene in Listeria monocytogenes inhibits anchoring of surface proteins and affects virulence. Mol Microbiol. 2002;43:869–881. doi: 10.1046/j.1365-2958.2002.02798.x. [DOI] [PubMed] [Google Scholar]
- Boneca IG, Dussurget O, Cabanes D, Nahori MA, Sousa S, Lecuit M, et al. A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc Natl Acad Sci USA. 2007;104:997–1002. doi: 10.1073/pnas.0609672104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bublitz M, Polle L, Holland C, Heinz DW, Nimtz M. Schubert WD. Structural basis for autoinhibition and activation of Auto, a virulence-associated peptidoglycan hydrolase of Listeria monocytogenes. Mol Microbiol. 2009;71:1509–1522. doi: 10.1111/j.1365-2958.2009.06619.x. [DOI] [PubMed] [Google Scholar]
- Bui NK, Gray J, Schwarz H, Schumann P, Blanot D. Vollmer W. The peptidoglycan sacculus of Myxococcus xanthus has unusual structural features and is degraded during glycerol-induced myxospore development. J Bacteriol. 2009;191:494–505. doi: 10.1128/JB.00608-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui NK, Eberhardt A, Vollmer D, Kern T, Bougault C, Tomasz A, et al. Isolation and analysis of cell wall components from Streptococcus pneumoniae. Anal Biochem. 2012;421:657–666. doi: 10.1016/j.ab.2011.11.026. [DOI] [PubMed] [Google Scholar]
- Cabanes D, Dussurget O, Dehoux P. Cossart P. Auto, a surface associated autolysin of Listeria monocytogenes required for entry into eukaryotic cells and virulence. Mol Microbiol. 2004;51:1601–1614. doi: 10.1111/j.1365-2958.2003.03945.x. [DOI] [PubMed] [Google Scholar]
- Carroll SA, Hain T, Technow U, Darji A, Pashalidis P, Joseph SW. Chakraborty T. Identification and characterization of a peptidoglycan hydrolase, MurA, of Listeria monocytogenes, a muramidase needed for cell separation. J Bacteriol. 2003;185:6801–6808. doi: 10.1128/JB.185.23.6801-6808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty T, Leimeister-Wächter M, Domann E, Hartl M, Goebel W, Nichterlein T. Notermans S. Coordinate regulation of virulence genes in Listeria monocytogenes requires the product of the prfA gene. J Bacteriol. 1992;174:568–574. doi: 10.1128/jb.174.2.568-574.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P. Toledo-Arana A. Listeria monocytogenes, a unique model in infection biology: an overview. Microbes Infect. 2008;10:1041–1050. doi: 10.1016/j.micinf.2008.07.043. [DOI] [PubMed] [Google Scholar]
- Daniel RA, Harry EJ. Errington J. Role of penicillin-binding protein PBP 2B in assembly and functioning of the division machinery of Bacillus subtilis. Mol Microbiol. 2000;35:299–311. doi: 10.1046/j.1365-2958.2000.01724.x. [DOI] [PubMed] [Google Scholar]
- Dell'Era S, Buchrieser C, Couve E, Schnell B, Briers Y, Schuppler M. Loessner MJ. Listeria monocytogenes L-forms respond to cell wall deficiency by modifying gene expression and the mode of division. Mol Microbiol. 2009;73:306–322. doi: 10.1111/j.1365-2958.2009.06774.x. [DOI] [PubMed] [Google Scholar]
- Freitag NE, Port GC. Miner MD. Listeria monocytogenes – from saprophyte to intracellular pathogen. Nat Rev Microbiol. 2009;7:623–628. doi: 10.1038/nrmicro2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard JL, Berche P, Frehel C, Gouin E. Cossart P. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell. 1991;65:1127–1141. doi: 10.1016/0092-8674(91)90009-n. [DOI] [PubMed] [Google Scholar]
- Gamba P, Veening JW, Saunders NJ, Hamoen LW. Daniel RA. Two-step assembly dynamics of the Bacillus subtilis divisome. J Bacteriol. 2009;191:4186–4194. doi: 10.1128/JB.01758-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, et al. Comparative genomics of Listeria species. Science. 2001;294:849–852. doi: 10.1126/science.1063447. [DOI] [PubMed] [Google Scholar]
- Glauner B. Separation and quantification of muropeptides with high-performance liquid chromatography. Anal Biochem. 1988;172:451–464. doi: 10.1016/0003-2697(88)90468-x. [DOI] [PubMed] [Google Scholar]
- Grant MH, Ravreby H. Lorber B. Cure of Listeria monocytogenes meningitis after early transition to oral therapy. Antimicrob Agents Chemother. 2010;54:2276–2277. doi: 10.1128/AAC.01815-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayo S, Join-Lambert O, Desroches MC. Le Monnier A. Comparison of the in vitro efficacies of moxifloxacin and amoxicillin against Listeria monocytogenes. Antimicrob Agents Chemother. 2008;52:1697–1702. doi: 10.1128/AAC.01211-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinane CM, Cotter PD, Ross RP. Hill C. Contribution of penicillin-binding protein homologs to antibiotic resistance, cell morphology, and virulence of Listeria monocytogenes EGDe. Antimicrob Agents Chemother. 2006;50:2824–2828. doi: 10.1128/AAC.00167-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbedel S, Hahn B, Daniel RA. Flieger A. DivIVA affects secretion of virulence-related autolysins in Listeria monocytogenes. Mol Microbiol. 2012;83:821–839. doi: 10.1111/j.1365-2958.2012.07969.x. [DOI] [PubMed] [Google Scholar]
- Halbedel S, Reiss S, Hahn B, Albrecht D, Mannala GK, Chakraborty T, et al. A systematic proteomic analysis of Listeria monocytogenes house-keeping protein secretion systems. Mol Cell Proteomics. 2014;13:3063–3081. doi: 10.1074/mcp.M114.041327. . doi: 10.1074/mcp.M114.041327; Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof H. Listeriosis: therapeutic options. FEMS Immunol Med Microbiol. 2003;35:203–205. doi: 10.1016/S0928-8244(02)00466-2. [DOI] [PubMed] [Google Scholar]
- Hof H. An update on the medical management of listeriosis. Expert Opin Pharmacother. 2004;5:1727–1735. doi: 10.1517/14656566.5.8.1727. [DOI] [PubMed] [Google Scholar]
- Holch A, Webb K, Lukjancenko O, Ussery D, Rosenthal BM. Gram L. Genome sequencing identifies two nearly unchanged strains of persistent Listeria monocytogenes isolated at two different fish processing plants sampled 6 years apart. Appl Environ Microbiol. 2013;79:2944–2951. doi: 10.1128/AEM.03715-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonquieres R, Bierne H, Fiedler F, Gounon P. Cossart P. Interaction between the protein InlB of Listeria monocytogenes and lipoteichoic acid: a novel mechanism of protein association at the surface of gram-positive bacteria. Mol Microbiol. 1999;34:902–914. doi: 10.1046/j.1365-2958.1999.01652.x. [DOI] [PubMed] [Google Scholar]
- Klumpp J, Staubli T, Schmitter S, Hupfeld M, Fouts DE. Loessner MJ. Genome sequences of three frequently used Listeria monocytogenes and Listeria ivanovii strains. Genome Announc. 2014;2:e00404-14. doi: 10.1128/genomeA.00404-14. , and: pii:.doi: 10.1128/genomeA.00404-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsak D, Vollmer W. Markiewicz Z. Listeria monocytogenes EGD lacking penicillin-binding protein 5 (PBP5) produces a thicker cell wall. FEMS Microbiol Lett. 2005;251:281–288. doi: 10.1016/j.femsle.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Korsak D, Markiewicz Z, Gutkind GO. Ayala JA. Identification of the full set of Listeria monocytogenes penicillin-binding proteins and characterization of PBPD2 (Lmo2812) BMC Microbiol. 2010;10:239. doi: 10.1186/1471-2180-10-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk-Balska A, Popowska M. Markiewicz Z. Re-evaluation of the significance of penicillin binding protein 3 in the susceptibility of Listeria monocytogenes to beta-lactam antibiotics. BMC Microbiol. 2012;12:57. doi: 10.1186/1471-2180-12-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun M, Mengaud J, Ohayon H, Nato F. Cossart P. Internalin must be on the bacterial surface to mediate entry of Listeria monocytogenes into epithelial cells. Mol Microbiol. 1996;21:579–592. doi: 10.1111/j.1365-2958.1996.tb02566.x. [DOI] [PubMed] [Google Scholar]
- Lemaire S, Van Bambeke F, Mingeot-Leclercq MP. Tulkens PM. Activity of three β-lactams (ertapenem, meropenem and ampicillin) against intraphagocytic Listeria monocytogenes and Staphylococcus aureus. J Antimicrob Chemother. 2005;55:897–904. doi: 10.1093/jac/dki094. [DOI] [PubMed] [Google Scholar]
- Lingnau A, Domann E, Hudel M, Bock M, Nichterlein T, Wehland J. Chakraborty T. Expression of the Listeria monocytogenes EGD inlA and inlB genes, whose products mediate bacterial entry into tissue culture cell lines, by PrfA-dependent and -independent mechanisms. Infect Immun. 1995;63:3896–3903. doi: 10.1128/iai.63.10.3896-3903.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machata S, Hain T, Rohde M. Chakraborty T. Simultaneous deficiency of both MurA and p60 proteins generates a rough phenotype in Listeria monocytogenes. J Bacteriol. 2005;187:8385–8394. doi: 10.1128/JB.187.24.8385-8394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson DC. Popham DL. Peptidoglycan synthesis in the absence of class A penicillin-binding proteins in Bacillus subtilis. J Bacteriol. 2003;185:1423–1431. doi: 10.1128/JB.185.4.1423-1431.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matano S, Satoh S, Harada Y, Nagata H. Sugimoto T. Antibiotic treatment for bacterial meningitis caused by Listeria monocytogenes in a patient with multiple myeloma. J Infect Chemother. 2010;16:123–125. doi: 10.1007/s10156-009-0014-3. [DOI] [PubMed] [Google Scholar]
- Monk IR, Gahan CG. Hill C. Tools for functional postgenomic analysis of Listeria monocytogenes. Appl Environ Microbiol. 2008;74:3921–3934. doi: 10.1128/AEM.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz P, Rojas L, Bunsow E, Saez E, Sanchez-Cambronero L, Alcala L, et al. Listeriosis: an emerging public health problem especially among the elderly. J Infect. 2012;64:19–33. doi: 10.1016/j.jinf.2011.10.006. [DOI] [PubMed] [Google Scholar]
- Murray T, Popham DL. Setlow P. Identification and characterization of pbpC, the gene encoding Bacillus subtilis penicillin-binding protein 3. J Bacteriol. 1996;178:6001–6005. doi: 10.1128/jb.178.20.6001-6005.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilgrim S, Kolb-Mäurer A, Gentschev I, Goebel W. Kuhn M. Deletion of the gene encoding p60 in Listeria monocytogenes leads to abnormal cell division and loss of actin-based motility. Infect Immun. 2003;71:3473–3484. doi: 10.1128/IAI.71.6.3473-3484.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popham DL. Setlow P. Phenotypes of Bacillus subtilis mutants lacking multiple class A high-molecular-weight penicillin-binding proteins. J Bacteriol. 1996;178:2079–2085. doi: 10.1128/jb.178.7.2079-2085.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quereda JJ, Pucciarelli MG, Botello-Morte L, Calvo E, Carvalho F, Bouchier C, et al. Occurrence of mutations impairing sigma factor B (SigB) function upon inactivation of Listeria monocytogenes genes encoding surface proteins. Microbiology. 2013;159:1328–1339. doi: 10.1099/mic.0.067744-0. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF. Maniatis T. Molecular Cloning: A Laboratory Manual. NY: Cold Spring Harbor Laboratory Press, Cold Spring Harbor; 1989. p. 3 v. [Google Scholar]
- Swaminathan B. Gerner-Smidt P. The epidemiology of human listeriosis. Microbes Infect. 2007;9:1236–1243. doi: 10.1016/j.micinf.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Van de Velde S, Carryn S, Van Bambeke F, Hill C, Tulkens PM. Sleator RD. Penicillin-binding proteins (PBP) and Lmo0441 (a PBP-like protein) play a role in Beta-lactam sensitivity of Listeria monocytogenes. Gut Pathog. 2009;1:23. doi: 10.1186/1757-4749-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente MF, Perez-Daz JC, Baquero F, Angel de Pedro M. Berenguer J. Penicillin-binding protein 3 of Listeria monocytogenes as the primary lethal target for beta-lactams. Antimicrob Agents Chemother. 1990;34:539–542. doi: 10.1128/aac.34.4.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer W. Tomasz A. The pgdA gene encodes for a peptidoglycan N-acetylglucosamine deacetylase in Streptococcus pneumoniae. J Biol Chem. 2000;275:20496–20501. doi: 10.1074/jbc.M910189199. [DOI] [PubMed] [Google Scholar]
- Zawadzka-Skomial J, Markiewicz Z, Nguyen-Disteche M, Devreese B, Frere JM. Terrak M. Characterization of the bifunctional glycosyltransferase/acyltransferase penicillin-binding protein 4 of Listeria monocytogenes. J Bacteriol. 2006;188:1875–1881. doi: 10.1128/JB.188.5.1875-1881.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Baumann U. Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.