

Abstract
How clathrin-mediated endocytosis (CME) retrieves vesicle proteins into newly formed synaptic vesicles (SVs) remains a major puzzle. Besides its roles in stimulating clathrin-coated vesicle formation and regulating SV size, the clathrin assembly protein AP180 has been identified as a key player in retrieving SV proteins. The mechanisms by which AP180 recruits SV proteins are not fully understood. Here, we show that following acute inactivation of AP180 in Drosophila, SV recycling is severely impaired at the larval neuromuscular synapse based on analyses of FM 1-43 uptake and synaptic ultrastructure. More dramatically, AP180 activity is important to maintain the integrity of SV protein complexes at the plasma membrane during endocytosis. These observations suggest that AP180 normally clusters SV proteins together during recycling. Consistent with this notion, SV protein composition and distribution are altered in AP180 mutant flies. Finally, AP180 co-immunoprecipitates with SV proteins, including the vesicular glutamate transporter and neuronal synaptobrevin. These results reveal a new mode by which AP180 couples protein retrieval to CME of SVs. AP180 is also genetically linked to Alzheimer's disease. Hence, the findings of this study may provide new mechanistic insight into the role of AP180 dysfunction in Alzheimer's disease.
Keywords: AP180, clathrin, Drosophila, endocytosis, exocytosis, synaptic vesicles, synaptobrevin, synaptotagmin, vesicular glutamate transporter
Synaptic vesicles (SVs) are principal functional units of the chemical synapse (1–3). SVs store and release neurotransmitters from presynaptic terminals in a manner that depends absolutely on precise control by synaptic trafficking machinery (4). SVs traffic in a cycle composed of exocytosis, recycling and spatial placement of SVs in the synapse to prepare the vesicle for repeated rounds of fusion and fission (5). Nerve cell communication is sustained only when SVs are properly regulated for rapid recycling (3,5,6). While a number of the molecules regulating SV trafficking have been discovered, many questions about the process remain unanswered.
A major unresolved issue for understanding SV recycling is how SVs maintain their identities and functionality after repeated cycles of fusion and fission. SVs differ significantly in composition from the plasma membrane (PM), notably with their unique sets of vesicular proteins such as vesicular transmitter transporter, calcium sensors and v-SNAREs (7), and SVs must retain their lipid and protein components in order to sustain continuous synaptic function (8–10). How SVs accomplish this task will depend on how they interact with the PM during exocytosis. Several complementary models exist to explain reuptake and reconstitution of SVs. One model is ‘kiss and run’, which postulates that SVs are recycled by closing a fusion pore formed between an SV and the PM following the release of transmitter (11). This mode is believed to operate during low levels of nerve activity (4). Because of the transient nature (∼<400 milliseconds) of the fusion pore, kiss and run may not have sufficient time for SV components to intermingle extensively with the PM and thus most likely intact SVs can be readily recycled.
A second model, and the one believed to predominate at most synapses and during high levels of neuronal activity, is that SVs completely fuse with PM during exocytosis (2). Under such circumstances, the recycling machinery must undergo at least three processes to faithfully reconstitute functional SVs: (i) correct recognition of SV components amid the myriad proteins and lipids in the PM; (ii) sorting and retrieving the SV proteins and (iii) regeneration of SVs on a rapid timescale (on the order of seconds). The general consensus is that synapses utilize clathrin-mediated endocytosis (CME) to reconstitute and reform SVs from the PM (4). CME can reform single SVs from the PM or from an endocytotic intermediate, such as endosomes or cisternae, which form typically via bulk uptake of the PM (12–14).
One clear challenge is to understand the mechanism by which CME couples SV reformation with SV protein retrieval. Some insights into this process were revealed in biochemical and genetic studies of proteins involved in CME. Two clathrin accessory proteins, Stoned and AP180, play a role in sorting and retrieving SV proteins (15–22). Neither protein is essential for SV recycling but they significantly modulate the rate and fidelity of SV recycling. Stoned is shown to bind directly with the SV protein synaptotagmin I (Syt1) and selectively retrieve it into newly formed vesicles (16,23). AP180 participates in the retrieval of the SV protein neuronal synaptobrevin (nSyb) (19,22,24,25) and may also be involved in retrieval of other SV proteins (22,24). In the Drosophila AP180 mutant, lap, nSyb and other SV proteins were mislocalized to axonal membranes where these proteins are usually absent (22). The function of these proteins has also been shown to be conserved in mammals in which perturbation of Stonin (the mammalian homolog of Stoned) and AP180 leads to vesicle protein retrieval defects (17,24,26).
A potential complication of studying the dynamic process of SV recycling in mutant animals is the effect of developmental compensation, which can allow for compensatory proteins to function in order to keep the mutant animal alive. This could mask the interpretation of the direct function of the protein of interest. To circumvent these problems, Davis and coworkers developed a technique to acutely and selectively inactivate molecules involved in CME using fluorescein-assisted light inactivation (FALI) (6,27). Here, we use the FALI acute inactivation technique to address the direct consequence of AP180 (LAP, Like-AP180) inactivation on SV recycling, protein sorting and retrieval. Our data show that LAP is necessary during active synaptic transmission for endocytosis, but not exocytosis. We further show that under normal conditions SV proteins are recycled together, but these same proteins diffuse apart from one another after LAP is acutely inactivated. This is likely owing to the absence of LAP binding to one or more of these proteins during endocytosis, as we show that LAP co-immunoprecipitates with a number of different vesicle proteins. We propose a model in which LAP functions to couple CME to the retrieval of multiple SV proteins.
Results
lap4C transgene rescues lap mutants
The synapse is a highly dynamic system with built-in mechanisms to compensate for deleterious perturbations to its function, particularly during development (4). The fundamental role for LAP in CME makes it difficult to separate basic developmental and maintenance functions from specific SV trafficking in lap mutants. Indeed, active zones are mislocalized, and neuromuscular junctions (NMJs) are overgrown in lap mutants, suggesting a developmental role for LAP in the synapse (22). Therefore, we sought to generate a tool that would allow for the assessment of the acute role of LAP in SV protein sorting and retrieval, separate from its developmental roles.
We took advantage of recent advances in Drosophila genetics to generate flies amenable to acute photoinactivation of a transgenic gene using FALI. This method is a genetically directed approach that enables one to study the immediate, acute inactivation of protein function with high spatial (<40 Å) and temporal (<30 seconds) resolution (28,29). Two fly labs have implemented FALI, mediated by the membrane-permeable reagent FlAsH-EDT2 (FlAsH-FALI), to address the role of putative regulators of SV cycling in Drosophila (6,27,30). We generated fly lines containing a transgenic lap consisting of its full upstream and downstream untranslated regions, all intact introns and exons, with either an N- or C-terminal tetracysteine tag amenable to FALI-mediated inactivation along with a FLAG tag (in short lap4C, Figure 1A). These transgenes, when placed in a lap mutant background, are capable of producing a tagged wild-type LAP protein [ Figure 1B, detectable at the NMJ (not shown)] and rescuing synaptic transmission defects (Figure 1C,D) and mutant larval lethal to adult viable.
Figure 1. Genetic rescue of lap mutant.
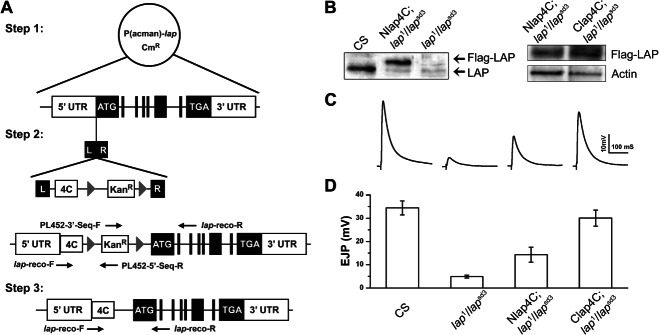
A) P(acman) vectors containing the full genomic sequence of LAP were modified by attaching a FALI-sensitive Flag-tetracysteine (4C) cassette to either the N- or C-terminus and incorporated into flies using PhiC-31-mediated transgenesis (see Materials and Methods for details). B) Left panel: Equal concentrations of total protein from adult fly head extracts as determined by BSA protein assay were loaded on SDS–PAGE gels and analyzed by Western blot using an antibody to the Drosophila LAP protein. Although endogenous LAP is present in control extracts (CS), its level is markedly reduced in both transgenic Nlap4C; lap1/lapsd3 and the mutant lap1/lapsd3 flies. Further, a band representing Flag-tagged LAP (with a slightly larger MW) is present only in the transgenic flies. Right panel: FLAG-tagged lap4C expression levels are examined using Western blot with actin as a loading control. Clap4C appears to be expressed at higher levels compared to Nlap4C, which may account for the better rescue of synaptic physiology shown below. C and D) The amplitude of EJPs in lap mutants is drastically reduced relative to control animals [down from 34.4 ± 3.0 mV (n = 8) in the control to 4.9 ± 0.7 mV (n = 7) in the lap mutant]. The Nlap4C; lap1/lapsd3 transgenic flies show partial rescue (14.3 ± 3.2 mV, n = 6) in the mutant background, whereas the Clap4C; lap1/lapsd3 transgenic flies exhibit almost complete rescue of physiology (30.0 ± 3.4 mV, n = 6).
SV recycling, but not exocytosis, is significantly blocked following acute LAP inactivation
LAP in flies (22,31,32), along with its homologs in mammals (24,25,33), the squid (34) and Caenorhabditis elegans (19), has been strongly implicated in SV recycling. However, previous data have relied on work in systems in which LAP or its homologs are either absent throughout development or where protein levels were substantially decreased for periods of hours or days prior to the experiment. Therefore, it has been difficult to separate non-SV endocytotic functions from the observed recycling defects. FM 1-43 is a dye that becomes incorporated into SVs upon stimulation and is often used as a live indicator of SV endocytosis (35). To determine whether LAP is indeed necessary for SV endocytosis, we performed experiments where we coupled acute inactivation of LAP with uptake of FM 1-43 following 1 min of stimulation with 90 mm potassium (Figure 2A). In control larval NMJs FM 1-43 is observed within synaptic boutons where SVs are localized. However, only small punctae of FM 1-43 dyes are detected within synaptic boutons in LAP-inactivated larvae. These results suggest that SV recycling is severely impaired in the absence of LAP function, a conclusion consistent with earlier studies (22,31,32).
Figure 2. Acute inactivation of LAP blocks SV recycling but does not affect SV exocytosis.
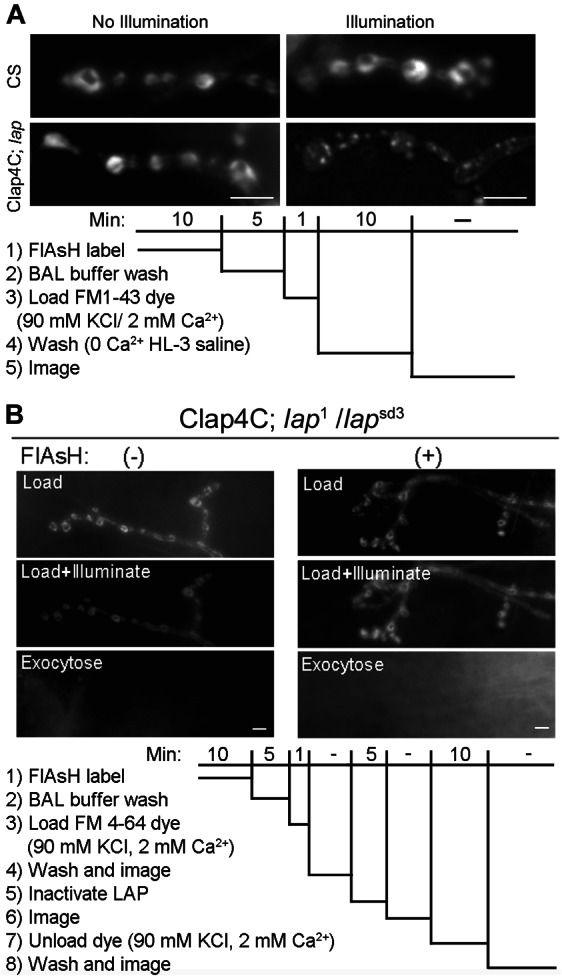
A) FM 1-43 dye uptake at third instar larval NMJs is normal in both wild-type or Clap4C-rescued lap mutant flies following 1-min nerve stimulation in the presence of the dye and without 488-nm light exposure. However, acute inactivation of LAP by 488-nm light illumination leads to the appearance of dye-positive punctate (note the normal uptake in wild-type NMJs following the light exposure). Presumably, these are large cisternae that form when CME is blocked. lap = lap1/lapsd3. B) For analysis of SV exocytosis we used the lipophilic dye FM 4-64 because it is not bleached by the 488-nm light used to inactivate LAP. FM 4-64 shows typical SV endocytosis in the presence of high K+. After the dye is loaded we inactivated LAP and then give a second round of stimulation in the absence of dye, FM 4-64 is exocytosed from the boutons and washed away. Inactivation of LAP has no effect on the ability of the FM dye to be exocytosed. A and B) Protocol for analysis of SV endocytosis and exocytosis using the lipophilic dye FM 1-43 or FM 4-64, respectively. For details, see Materials and Methods. Scale bars = 10 µm.
We next addressed whether the defect in SV recycling could be a secondary consequence of impaired SV exocytosis following acute LAP inactivation. In this set of experiments, we used the FM dye variant, FM 4-64, to label SVs as it does not bleach during the 5-min exposure to the 488-nm light used to inactivate LAP. As shown in Figure 2B, potassium stimulation of the motor nerve was sufficient to unload FM 4-64 from synaptic boutons at the larval NMJ following acute inactivation of LAP. Consistently, electrophysiological recordings from the larval body wall muscle showed that evoked transmitter release remained normal immediately following LAP acute inactivation (Figure 3). We note that the 488-nm light treatment caused a significant depolarization of the muscle resting potential, which likely accounts for the small but statistically insignificant reduction in excitatory junction potential (EJP) amplitude in both FlAsH-EDT2-treated or untreated larvae (both groups were Clap4C; lap1/lapsd3). These imaging and electrophysiological results provide the first evidence that LAP is not required for SV exocytosis, but it functions to sustain SV recycling.
Figure 3. Acute inactivation of LAP does not affect transmitter release.
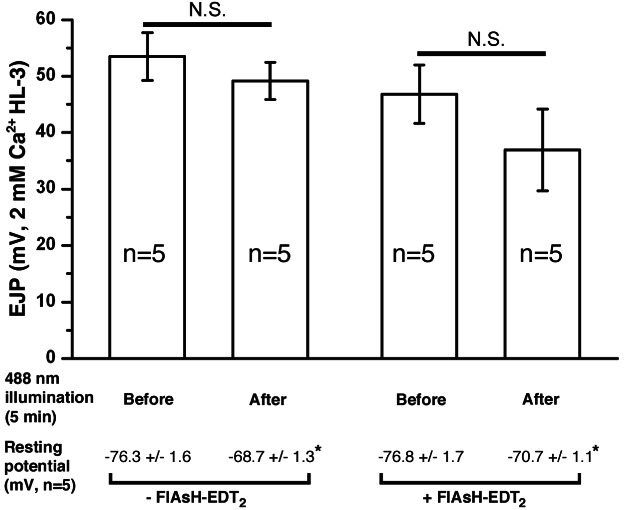
Third instar larval NMJ preparations from Clap4C; lap1/lapsd3 were incubated with or without FlAsH-EDT2 followed by exposure to 488-nm light for 5 min to inactivate LAP. The membrane resting and EJP were recorded just prior to and immediately following LAP inactivation. The NMJ preparation was bathed in HL-3 saline containing 2 mm CaCl2. Note that 488-nm light exposure slightly but significantly depolarized the resting potential of muscles without significantly reducing EJP amplitude.
Immunohistochemical analysis shows stimulation time-dependent membrane protrusions in the absence of LAP activity
Establishment of the capability of FlAsH-FALI techniques positioned us to address questions regarding the fate of SV proteins in real time during SV recycling. Specifically: (i) How are SV proteins distributed at synapses? (2) How are they repacked and internalized? (3) Are SVs recycled en mass in one packet or through separate routes? To observe the initial fate of SV proteins, we performed acute inactivation of LAP in third instar larval NMJs just prior to nerve stimulation. Immediately following exposure to fluorescent light to inactivate LAP, the larval NMJs were stimulated with high potassium for either 1 or 10 min, and immediately fixed and prepared for immunohistochemistry. We stained the NMJ with anti-horseradish peroxidase (HRP) to mark the neuronal PM (36) and with anti-Syt1 to indicate SV location (37). In control flies that do not express the lap4C transgene, synaptic boutons show a typical ‘beads on a string’ phenotype, and Syt1 is localized within the boundaries of these boutons. The duration of stimulation has little effect on the distribution of Syt1, as at both 1 and 10 min, Syt1 is dispersed within the boutons, with higher immunoreactivity in smaller boutons compared with the larger boutons (Figure 4A,B).
Figure 4. Acute inactivation of LAP results in the appearance of abnormal membrane structures that contain SV proteins.
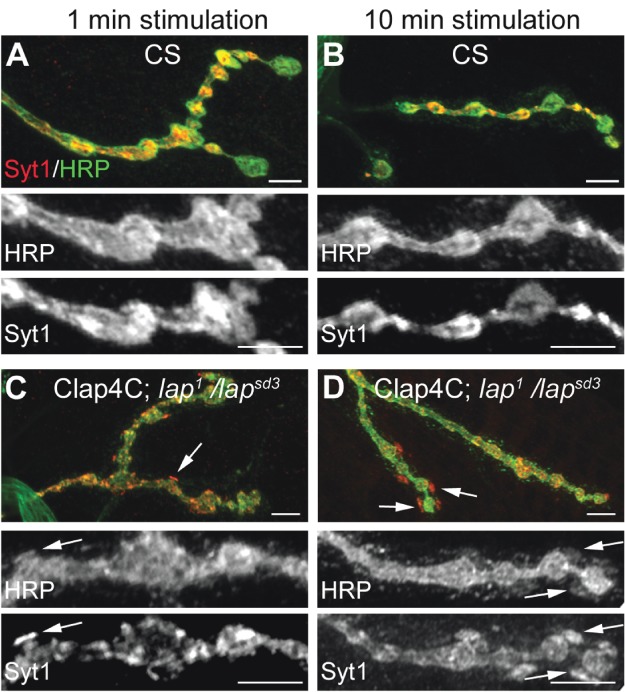
Third instar larval NMJ preparations from the indicated genotypes were dissected, incubated with FlAsH-EDT2 and muscle 4 from segment A2 or A3 was exposed to 488-nm-filtered fluorescent light for 5 min. Following illumination, the larval NMJs were stimulated with high K+ (90 mm) Jan's solution for either 1 min (left column) or 10 min (right column). Following nerve stimulation, the larval body wall NMJ preparation was washed rapidly with Ca2+-free HL-3 saline and fixed. The larval NMJs were then stained with antibodies to HRP (neuronal membrane) and to the SV protein, Syt1. In control flies (A and B), Syt1 is found to be localized within the boutons in a typical ‘doughnut’ pattern. However, when LAP is inactivated (C and D), Syt1 distribution becomes more punctate. These punctate are sometimes localized to HRP-positive areas that appear to protrude outward from the boutons (arrows). Scale bars = 10 µm.
In contrast, acute inactivation of LAP greatly alters the distribution of Syt1. Within 1 min, and proceeding through 10 min of stimulation, Syt1 distribution in LAP-inactivated boutons is tightly packed in discrete pockets around the membrane (Figure 4C,D) rather than the diffuse appearance of Syt1 in controls. Strikingly, these pockets occasionally also appear to be protruding outward of the bouton (Figure 4C,D, white arrows). A close examination reveals that the Syt1-intense packet is not outside the synaptic bouton, as these areas of protrusion are also stained positive for HRP, indicating that they are contiguous with the presynaptic membrane. However, the HRP signal is greatly reduced compared with the bouton proper. To our knowledge, this is the first observation of such outward membrane protrusions following acute inactivation of SV recycling in any synapses. We also observed similar protrusions at NMJs following acute inactivation of clathrin heavy chain (data not shown), suggesting that they are caused by accumulation of SV proteins after initial SV collapse into the PM. We note that electron microscopic studies of the electric ray electric organ and of the rat brain have shown similar protrusions termed pseudopodia, following exocytosis under conditions of rapid stimulation (38–40). Hence, pseudopodia or acute synaptic membrane protrusions are likely conserved synaptic membrane transients in both invertebrate and vertebrate animals.
Further evidence that in the absence of LAP activity, SV proteins diffuse away from each other following exocytosis
To better understand the relative distribution of actively cycling SV proteins upon LAP inactivation, we co-labeled synaptic terminals with Syt1 and a second SV protein, cysteine string protein [CSP (41)], following 1 min of nerve stimulation. In control terminals, Syt1 and CSP staining mostly overlap (although they do not fully overlap; Table 1) and show a similar general pattern throughout the NMJ, indicating that these proteins likely traffic as clusters of multiple SV proteins following SV fusion (42) (Figure 5A–A″). However, in stimulated terminals in which LAP is acutely inactivated, Syt1 and CSP show several unique staining patterns (Figure 5B–B″). For instance, CSP and Syt1 appear as small punctate at LAP-inactivated terminals, whereas both proteins have a typical diffuse, ‘donut-like’ appearance in control terminals. Moreover, in LAP-inactivated terminals where Syt1 and CSP overlap, they show striking differences in their respective intensities. Finally, there are areas of the membrane that appear positive for only one of the two proteins, indicating SV proteins codependent distribution may be rapidly disrupted in the absence of LAP activity (Figure 5B; arrows in B′ and B″; Table 1). We also examined the distribution of nSyb and CSP after a period of 1-min stimulation and found that in the LAP-inactivated synaptic terminals these two SV proteins also diffused more independently than in control (wild-type) boutons (Figure 5C–D″; Table 1).
Table 1.
Colocalization of synaptic vesicle proteins at synaptic boutons
| SV protein 1 | SV protein 2 | Stimulation (min) | Recovery (min) | % Colocalization: control (n) | % Colocalization: LAP inactivated (n) | p-Value |
|---|---|---|---|---|---|---|
| Synaptotagmin | CSP | 1 | – | 86 (25) | 76 (23) | <0.0001 |
| n-Synaptobrevin | CSP | 1 | – | 75 (34) | 66 (42) | <0.0001 |
| Synaptotagmin | CSP | 10 | – | 78 (20) | 68 (20) | <0.0001 |
| vGlut | Synapsin | 5 | – | 94 (23) | 85 (20) | <0.0001 |
| vGlut | Synapsin | 30 | – | 94 (21) | 77 (25) | <0.0001 |
| Synaptotagmin | Synapsin | 10 | 20 | 77 (29) | 65 (28) | <0.0001 |
Figure 5. SV protein distribution is altered in stimulated nerve terminals where LAP is inactivated.
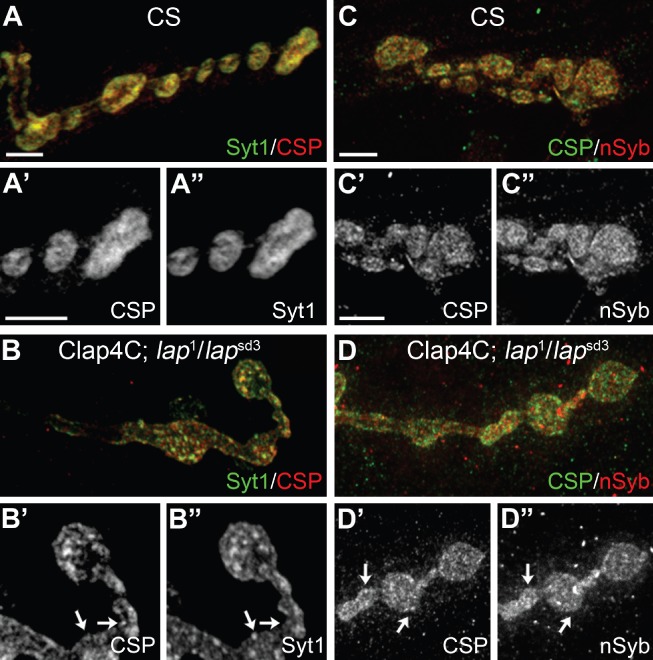
Third instar larval NMJ preparations from the indicated genotypes were dissected, incubated with FlAsH-EDT2 and muscle 4 from segment A2 or A3 was exposed to 488-nm-filtered fluorescent light for 5 min. Following illumination, the larval NMJs were stimulated with high K+ (90 mm) Jan's solution for 1 min and immediately fixed with 4% PFA. The larval NMJs were then stained with antibodies to the SV proteins Syt1 (green) and CSP (red) or CSP (green) and nSyb (red). A and C) In control larvae, a higher percentage of SV proteins overlaps in boutons following 1-min stimulation. B and D) When LAP is inactivated, SV proteins appear to form discrete punctate. Further, close observation of the staining pattern reveals that many of these punctate are positive for only one SV protein (arrows). Overall, SV proteins no longer colocalized in the same punctate in LAP-inactivated synaptic boutons. The protocol for both control and experimental samples was identical. Scale bars = 10 µm.
Synaptic membrane protrusions internalize following a period of resting
Our preceding data indicate that LAP is necessary for appropriate distribution of SV proteins at early time points during SV recycling. To understand the time-dependent role of LAP in active nerve terminals, we stained for additional SV proteins over an extended period following stimulation. We examined whether independent diffusion was more pronounced during extended periods (5, 10 and 30 min) following nerve stimulation after LAP had been acutely inactivated. We show that the distribution of synapsin [Syn (43)] and the vesicular glutamate transporter [vGlut (44)] is also altered following 5- and 30-min stimulation periods. Syn and vGlut diffuse independently in nerve terminals where LAP is inactivated (Figure 6B,D; white arrows), but not in controls (Figure 6A,C). Following 10-min stimulation Syt1 and CSP overlapping in LAP-inactivated synaptic boutons is also significantly reduced compared to those in the wild-type synapses (Figure 6E,F; Table 1). Overall, there are more punctate formed in LAP-inactivated synaptic boutons and these punctate often do not show same levels of SV proteins.
Figure 6. Independent diffusion of SV proteins following nerve stimulation is sustained for long periods following LAP inactivation.

Third instar larval NMJ preparations from the indicated genotypes were dissected, incubated with FlAsH-EDT2 and muscle 4 from segment A2 or A3 was exposed to 488-nm-filtered fluorescent light for 5 min. Clap4C; lap = Clap4C; lap1/lapsd3. Following illumination, the larval NMJs were stimulated continuously with high K+ (90 mm) Jan's solution for 5 min (A and B) or 30 min (C and D) or 10 min (E and F). Following nerve stimulation, the larvae were washed rapidly with Ca2+-free HL-3 saline and fixed. The NMJs were then stained for a pair of SV proteins Syn and vGlut or Syt1 and CSP. Panels A–D also show HRP-stained NMJs (blue) to mark the neuronal membrane. The stimulation paradigm does not affect SV protein colocalization in wild-type control larval NMJs in which most SV proteins are colocalized although we note that not all SV proteins are fully colocalized even in the wild-type larval NMJs. In contrast, there are more punctate in LAP-inactivated synaptic boutons and SV proteins are often not colocalized in these punctate (arrows). G and H) Following 10 min of stimulation with high K+, nerve terminals were allowed to rest in Ca2+-free HL-3 saline for an additional 20 min prior to fixation. Following the resting period, dramatically large punctate can be observed positive for both Syt1 and Syn (arrowheads) in larval NMJs whose LAP is inactivated, while some portions of the punctate within the bouton are positive for only one of the two SV proteins (arrows). The protocol for both control and experimental samples was identical. Scale bars = 10 µm.
We next examined the localization of SV proteins in synapses that had been stimulated for 10 min and allowed to rest for 20 min prior to fixation. In synapses in which LAP is active (i.e. with FlAsH), there is a high degree of colocalization between Syn and Syt1 with an SV distribution typical of control synapses (i.e. without FlAsH) (Figure 6G). However, in LAP-inactivated synapses the synaptic protrusions are no longer detectable and instead Syn and Syt1 are found in large punctate inside the synaptic boutons, which may represent bulk uptake of SVs (Figure 6H, arrowheads). There are also membrane areas in which these two proteins do not colocalize well at LAP-inactivated NMJs (Figure 6H, arrows). These results confirm that LAP is required for co-trafficking of SV proteins during endocytosis and suggest that over time membrane protrusions are internalized. Hence, LAP is critical for the appropriate localization of SV proteins following nerve stimulation, most likely through coupling SV protein uptake to CME.
Large cisternae form in the absence of LAP activity following nerve stimulation
To better understand the nature of synaptic membrane protrusions and cisternae, we examined the ultrastructure of stimulated NMJs following LAP acute inactivation. In control boutons stimulated with high potassium for 5 min, we observed densely packed SVs along with multiple cisternae (Figure 7A). The presence of these cisternae has been previously reported as a phenomenon specific to potassium stimulation paradigm (45). Conversely, in boutons in which LAP was inactivated, we rarely observed normal-sized SVs (∼40 nm). What we did observe falls generally into two categories: (i) boutons packed with multiple intermediate-sized cisternae (Figure 7B) or (ii) boutons exhibiting massive SV depletion, but containing very large cisternae and membrane infoldings (Figure 7C). We were unable to observe specific outward synaptic protrusions as we observed by immunohistochemistry, but a closer examination of the large cisternae forming from the PM revealed interesting features (Figure 7D–F). The membrane formations we see are different from the bulk membrane formations that form upon clathrin inactivation, which can reach up to 1 µm in diameter and form sheets that appear to fold in upon themselves (30,27). By distinction, the formations that appear upon LAP inactivation are single large membrane-bound cisternae. Although much larger than a typical SV, the membrane does not fold in upon itself, but rather balloons inward. Further, these cisternae appear contiguous with the presynaptic PM (Figure 7E,F), likely explaining the appearance of FM dye-positive punctate in LAP-inactivated boutons in Figure 2. Most intriguingly, we observed cisternae that contained smaller, electron-dense vesicles that are similar to SVs in size (Figure 7E). These observations are consistent with the notion that LAP function is necessary for the reformation of SVs, and in the absence of its function, membrane reuptake is severely altered.
Figure 7. Ultrastructural analysis of synaptic boutons following acute inactivation of LAP.

Third instar larval NMJ preparations from Clap4C; lap1/lapsd3 or control (lap1/TM6B,Tb) flies were dissected, incubated with FlAsH-EDT2 and muscles 6/7 from segment A2 or A3 were exposed to 488-nm-filtered fluorescent light for 5 min. Following illumination, the larval NMJs were stimulated with high K+ (90 mm) Jan's solution for 5 min. Following stimulation, the larval NMJs were washed rapidly with Ca2+-free HL-3 saline. The tissue was fixed and prepared for electron microscopy. A) Control synaptic boutons show densely packed SVs along with cisternae which, as noted in the text, are characteristics of the stimulation paradigm. Boutons in which LAP is inactivated show two primary phenotypes: (i) the absence of typically sized SVs, but the presence of large cisternae (B) or (ii) nearly complete absence of SVs, and the appearance of large membrane infoldings (C). When LAP is acutely inactivated, we also occasionally observe large balloon-like membrane infoldings that remain contiguous with the PM (D–F, arrows) and may contain smaller vesicle-like structures (E). Scale bars = 500 nm.
A subset of SV protein distribution is altered in lap mutants
Our previous work and those by others suggest that AP180 plays a role in retrieving the SV protein nSyb into newly formed SVs (19,22,24,35). Our data from acute inactivation of LAP further suggest that LAP may be coupling SV protein clustering at the PM to endocytosis. These observations raise the possibility that SV protein composition may be altered in the absence of LAP. To test this hypothesis, we separated protein lysate obtained from either control (Canton S, CS) or lap mutant adult heads (from 3- to 5-day-old flies) over 5–25% glycerol density gradients using high-speed centrifugation. The gradients were fractionated into 14 equal volume fractions, and proteins from the individual fractions were probed for SV vesicle components. In gradients obtained from control lysates, SV proteins migrate primarily to fractions 3–6 with a sharp peak (Figure 8A). This pattern is consistent with previously published results using a similar protocol (16).
Figure 8. Distinct populations of SVs with different levels of SV proteins are present in lap mutants.

Total brain lysate was isolated from 3- to 5-day-old adult wild-type (CS) or lap (lap1/lapsd3) mutant flies. The lysate was separated over a 5–25% glycerol gradient. A total of 14 fractions were collected from the top, diluted in SDS sample buffer, separated over an SDS–PAGE gel, transferred to nitrocellulose and probed with the indicated antibodies. A and B) In control flies, SVs proteins are enriched primarily in fractions 3–6. While these fractions also contain SV proteins in lap mutants, these proteins are also seen to migrate into the less dense fractions 1–7. C–F) Graphical representation of data in A and B. Protein density was measured using ImageJ. Data are plotted as the percentage of protein in each fraction to total protein level for each respective protein. G–J) To determine the percentage of protein found in either peak (4–6) or non-peak (1–2) fractions, the protein density was summed for their respective fractions and plotted as a percentage of total protein in all fractions. Although ∼70–80% of the total SV protein is found in peak fractions of control lysates, only ∼50% of total SV protein is found in these fractions of lap mutants. Conversely, less than 20% of total SV protein is found in non-peak fractions of control flies, whereas ∼40% is found in these fractions of lap mutants. K) Histogram plots of percentage of SV proteins in fractions 5–7. These data show clear differences in SV protein compositions between the control and the lap mutant.
When we probed for SV proteins in gradient fractions obtained from lap mutants, a striking difference emerged. The SV protein migration range is much wider than is the case for controls such that the proteins can be seen to less evenly distribute to fractions 2–7 (Figure 8B). We graphed the average migration pattern for individual SV proteins from three independent experiments (Figure 8C–F). Again, a sharp peak in fractions 4 and 5 can be seen in controls (solid lines) for each individual protein. In lap mutants, SV protein migration pattern is varied, with no identifiable peak. To better understand the differences, we used an alternative means to graph the SV distribution over gradients. We divided the fractions into two groups: peak (fractions 4–6) and non-peak (fractions 1–2) (Figure 8G–J). We excluded other fractions because the presence of SV protein is negligible in these fractions. In control lysates, for individual SV proteins, 70–80% of the total protein is found in peak fractions, while less than 20% is found in the non-peak fractions. Conversely, in lap mutant lysates, individual SV proteins are primarily evenly distributed between peak and non-peak fractions. Control boutons have uniformly sized SVs, whereas lap mutants display a range of sizes at the ultrastructure level, with a significant proportion of large vesicles (31). It is possible that the non-peak fractions on our gradients represent these large vesicles if these vesicles contain proportionally less content. These would result in less dense vesicles that would migrate into low-density fractions.
We also plotted the percentage of each SV proteins in three fractions (5, 6 and 7) and observed striking differences in SV protein composition between the wild-type and the lap mutant flies (Figure 8K). In fraction 5 the lap mutant has a considerable reduction in nSyb and vGlut but an increase in Syt1, Syn and CSP compared with the control. In fractions 6 and 7, the mutant has severely reduced levels of nSyb, a mild reduction in vGlut but a drastic increase in CSP. Hence, at the population level SVs have significantly altered their protein composition in the lap mutant.
LAP interacts with SV proteins in a salt- and detergent-sensitive manner
Our data to this point are consistent with a model in which LAP functions to sort and retrieve SV proteins in the endocytic formation of SVs. SV proteins have been shown to remain clustered upon SV fusion with the PM and recycling (42,46,47). We wondered if LAP could recognize membrane areas that are enriched for recently fused SV proteins, perhaps through direct association with these proteins. To determine whether LAP interacts with SV proteins, we used anti-Flag beads to perform co-immunoprecipitation (IP) from 3- to 5-day adult head protein lysates obtained from either control (CS) or flies expressing a genomic Flag-tagged LAP in the absence of detergent. In this method (i.e. without detergent), we should be able to detect all possible LAP interactions taking place either on SVs themselves, on the PM, or in soluble cytosol. We probed the IP pellet with antibodies to several SV proteins, including Syt1, CSP, nSyb, vGlut and Syn. Our experiments show an interaction between LAP and all SV proteins observed in the presence of 150 mm NaCl (Figure 9A). We also show that LAP does not co-IP with the SV protein Syn, suggesting that LAP interactions with other SV proteins are specific.
Figure 9. LAP interacts with SV proteins in a detergent- and salt-sensitive manner.
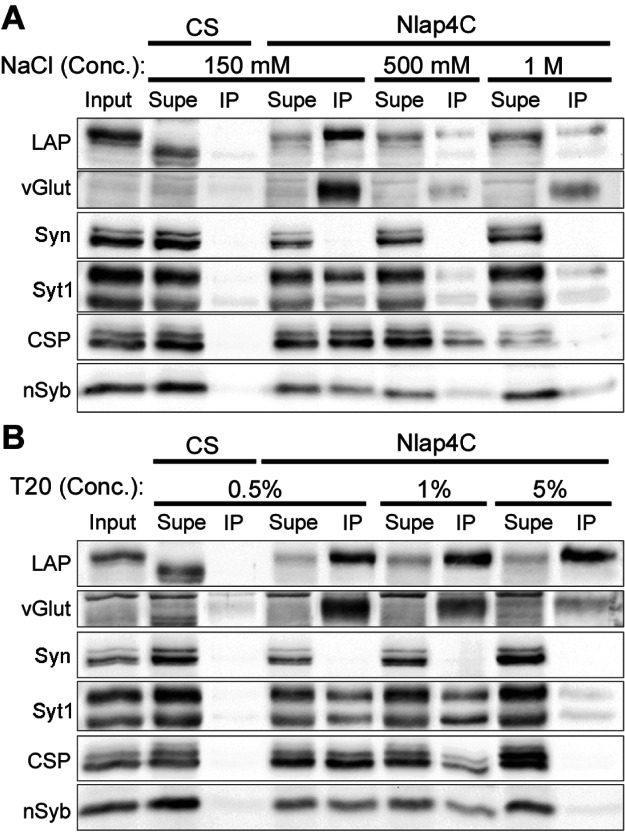
A and B) Total brain lysate was isolated from 3- to 5-day-old adult flies from the indicated genotypes, and subjected to co-IP using anti-Flag agarose beads. Samples from input (total brain lysate), supernatant (lysate washed over the beads) and IP (eluted from beads) fractions were collected, diluted in SDS sample buffer, separated over an SDS–PAGE gel, transferred to nitrocellulose and probed with the indicated antibodies. Input for both A and B was taken from Nlap4C-Flag transgenic fly brain lysates (and hence the higher MW for LAP). A) Co-immunoprecipitates with varying concentrations of NaCl but without detergent: Following incubation of the lysate with beads, unbound proteins were washed in the presence of increasing concentrations of salt (NaCl) to disrupt protein–protein interactions. At the lowest concentration of salt, LAP interacts with all SV proteins probed with the exception of Syn. However, these interactions are disrupted by increasing salt concentrations. At 1 m NaCl, LAP interaction with vGlut, Syt1 and nSyb remains albeit at weak levels. B) Co-IP with detergent: Following incubation of the lysate with beads, unbound proteins were washed in the presence of increasing concentrations of the detergent, Tween-20, to disrupt membranes. At the lowest concentration of the detergent (0.5%), LAP co-immunoprecipitates all SV proteins probed, with the exception of Syn. Increasing the concentration of the detergent to 1% does little to disrupt these interactions. However, a further fivefold increase in the detergent disrupts LAP interaction with all SV proteins except vGlut, Syt1 and nSyb (weakly). These interactions are also significantly weakened by higher detergent concentrations.
Our co-IP conditions favor LAP pulling down a complex of SV proteins, consisting of either direct protein–protein interactions or as membrane fractions. To determine if LAP interacts with SV proteins in a protein complex, we then increased salt concentrations to disrupt electrostatic interactions between SV proteins (Figure 9A). While increasing the concentration of NaCl to 500 mm decreases the presence of SV proteins in the immunoprecipitate, all interactions are still in place. Raising the salt concentration to 1 m only disrupts the interaction between LAP and CSP, but not other SV proteins. These results suggest that LAP interacts with each of these proteins individually or the interaction is membrane dependent.
One concern is the absence of Syn in the LAP co-IP. Syn is not an integral vesicular membrane protein and thus it is possible that Syn has fully dissociated from SVs in our lysates. We deem this unlikely as our fractionation data in Figure 8 show that Syn co-migrates closely with other SV proteins. To address this concern and to examine the protein interactions without the influence of the plasma and vesicular membrane, we repeated our co-IP experiments with increasing concentrations of Tween-20 in the wash buffer (Figure 9B). We chose Tween-20 as a detergent because it is strong enough to solubilize membranes but gentle enough to still retain native protein function and structure. Tween-20 at concentrations of 0.5 and 1% did little to disrupt LAP interactions with SV proteins. However, raising the detergent concentration to 5% disrupted or severely reduced all LAP interactions, except with LAP itself, vGlut and Syt1. These results suggest biochemical mechanisms by which LAP helps retrieve SV proteins during endocytosis.
Discussion
Our studies of SV trafficking following acute inactivation of LAP have revealed a number of important findings, some of which have also confirmed our previous reports. First, we show for the first time that inactivation of LAP does not affect SV exocytosis. This is not unexpected but important because previously we showed that SV recycling as well as exocytosis were dramatically impaired in the lap mutant. But we could not discern whether the defect in endocytosis caused the reduction in transmitter release or the other way around. Our new data obtained from acute inactivation experiments clearly show that LAP does not function directly in SV exocytosis and lend a strong support to our early conclusion that lap mutations indirectly impact SV exocytosis.
Second, we present the evidence that in the absence of LAP function SVs fused into the PM have dramatic effects on the morphology of the PM. For the first time we observed the outward membrane protrusion formed after blocking LAP function, suggesting that SVs collapse into the PM and some of the newly added membranes have to expand outward. Earlier electron microscopic studies of electric ray and rat brain synapses have reported pseudopodia formation following high activity of neurons (39). In these studies, the investigators could not discern why pseudopodia formed, but our study suggests that pseudopodia are a temporary consequence of inefficient SV recycling.
Third, and most interestingly, we have observed dissociation of SV proteins following LAP inactivation and the formation of large punctate that may correspond to the large cisternae observed in our ultrastructural examinations. The loss of SV protein clustering in the absence of LAP supports our earlier observations that some SV proteins are mislocalized to axonal membranes where SV proteins are usually absent. It also raises the possibility that LAP plays a role in clustering, sorting and retrieving a subset of SV proteins during endocytosis. This conclusion is consistent with the finding that mammalian AP180 binds and sorts synaptobrevin/VAMP2 (24). Further, membrane bending requires the direct action of proteins (48). The ANTH domain of LAP has long been postulated to have membrane bending properties (49–51). More recently, AP180 has been suggested to accomplish the task of membrane bending through the novel mechanism of protein crowding (52). Protein crowding, combined with our protein interaction data herein, fits a model in which LAP binding to multiple proteins helps stabilize clathrin-coated pit assembly to overcome a high energy barrier that may otherwise preclude productive endocytosis during SV recycling (53). Despite the stringency of our co-IP conditions, we cannot discount the possibility that LAP does not directly bind all the SV proteins we identified. Koo et al. identified only synaptobrevin/VAMP2 as an interacting partner of mammalian AP180 (24), and this may also be the case in C. elegans (19). Nonetheless, we clearly show that SV protein clustering is more disrupted in the absence of LAP activity, and we postulate that this is owing to the absence of interaction between LAP and one or several SV proteins (Figure 10).
Figure 10. Proposed modes by which LAP may sort and retrieve SV proteins into newly formed SVs during CME.
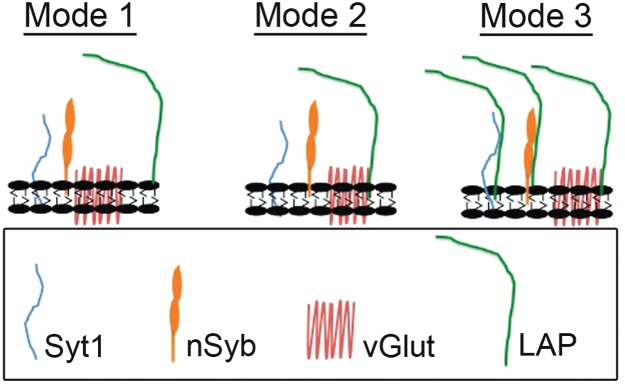
In mode 1, LAP could retrieve a subset of SV proteins through interaction with a membrane patch (equivalent to lipid rafts). In mode 2, LAP could directly bind to vGlut, which in turn binds to other SV proteins or a membrane patch. In mode 3, LAP could directly interact with a subset of individual proteins. Our data with increased salt and detergent concentrations seem to favor mode 2.
It is also interesting to note that there are some differences in SV protein localization in the lap mutant (22,31) compared to those undergone acute LAP inactivation (this study). Specifically, there is no protein aggregate observed in the lap mutant. We believe that these aggregates are bulk membrane uptake of SVs, which are detected in transmission electron microscopy micrographs. The formation of protein aggregates likely reflects nerve activity levels. As shown in Figure 6G,H, following a 20-min resting period SV proteins become more evenly distributed inside synaptic boutons compared to the images obtained immediately following nerve stimulation in Figure 6B,D,F. These results suggest that over time, after intense nerve activity, aggregates are reorganized into individual SVs (a major pathway for bulk uptake of SVs). In previous publications, we probed SV protein localization in synaptic boutons of the lap mutant, which also has severe exocytotic defects. Alternatively, developmental compensation may reduce the chance for the formation of such aggregates in the lap mutant. In the lap mutant we observed a small increase in the number of cisternae but frequency of cisternae is lower and the size is generally smaller than what we have observed, shown in Figure 7, in which LAP is acutely inactivated. These differences may account for the lack of aggregates in the lap mutant.
Fourth, it should not be a complete surprise that the nerve terminal fails to recycle certain SV proteins in the absence of LAP if LAP is required to retrieve them into newly formed vesicles. Our membrane fractionation studies suggest a change in vesicle protein distribution, and particularly the altered stoichiometry of SV proteins in lap mutants. These results suggest that the SV composition may indeed be altered without LAP. This change in SV identity could explain the dramatic defect in SV functionality.
Finally, we have obtained biochemical data suggesting that LAP has high affinity binding to the SV protein vGlut, relatively moderate affinity for the SV protein Syt1 and nSyb, low affinity for the SV protein CSP and no interaction at all with the SV protein Syn. Although we have not identified which specific interaction or interactions are directly responsible for protein bundling/crowding, we postulate three different modes by which LAP may interact with these SV proteins (Figure 10). In mode 1, LAP could retrieve a subset of SV proteins through interaction with a membrane patch (equivalent to lipid rafts). In mode 2, LAP could directly bind to vGlut, which in turn binds to other SV proteins or a small patch of membrane. In mode 3, LAP could directly interact with a subset of individual proteins. Our data with increased salt and detergent concentrations seem to favor mode 2. These LAP and SV protein interactions likely provide the molecular mechanisms by which LAP sorts and retrieves SV proteins to reconstitute and recycle SVs through CME.
Our data show that SV proteins are not fully colocalized even in wild-type synapses. Although we cannot claim that our observation is not partly due to our experimental conditions (e.g. an artifact of fixation), we rather speculate that the dissociation of SV proteins normally occurs following intense nerve stimulation owing to saturation of the retrieval machinery. We believe that this interpretation is consistent with earlier reports that non-heterogeneous pools of SV proteins reside on the PM (54–56), and that this observation may be partly due to intense stimulation conditions (42). We would like to emphasize that our colocalization data are less than ideal and may have alternative explanations. By no means do we imply the change in SV protein colocalization in the LAP-inactivated boutons correlates with single vesicle changes. Other approaches (such as super-high resolution microscopy or live imaging) would offer more definitive evidence. Nonetheless, our analysis shows that following acute inactivation of LAP the degree of colocalization of SV proteins is significantly further reduced. We believe that such results support a role for LAP in helping cluster SV proteins during endocytosis. Importantly, Ramaswami and coworkers earlier showed that SV proteins such as Syt1 and CSP are localized to the PM but still colocalize in shibirets mutant following blockage of SV recycling (57). Hence, blocking SV recycling alone is insufficient to cause dissociation of SV proteins.
Our study reveals the first in vivo evidence that SV composition may be altered in the absence of AP180 and offers additional molecular mechanisms by which AP180 retrieves SV proteins during reformation of SVs through CME. We do not understand why LAP has the strongest interaction with vGlut in flies, whereas AP180 or CALM does not interact with vGlut in mammals. Nonetheless, our results reinforce the importance of AP180 in maintaining SV protein composition and hence SV functionality in the vesicle cycle.
We show for the first time that acute disruption of a critical endocytic protein, prior to potential compensatory actions, immediately disrupts SV recycling and SV composition. This finding provides new insight into the link between AP180 and Alzheimer's disease progression (22,58). AP180 levels are reduced in the brains of Alzheimer's patients (59,60), and a recent genome-wide association study linked the AP180 homolog PICALM as a risk factor in Alzheimer's disease (61). We believe that our findings here may help explain the earliest links between onset of neurodegenerative disease and long-term cognitive dysfunction, and therefore the study of AP180 in SV protein retrieval can help advance the understanding of the synaptic defect in Alzheimer's disease.
Materials and Methods
Fly strains
All stocks were maintained on standard cornmeal medium at 25°C. The following Drosophila stocks were used: lapsd3/TM6B,Tb (22,62); lap1/TM6B,Tb (31); CS and Nlap4C or Clap4C (see below for details on generation of these transgenic lines). All stocks were maintained in the PI lab and some were also available at the Bloomington Drosophila stock center.
Generation of lap4C lines
The Clap4C and Nlap4C parental stocks were generated via recombineering as follows: A P(acman) clone containing the full-length genomic lap [P(acman)-lap], including an extra 6 kb upstream and downstream to include potential regulatory elements, and a w+ transgene, was identified using the online resource, http://www.pacmanfly.org, and obtained from CHORI (Oakland, CA). A polymerase chain reaction fragment coding for Flag and a tetracysteine cassette (4C) was generated and, using homologous recombination, inserted into either the N- or C-terminal of LAP (Nlap4C and Clap4C, respectively) as previously described (63). The modified lap transgenes were injected into w− flies and incorporated into the left arm of chromosome 2 using site-specific integration via PhiC31 integrase-mediated site-specific transgenesis (Genetivision). Positive transgene insertions were identified by the presence of rosy eyes and maintained as stocks. Clap4C and Nlap4C flies were balanced on the third chromosome and crossed into a lap null mutant background to generate the following flies: Nlap4C; lap1/lapsd3 and Clap4C; lap1/lapsd3.
FlAsH-FALI
We followed the methods described by Davis and coworkers (6,27) and Verstreken and coworkers (30). Briefly, third instar larvae were dissected in Ca2+-free HL-3 saline [110 mm NaCl, 5 mm KCl, 10 mm NaHCO3, 5 mm Hepes, 30 mm sucrose, 5 mm trehalose and 10 mm MgCl2, pH 7.2 (64)] and loaded with 1 µm FlAsH-EDTA (Invitrogen) in Ca2+-free HL-3 saline for 10 min in the dark at room temperature. The preparations were then washed with the manufacturer-supplied wash buffer (BAL wash buffer, 2,3-dimercapto-1-propanol, Invitrogen; BAL was diluted in Ca2+-free HL-3 saline) for 5 min to remove unbound FlAsH and returned to Ca2+-free HL-3 saline. Photoinactivation was achieved by illuminating either muscle 4 or muscles 6/7, in segments A2 or A3, with epifluorescent light filtered at 488 nm for 5 min using a 63× water immersion lens on a Zeiss AxioImager.Z1m microscope. Following photoactivation the synapses were stimulated for 1, 5, 10 or 30 min in 90 mm KCl, 2 mm Ca2+ Jan's solution (25 mm NaCl, 90 mm KCl, 10 mm NaHCO3, 5 mm Hepes, 30 mm sucrose, 5 mm trehalose, 10 mm MgCl2 and 1.5 mm CaCl2, pH 7.2) (65) and subsequently washed thrice with ice cold Ca2+-free HL-3 saline and proceeded immediately to fixation. In all the control experiments for LAP inactivation, including the electrophysiological studies, the controls NMJs were also exposed to the 488-nm light for 5 min.
Electrophysiology
The standard third instar larval body wall preparation was used for electrophysiological recordings (31,61). The preparation was bathed in HL-3 solution. Muscle synaptic potentials were recorded using an Axon Clamp 2B amplifier (Axon Instruments) and acquired by a Dell PC computer equipped with pclamp software. For photoinactivation, the preparations were exposed to epifluorescent light filtered at 488 nm through a 60× water immersion objective (Olympus).
FM dye experiments
Third instar larval NMJ preparations were dissected and labeled with FlAsH as described above. For experiments measuring endocytosis, following photoinactivation, larval NMJ preparations were stimulated by immersion in 90 mm KCl Jan's solution with 4 µm FM 1-43 (Invitrogen) for 1 min. Free dye was washed by 5 × 2 min washes in Ca2+-free HL-3 saline, and the remaining dye was imaged using a 63× water immersion objective on a Zeiss AxioImager.Z1m microscope using a GFP filter set.
For experiments measuring exocytosis of FM dye, 10 µm FM 4-64 was loaded for 1 min using 90 mm KCl Jan's solution prior to photoinactivation. Following photoinactivation with epifluorescent light filtered at 488 nm, the preparation was quickly imaged to ensure the FM 4-64 had not bleached and then unloaded by 5 × 2 min washes in Ca2+-free HL-3 saline. The preparation was then imaged a second time to measure any remaining dye.
Generation of dVGlut and nSyb antibodies
Synthetic peptides against dVGlut (amino acids 614-632: C YGYTQGQMPSYDPQGYQQQ) and nSyb (amino acids 26-44: C PAGEGGDGEIVGGPHNPQQ–amide) were generated commercially and used to generate polyclonal antibodies in rabbits through YenZym™ antibodies, LLC (http://www.YenZym.com). Crude sera were affinity-purified and quality controlled by western blot and immunocytochemistry.
Immunohistochemistry
Third instar larval NMJ preparations were prepared as indicated for FM 1-43 experiments, except that the 90 mm KCl did not contain FM 1-43. Following nerve stimulation, the larvae NMJ preparation was washed rapidly thrice with Ca2+-free HL-3 saline and immediately fixed in 4% paraformaldehyde (PFA) for 15 min. The preparations were then incubated overnight at 4°C with primary antibodies in 1× PBX (1× PBS containing 0.1% Triton-X) using the following dilutions: rabbit anti-Syt1 (1:500, courtesy of Noreen Reist), mouse anti-CSP (1:50, ab49, Developmental Studies Hybridoma Bank, DSHB), rabbit anti-vGlut (44) and newly generated rabbit-anti vGlut, mouse anti-Syn (1:100, DSHB), nSyb (1:100) or anti-HRP-488 (1:200, Invitrogen). Alexafluor-594 (Invitrogen) secondary antibody was used at 1:200. Fixed specimens were mounted in Vectashield (Vector Laboratories) and imaged using an LSM Olympus FV1000 confocal microscope with a Plan-Apochromat 60× (1.42 NA) objective. Images in Figure 5C,D were obtained using Leica SP8 confocal microscope with a HC PL APO 63× (1.40 OIL CS2) objective. Images were taken from muscle 4 or muscle 6/7, segments A2 or A3 and processed with ImageJ (NIH) and Photoshop CS4.
Electron microscopy and analysis
Third instar NMJ tissues were processed as previously described (66). Briefly, they were dissected in Ca2+-free saline [HL-3, pH 7.2 (64)], processed as described for FlAsH-FALI and fixed for 1 h in 1% acrolein and 2.5% glutaraldehyde in 0.1 m sodium cacodylate (Cac) buffer (pH 7.2). After washing, larvae were post-fixed for 1 h in 0.5% OsO4, 0.8% KFeCn in 0.1 m Cac, incubated in 5% uranyl acetate for 1 h to overnight, dehydrated with graded ethanols, transitioned with two changes of propylene oxide and embedded in Eponate 12. Sections of 60–90 nm were cut using a diamond knife and Reichert Ultracut E ultramicrotome and post-stained with uranyl acetate and Reynold's lead citrate. Images were captured using a JEOL JEM 2000 EX-II TEM operated at 100 kV and 12 000× magnification. Negatives were digitized using an Epson Perfection V700 scanner at 1500 dpi. Figures were prepared from raw images and adjusted for brightness and contrast, scale calibration, using Photoshop (Adobe).
Co-immunoprecipitation
Adult fly heads were isolated from wild-type CS and transgenic Nlap4C flies. A total of 2.5 mL of whole flies was flash frozen in liquid nitrogen, vortexed and passed first through a small sieve (no. 25, 710 µm, Precision Eforming, LLC) to allow for separation of fly heads and then through a second sieve (no. 40, 425 µm, Precision Eforming, LLC) to isolate heads from wings and legs. The heads were transferred to a 1.5-mL conical tube and, using a pestle, homogenized in 100 μL of lysis buffer [10 mm Hepes, 0.1 mm MgCl2, 150 mm NaCl, 5 mm NEM, 2 mm phenylmethylsulfonyl fluoride and Protease Inhibitor Cocktail (Roche)] and spun at 13 000 rpm (17,949 × g) for 15 min at 4°C. The spin was repeated, and 50 μL of the supernatant was taken for input, while the remaining lysate was subjected to IP. The lysates were precleared with mouse IgG agarose beads (Sigma) for 2 h at 4°C followed by incubation with anti-Flag M2 affinity gel (Sigma) overnight at 4°C. Anti-Flag beads were spun at 5000× g for 30 seconds and 100 μL of supernatant was removed with a narrow-end pipette. The gel was washed thrice with 500 μL lysis buffer with protease inhibitors allowing time for beads to settle after each spin. Elution of Flag proteins was performed by competition with a Flag peptide. Hundred microliters of 3× Flag elution solution (150 µg/mL) was added to resin and incubated for 30 min with gentle shaking at 4°C. The sample was centrifuged at 8200× g for 30 seconds, supernatant was removed and diluted 20 μL in sample buffer. Input, supernatant and immunoprecipitate samples were diluted in SDS sample buffer to a 1× final concentration. NaCl or Tween-20 was added to the lysis buffer to reach the final concentrations used in our experiments.
Density gradient fractionation
The fractionation protocol was adapted from Phillips et al. (16). Briefly, ∼0.1 g of heads was collected from 3- to 5-day-old adult CS flies or 3- to 5-day-old lap mutant flies (lap1/lapsd3). The heads were homogenized in 500 μL of buffer A (150 mm NaCl, 10 mm Hepes, pH 7.4, 1 mm EGTA and 0.1 mm MgCl2) using 20 strokes in a glass–glass homogenizer. The lysate was rotated end-over-end for 20 min at 4°C and centrifuged at 1000× g for 10 min at 4°C. A total of 150 μL of the resulting supernatant was layered over a 5–25% glycerol gradient on a 50% sucrose pad and centrifuged at 50 000 rpm (213,818 × g) for 30 min at 4°C in a TLS-55 rotor (Beckman Coulter). Fifteen fractions (∼133 μL/fraction) were collected from the bottom and diluted in SDS sample buffer.
Western blot
Following dilution in SDS sample buffer, proteins were separated by SDS–PAGE, transferred to nitrocellulose and detected using the following antibodies: goat anti-LAP (1:2000); rabbit anti-vGlut (1:10 000, courtesy of Aaron DiAntonio, and this study); mouse anti-Syn (1:200); rabbit anti-Syt1 (1:1000, Noreen Reist); mouse anti-CSP (1:200, DSHB) and rabbit anti-nSyb (1:1000), followed by probing with HRP-conjugated secondary antibodies (1:1000, Jackson Immunoresearch). The HRP signal was detected with ECL Plus (GE Healthcare) and imaged on a Chemi Doc XRS+ Imaging System (BioRad).
Colocalization analysis
The original acquisition images were opened in ImageJ in separate channels, and a region of analysis (ROI) was drawn around individual boutons. Following input of the point spread function, colocalization analysis was performed using the Coloc 2 plugin within the ROI in separate Z-sections. The percentage of colocalization for each individual bouton (20–30 per condition) was input into GraphPad Prism 5 and the software was used to run one-way analysis of variance (anova) with a Bonferroni post-test to evaluate statistical significance.
Statistical analysis
Statistical analysis was performed using GraphPad Prism Software. Statistical significance was calculated using one-way anova followed by Tukey's post-test. Significance is indicated with asterisks: *p < 0.05; **p < 0.01; ***p < 0.001.
Acknowledgments
We thank Marie Phillips for her (advice) on the membrane fractionation experiments; Patrik Verstreken, Koen Venken and Hugo Bellen for their advice on the cloning of p[ACMAN]; Richard Daniels and Aaron DiAntonio for the vGlut antibody; the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA 52242, for the monoclonal antibodies to CSP and Syn; and YenZym for the production of polyclonal antibodies to vGlut and nSyb. We thank Jody Rada for her generosity of letting us to use her confocal microscope and Emily Kumimoto for assistance in fly genetics and biochemistry. We also thank Thomas Phillips for discussion on pseudopodia. This research was supported by a grant from NSF (IOS-0822236 to B. Z.) and in part by a grant from NIH/NINDS (RO1-NS060878 to B. Z.) and NSF (IOS-1025966 to N. E. R). The authors declare no conflict of interest.
References
- 1.Katz B. Quantal mechanism of neural transmitter release. Science. 1971;173:123–126. doi: 10.1126/science.173.3992.123. [DOI] [PubMed] [Google Scholar]
- 2.Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cremona O, De Camilli P. Synaptic vesicle endocytosis. Curr Opin Neurobiol. 1997;7:323–330. doi: 10.1016/s0959-4388(97)80059-1. [DOI] [PubMed] [Google Scholar]
- 4.Dittman J, Ryan TA. Molecular circuitry of endocytosis at nerve terminals. Annu Rev Cell Dev Biol. 2009;25:133–160. doi: 10.1146/annurev.cellbio.042308.113302. [DOI] [PubMed] [Google Scholar]
- 5.Südhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 6.Marek KW, Davis GW. Transgenically encoded protein photoinactivation (FlAsH-FALI): acute inactivation of synaptotagmin I. Neuron. 2002;36:805–813. doi: 10.1016/s0896-6273(02)01068-1. [DOI] [PubMed] [Google Scholar]
- 7.Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 8.Yao C-K, Lin YQ, Ly CV, Ohyama T, Haueter CM, Moiseenkova-Bell VY, Wensel TG, Bellen HJ. A synaptic vesicle-associated Ca2+ channel promotes endocytosis and couples exocytosis to endocytosis. Cell. 2009;138:947–960. doi: 10.1016/j.cell.2009.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gordon SL, Leube RE, Cousin MA. Synaptophysin is required for synaptobrevin retrieval during synaptic vesicle endocytosis. J Neurosci. 2011;31:14032–14036. doi: 10.1523/JNEUROSCI.3162-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poskanzer KE, Fetter RD, Davis GW. Discrete residues in the c(2)b domain of synaptotagmin I independently specify endocytic rate and synaptic vesicle size. Neuron. 2006;50:49–62. doi: 10.1016/j.neuron.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 11.Ceccarelli B, Hurlbut WP, Mauro A. Turnover of transmitter and synaptic vesicles at the frog neuromuscular junction. J Cell Biol. 1973;57:499–524. doi: 10.1083/jcb.57.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clayton EL, Evans GJO, Cousin MA. Bulk synaptic vesicle endocytosis is rapidly triggered during strong stimulation. J Neurosci. 2008;28:6627–6632. doi: 10.1523/JNEUROSCI.1445-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uytterhoeven V, Kuenen S, Kasprowicz J, Miskiewicz K, Verstreken P. Loss of skywalker reveals synaptic endosomes as sorting stations for synaptic vesicle proteins. Cell. 2011;145:117–132. doi: 10.1016/j.cell.2011.02.039. [DOI] [PubMed] [Google Scholar]
- 14.Paillart C, Li J, Matthews G, Sterling P. Endocytosis and vesicle recycling at a ribbon synapse. J Neurosci. 2003;23:4092–4099. doi: 10.1523/JNEUROSCI.23-10-04092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fergestad T, Davis WS, Broadie K. The stoned proteins regulate synaptic vesicle recycling in the presynaptic terminal. J Neurosci. 1999;19:5847–5860. doi: 10.1523/JNEUROSCI.19-14-05847.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips AM, Smith M, Ramaswami M, Kelly LE. The products of the Drosophila stoned locus interact with synaptic vesicles via synaptotagmin. J Neurosci. 2000;20:8254–8261. doi: 10.1523/JNEUROSCI.20-22-08254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diril MK, Wienisch M, Jung N, Klingauf J, Haucke V. Stonin 2 is an AP-2-dependent endocytic sorting adaptor for synaptotagmin internalization and recycling. Dev Cell. 2006;10:233–244. doi: 10.1016/j.devcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 18.Mohrmann R, Matthies HJ, Woodruff E, Broadie K. Stoned B mediates sorting of integral synaptic vesicle proteins. Neuroscience. 2008;153:1048–1063. doi: 10.1016/j.neuroscience.2008.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nonet ML, Holgado AM, Brewer F, Serpe CJ, Norbeck BA, Holleran J, Wei L, Hartwieg E, Jorgensen EM, Alfonso A. UNC-11, a Caenorhabditis elegans AP180 homologue, regulates the size and protein composition of synaptic vesicles. Mol Biol Cell. 1999;10:2343–2360. doi: 10.1091/mbc.10.7.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stimson DT, Estes PS, Smith M, Kelly LE, Ramaswami M. A product of the Drosophila stoned locus regulates neurotransmitter release. J Neurosci. 1998;18:9638–9649. doi: 10.1523/JNEUROSCI.18-23-09638.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stimson DT, Estes PS, Rao S, Krishnan KS, Kelly LE, Ramaswami M. Drosophila stoned proteins regulate the rate and fidelity of synaptic vesicle internalization. J Neurosci. 2001;21:3034–3044. doi: 10.1523/JNEUROSCI.21-09-03034.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao H, Daniels RW, MacLeod GT, Charlton MP, Atwood HL, Zhang B. AP180 maintains the distribution of synaptic and vesicle proteins in the nerve terminal and indirectly regulates the efficacy of Ca2+-triggered exocytosis. J Neurophysiol. 2005;94:1888–1903. doi: 10.1152/jn.00080.2005. [DOI] [PubMed] [Google Scholar]
- 23.Fergestad T, Broadie K. Interaction of stoned and synaptotagmin in synaptic vesicle endocytosis. J Neurosci. 2001;21:1218–1227. doi: 10.1523/JNEUROSCI.21-04-01218.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koo SJ, Markovic S, Puchkov D, Mahrenholz CC, Beceren-Braun F, Maritzen T, Dernedde J, Volkmer R, Oschkinat H, Haucke V. SNARE motif-mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proc Natl Acad Sci U S A. 2011;108:13540–13545. doi: 10.1073/pnas.1107067108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petralia RS, Wang Y-X, Indig FE, Bushlin I, Wu F, Mattson MP, Yao PJ. Reduction of AP180 and CALM produces defects in synaptic vesicle size and density. Neuromolecular Med. 2013;15:49–60. doi: 10.1007/s12017-012-8194-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walther K, Krauss M, Diril MK, Lemke S, Ricotta D, Honing S, Kaiser S, Haucke V. Human stoned B interacts with AP-2 and synaptotagmin and facilitates clathrin-coated vesicle uncoating. EMBO Rep. 2001;2:634–640. doi: 10.1093/embo-reports/kve134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heerssen H, Fetter RD, Davis GW. Clathrin dependence of synaptic-vesicle formation at the Drosophila neuromuscular junction. Curr Biol. 2008;18:401–409. doi: 10.1016/j.cub.2008.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 29.Beck S, Sakurai T, Eustace BK, Beste G, Schier R, Rudert F, Jay DG. Fluorophore-assisted light inactivation: a high-throughput tool for direct target validation of proteins. Proteomics. 2002;2:247–255. doi: 10.1002/1615-9861(200203)2:3<247::aid-prot247>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 30.Kasprowicz J, Kuenen S, Miskiewicz K, Habets RLP, Smitz L, Verstreken P. Inactivation of clathrin heavy chain inhibits synaptic recycling but allows bulk membrane uptake. J Cell Biol. 2008;182:1007–1016. doi: 10.1083/jcb.200804162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang B, Koh YH, Beckstead RB, Budnik V, Ganetzky B, Bellen HJ. Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron. 1998;21:1465–1475. doi: 10.1016/s0896-6273(00)80664-9. [DOI] [PubMed] [Google Scholar]
- 32.Karunanithi S, Marin L, Wong K, Atwood HL. Quantal size and variation determined by vesicle size in normal and mutant Drosophila glutamatergic synapses. J Neurosci. 2002;22:10267–10276. doi: 10.1523/JNEUROSCI.22-23-10267.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harel A, Wu F, Mattson MP, Morris CM, Yao PJ. Evidence for CALM in directing VAMP2 trafficking. Traffic. 2008;9:417–429. doi: 10.1111/j.1600-0854.2007.00694.x. [DOI] [PubMed] [Google Scholar]
- 34.Morgan JR, Zhao X, Womack M, Prasad K, Augustine GJ, Lafer EM. A role for the clathrin assembly domain of AP180 in synaptic vesicle endocytosis. J Neurosci. 1999;19:10201–10212. doi: 10.1523/JNEUROSCI.19-23-10201.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- 36.Jan LY, Jan YN. Antibodies to horseradish peroxidase as specific neuronal markers in Drosophila and in grasshopper embryos. Proc Natl Acad Sci U S A. 1982;79:2700–2704. doi: 10.1073/pnas.79.8.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loewen CA, Mackler JM, Reist NE. Drosophila synaptotagmin I null mutants survive to early adulthood. Genesis. 2001;31:30–36. doi: 10.1002/gene.10002. [DOI] [PubMed] [Google Scholar]
- 38.Boyne AF, McLeod S. Ultrastructural plasticity in stimulated nerve terminals: pseudopodial invasions of abutted terminals in Torpedine ray electric organ. Neuroscience. 1979;4:615–624. doi: 10.1016/0306-4522(79)90138-6. [DOI] [PubMed] [Google Scholar]
- 39.Boyne AF, Phillips TE. Exocytosis and nerve terminal pseudopodia. Fed Proc. 1982;41:2188–2192. [PubMed] [Google Scholar]
- 40.Boyne AF, Tarrant SB. Pseudopodial interdigitations between abutted nerve terminals: diffusion traps which occur in several nuclei of the rat limbic system. J Neurosci. 1982;2:463–469. doi: 10.1523/JNEUROSCI.02-04-00463.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zinsmaier KE, Hofbauer A, Heimbeck G, Pflugfelder GO, Buchner S, Buchner E. A cysteine-string protein is expressed in retina and brain of Drosophila. J Neurogenet. 1990;7:15–29. doi: 10.3109/01677069009084150. [DOI] [PubMed] [Google Scholar]
- 42.Opazo F, Punge A, Bückers J, Hoopmann P, Kastrup L, Hell SW, Rizzoli SO. Limited intermixing of synaptic vesicle components upon vesicle recycling. Traffic. 2010;11:800–812. doi: 10.1111/j.1600-0854.2010.01058.x. [DOI] [PubMed] [Google Scholar]
- 43.Klagges BR, Heimbeck G, Godenschwege TA, Hofbauer A, Pflugfelder GO, Reifegerste R, Reisch D, Schaupp M, Buchner S, Buchner E. Invertebrate synapsins: a single gene codes for several isoforms in Drosophila. J Neurosci. 1996;16:3154–3165. doi: 10.1523/JNEUROSCI.16-10-03154.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akbergenova Y, Bykhovskaia M. Enhancement of the endosomal endocytic pathway increases quantal size. Mol Cell Neurosci. 2009;40:199–206. doi: 10.1016/j.mcn.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bennett MK, Calakos N, Kreiner T, Scheller RH. Synaptic vesicle membrane proteins interact to form a multimeric complex. J Cell Biol. 1992;116:761–775. doi: 10.1083/jcb.116.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willig KI, Rizzoli SO, Westphal V, Jahn R, Hell SW. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nat Cell Biol. 2006;440:935–939. doi: 10.1038/nature04592. [DOI] [PubMed] [Google Scholar]
- 48.Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJG, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 49.Legendre-Guillemin V, Wasiak S, Hussain NK, Angers A, McPherson PS. ENTH/ANTH proteins and clathrin-mediated membrane budding. J Cell Sci. 2004;117:9–18. doi: 10.1242/jcs.00928. [DOI] [PubMed] [Google Scholar]
- 50.Stahelin RV, Long F, Peter BJ, Murray D, De Camilli P, McMahon HT, Cho W. Contrasting membrane interaction mechanisms of AP180 N-terminal homology (ANTH) and epsin N-terminal homology (ENTH) domains. J Biol Chem. 2003;278:28993–28999. doi: 10.1074/jbc.M302865200. [DOI] [PubMed] [Google Scholar]
- 51.Ford MG, Pearse BM, Higgins MK, Vallis Y, Owen DJ, Gibson A, Hopkins CR, Evans PR, Mcmahon HT. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science. 2001;291:1051–1055. doi: 10.1126/science.291.5506.1051. [DOI] [PubMed] [Google Scholar]
- 52.Stachowiak JC, Schmid EM, Ryan CJ, Ann HS, Sasaki DY, Sherman MB, Geissler PL, Fletcher DA, Hayden CC. Membrane bending by protein-protein crowding. Nat Cell Biol. 2012;14:944–949. doi: 10.1038/ncb2561. [DOI] [PubMed] [Google Scholar]
- 53.Banerjee A, Berezhkovskii A, Nossal R. Stochastic model of clathrin-coated pit assembly. Biophys J. 2012;102:2725–2730. doi: 10.1016/j.bpj.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernández-Alfonso T, Kwan R, Ryan TA. Synaptic vesicles interchange their membrane proteins with a large surface reservoir during recycling. Neuron. 2006;51:179–186. doi: 10.1016/j.neuron.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 55.Wienisch M, Klingauf J. Vesicular proteins exocytosed and subsequently retrieved by compensatory endocytosis are nonidentical. Nat Neurosci. 2006;9:1019–1027. doi: 10.1038/nn1739. [DOI] [PubMed] [Google Scholar]
- 56.Fernández-Alfonso T, Ryan TA. A heterogeneous “resting” pool of synaptic vesicles that is dynamically interchanged across boutons in mammalian CNS synapses. Brain Cell Biol. 2008;36:87–100. doi: 10.1007/s11068-008-9030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Estes PS, Roos J, van der Bliek A, Kelly RB, Krishnan KS, Ramaswami M. Traffic of dynamin within individual Drosophila synaptic boutons relative to compartment-specific markers. J Neurosci. 1996;16:5443–5456. doi: 10.1523/JNEUROSCI.16-17-05443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maritzen T, Koo SJ, Haucke V. Turning CALM into excitement: AP180 and CALM in endocytosis and disease. Biol Cell. 2012;104:588–602. doi: 10.1111/boc.201200008. [DOI] [PubMed] [Google Scholar]
- 59.Yao PJ, Coleman PD. Reduced O-glycosylated clathrin assembly protein AP180: implication for synaptic vesicle recycling dysfunction in Alzheimer's disease. Neurosci Lett. 1998;252:33–36. doi: 10.1016/s0304-3940(98)00547-3. [DOI] [PubMed] [Google Scholar]
- 60.Cao Y, Xiao Y, Ravid R, Guan Z-Z. Changed clathrin regulatory proteins in the brains of Alzheimer's disease patients and animal models. J Alzheimers Dis. 2010;22:329–342. doi: 10.3233/JAD-2010-100162. [DOI] [PubMed] [Google Scholar]
- 61.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Babcock MC, Stowers RS, Leither J, Goodman CS, Pallanck LJ. A genetic screen for synaptic transmission mutants mapping to the right arm of chromosome 3 in Drosophila. Genetics. 2003;165:171–183. doi: 10.1093/genetics/165.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Venken KJ, He Y, Hoskins RA, Bellen HJ. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–1751. doi: 10.1126/science.1134426. [DOI] [PubMed] [Google Scholar]
- 64.Stewart BA, Atwood HL, Renger JJ, Wang J, Wu C-F. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol A. 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- 65.Jan LY, Jan YN. Properties of the larval neuromuscular junction in Drosophila melanogaster. J Physiol. 1976;262:189–214. doi: 10.1113/jphysiol.1976.sp011592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reist NE, Buchanan J, Li J, Diantonio A, Buxton EM, Schwarz TL. Morphologically docked synaptic vesicles are reduced in synaptotagmin mutants of Drosophila. J Neurosci. 1998;18:7662–7673. doi: 10.1523/JNEUROSCI.18-19-07662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]