

Abstract
Sickle cell disease (SCD) produces many structural and functional abnormalities in the kidney, including glomerular abnormalities. Albuminuria is the most common manifestation of glomerular damage, with a prevalence between 26 and 68% in adult patients. The pathophysiology of albuminuria in SCD is likely multifactorial, with contributions from hyperfiltration, glomerular hypertension, ischemia-reperfusion injury, oxidative stress, decreased nitric oxide (NO) bioavailability, and endothelial dysfunction. Although its natural history in SCD remains inadequately defined, albuminuria is associated with increased echocardiography-derived tricuspid regurgitant jet velocity, systemic blood pressure, and hypertension, as well as history of stroke, suggesting a shared vasculopathic pathophysiology. While most patients with albuminuria are treated with angiotensin converting enzyme inhibitors/angiotensin receptor blockers, there are no published long-term data on the efficacy of these agents. With the improved patient survival following kidney transplantation, SCD patients with end-stage renal disease should be considered for this treatment modality. Given the high prevalence of albuminuria and its association with multiple SCD-related clinical complications, additional studies are needed to answer several clinically important questions in a bid to adequately elucidate its pathophysiology, natural history, and treatment.
Introduction
Sickle cell disease (SCD), one of the most common monogenic disorders worldwide, results in many structural and functional abnormalities in the kidney, including abnormalities of tubular function, hematuria, and glomerular abnormalities [1]. The kidney is particularly sensitive to the effects of hypoxia because of its high rate of oxygen consumption [2]. Due to the presence of acidosis, hypertonicity and hypoxia in the environment of the renal medulla, this portion of the kidney is very susceptible to changes in oxygen delivery. As blood traverses the slow-moving circuit of the medullary vasa recta, the hyperosmolar milieu may enhance dehydration of red blood cells (RBCs), allowing polymerization of sickle hemoglobin (HbS) and resulting in vaso-occlusion and medullary microinfarction [3,4]. Indeed, microangiopathic studies show the loss of vasa recta in older patients with SCD, with those that remain being abnormally dilated or blunted [5]. This review will focus on albuminuria, the most common clinical manifestation of glomerular damage in SCD.
Renal Pathology in SCD
In young SCD patients with normal renal function, the kidneys are enlarged, with a smooth capsular surface [6]. With advancing age and the development of chronic renal failure, the kidneys become scarred and shrunken, with the capsular surface ranging from coarsely granular to grossly distorted and scarred [6]. Unlike in normal individuals, glomerular size increases with age in SCD [6]. These enlarged, markedly hypercellular glomeruli exhibit lobulation of the glomerular tuft on histological examination. In the early stages of sickle cell nephropathy, renal biopsy shows glomerular hypertrophy, hemosiderin deposits, and focal areas of hemorrhage or necrosis [7,8]. In later stages, interstitial inflammation, edema, fibrosis, tubular atrophy, and papillary infarcts are commonly observed, with these changes mostly due to vascular dropout [7,8]. A multicenter retrospective survey of 18 SCD patients (HbSS—16; HbSC—1; 1 HbSβ+ thalassemia—1) who underwent renal biopsies for isolated proteinuria or in association with acute or progressive impairment of renal function showed focal segmental glomerulosclerosis (FSGS) in seven cases, membranoproliferative glomerulonephritis (MPGN) in five cases, thrombotic microangiopathic glomerulopathy in three cases, and glomerular hypertrophy with or without mesangial hypercellularity (early sickle cell disease glomerulopathy) in three cases, suggesting a wide spectrum of glomerular lesions in SCD [9]. Regardless of the observed morphologic lesion, glomeruli were enlarged and the capillaries were distended by sickled RBCs. Of the seven FSGS cases, exclusive not otherwise specified (NOS) lesions were seen in three cases, NOS lesions associated with tip lesions in two cases and two cases showed concomitant tip lesions and perihilar lesions [9]. A collapsing pattern of FSGS has also been reported [10]. Glomerular changes that are indistinguishable from those of proliferative glomerulonephritis may be seen in SCD patients with no apparent renal disease [6]. Reduplication of the basement membrane and mesangial proliferation are also seen in SCD, especially as patients' age. Immunofluorescence microscopy in patients with FSGS type lesions demonstrates irregular staining for IgM and C3 in areas of sclerosis [7,8,10], and MPGN lesions demonstrate capillary wall staining for IgG, IgM, IgA, C3, and C1q [10]. Electron microscopy of glomeruli in SCD patients with proteinuria or the nephrotic syndrome show effacement of the podocyte foot processes, with occasional wrinkling of the capillary wall, usually associated with partial or complete mesangial interposition [8].
Transgenic mouse models of SCD show findings similar to those observed in human sickle cell glomerulopathy. The transgenic SAD mouse bears the human α-globin gene and the HbS mutation, βS, as well as βAntilles and βD-Punjab, which greatly enhance the tendency of its hemoglobin to polymerize. The renal pathology of the SAD mouse shows glomerular hypertrophy and mesangial sclerosis, which increase in frequency and severity as the mice age [11]. Glomerular damage is associated with increased levels of blood urea nitrogen and nonselective proteinuria. Other renal changes in these mice include hemosiderosis, cortical infarcts, and papillary necrosis. In sickle mice that exclusively express the human α- and βS-globin (Berkeley mice), kidney weights are increased 2-fold, and histologic analysis shows fibrosis, atrophy, infarcts, and cysts, with increased iron deposits in the tubular epithelium [12]. Hemizygous sickle mice expressing human α- and βS- transgenes in combination with mouse β+/−, show mild to severe renal lesions including glomerulonephritis, dilated tubules, atypical tubules, tubular degeneration, mild necrosis, and interstitial fibrosis in about 50% of mice [13]. The renal tubular epithelium shows increased expression of inducible nitric oxide (NO) synthase and 3-nitrotyrosine, as well as increased staining for vascular cell adhesion molecule-1 (VCAM-1) in the interstitial capillary cells and tubular epithelial cells compared with normal control mice.
Pathophysiology
The relative hypoxia, acidosis, and hyperosmolarity of the inner medulla favor polymerization of deoxygenated HbS and subsequent sickling of RBCs. These repeated cycles of sickling are thought to lead to ischemic injury and microinfarction, which ultimately result in reduced medullary blood flow. Worsening hypoxia results in localized prostaglandin release and marked vasodilation, which increases renal blood flow and hence glomerular filtration rate (GFR). Indirect measurements of renal prostaglandin activity have been performed by observing the effects of the prostaglandin synthesis inhibitor, indomethacin. Following administration of indomethacin, GFR and effective renal plasma flow (ERPF) fell significantly in HbSS patients but remained unchanged in control subjects [14], suggesting that prostaglandins are important in maintaining the supranormal levels of GFR and ERPF observed in SCD. The association of increased GFR in HbSS patients with albuminuria raises the possibility that hyperfiltration may contribute to renal damage [15–18] (TableI). If, indeed, hyperfiltration is a necessary precursor of glomerulopathy in SCD, then endogenous perturbation of the prostaglandin systems could explain the early development of FSGS in this setting. Hyperfiltration also may lead to endothelial hyperplasia and ultimately glomerular fibrosis [31,32].
TABLE I.
Pathophysiology of Albuminuria in Sickle Cell Disease
| Proposed mechanisms | References |
|---|---|
| Hyperfiltration | [15–18] |
| Glomerular hypertension | [7,8,19,20] |
| Ischemia-reperfusion injury/oxidative stress | [21–24] |
| Decreased nitric oxide bioavailability due to hemolysis and/or increased levels of soluble fms-like tyrosine kinase-1 | [25–29] |
| Chronic treatment with opioid analgesics | [30] |
The pathophysiology of sickle cell glomerulopathy has been reported to resemble that which develops in rodents following subtotal nephrectomy [7,8,19]. In these animals, glomerular hypertension in the remaining hypertrophied glomeruli may result in glomerular sclerosis [20]. The observation that glomerular hypertension is attenuated by angiotensin converting enzyme inhibitors in rodents, combined with the quick and reversible decrease in protein excretion in HbSS patients receiving these agents gives further credence to the possibility that this mechanism contributes to SCD-related glomerulopathy [7,20].
Transgenic mice homozygous for the murine β-globin deletion that carry two transgenes, αHβS and αHβS-Antilles, exhibit oxidative stress in the kidney as indicated by increased amounts of lipid peroxidation [21]. Exacerbation of oxidative stress increased sickling of RBCs, extending from the medulla into the cortical capillaries and glomeruli. In these sickle mice, renal ischemia following total renal artery occlusion for 15 min resulted in a significant increase in creatinine and blood urea nitrogen levels that was associated with diffuse acute tubular necrosis extending across the entire width of the cortex, with large segments of cortical necrosis [22]. Six hours after bilateral renal ischemia for 22.5 min, the sickle mice showed greater renal histological injury than wild-type mice with extensive tubular necrosis extending from the corticomedullary junction through the full thickness of the cortex accompanied by marked congestion in peritubular capillaries and an acute glomerulopathy consisting of endothelialitis and mesangiolysis [22]. These latter changes along with accompanying reparative responses, especially if incurred through repetitive cycles of renal ischemia, may contribute to a chronic glomerulopathy in SCD (Fig. 1).
Figure 1.
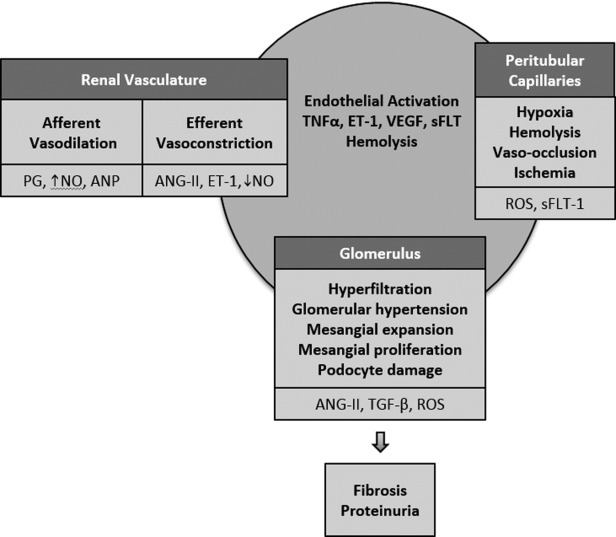
Proposed mechanisms of glomerulopathy in sickle cell disease. Multiple mechanisms may contribute to the pathogenesis of glomerular damage in sickle cell disease. These may occur due to changes in the renal vasculature, peritubular capillaries and the glomerulus. PG: prostaglandins; NO: nitric oxide; ANP: atrial natriuretic peptide; ROS: reactive oxygen species; sFLT-1: soluble fms-like tyrosine kinase-1; VEGF: vascular endothelial growth factor; ANG-II: angiotensin II; TGF-β: transforming growth factor-β; TNFα: tumor necrosis factor-α; ET-1: endothelin-1.
Renal ischemia results in an impairment of GFR, as well as a reduction in renal blood flow in sickle mice [23]. Ischemia results in a failure of phosphorylation of MAPK/Akt signaling proteins, ERK1/2 (p42/44), JNK (p44/46), p38, and Akt in sickle mice. These proteins influence inflammatory responses and cell survival, proliferation, migration, and differentiation following ischemia, and thus modulate the renal response to acute ischemic injury [23]. As the phosphorylated form of these signaling proteins usually determines the active moiety, the recruitment of signaling processes following ischemia is markedly impaired in the kidney of sickle mice. The impaired phosphorylation of mitogen-activated protein kinase (MAPK) and Akt proteins in the kidney following ischemia may partly be due to decreased renal ATP content and correlates with increased expression of TNF-α, an inflammatory cytokine which may contribute to the increased sensitivity of the kidney to ischemia in sickle mice [23].
More recent data appear to confirm the role of oxidative stress in sickle cell glomerulopathy. Reactive oxygen species (ROS) increase the conversion of oxidized angiotensinogen to angiotensin II (ATII), and increase secondary angiotensin receptor 1 (AT1R)-mediated generation of transforming growth factor-beta 1 (TGF-β1) in the kidney in SCD, which then phosphorylates Smad 2/3 [24]. In young sickle mice, blockade of the AT1R with losartan, or its ligand ATII by an angiotensin converting enzyme inhibitor, captopril, prevented albuminuria and FSGS development but worsened hyposthenuria, suggesting that excessive AT1R signaling causes sickle cell glomerulopathy, while AT1R promotes urine concentrating ability. Furthermore, AT1R signaling is known to activate NADPH oxidase to generate ROS and mice treated with captopril and losartan had reduced ROS in RBC, platelets as well as the kidneys, and consequently reduced renin-angiotensin system activation.
The level of soluble fms-like tyrosine kinase-1 (sFLT-1) is elevated in patients with SCD patients compared to healthy, race-matched control subjects [33,34]. sFLT-1 is a member of the vascular endothelial growth factor receptor (VEGFR) family [35] and is a splice variant of VEGFR1. VEGF, a well-known promoter of angiogenesis and an endogenous regulator of endothelial integrity promotes endothelial cell survival and angiogenesis through activation of the protein kinase, Akt, which in turn activates endothelial NO synthase and increases NO generation [36]. By antagonizing VEGF action and inhibiting Akt phosphorylation, sFLT-1 decreases NO bioavailability, thus leading to endothelial dysfunction. sFLT-1 has been linked with preeclampsia, a condition characterized by hypertension, proteinuria, and endothelial dysfunction [37]. We have shown that SCD patients with macroalbuminuria have significantly higher levels of sFLT-1 and soluble VCAM-1 compared to patients with lower levels of albuminuria [25]. The association of albuminuria with soluble VCAM-1, combined with the association of sFLT-1 with soluble VCAM-1, suggests that sFLT-1 may contribute to the pathogenesis of albuminuria in SCD by promoting endothelial dysfunction [25].
Multiple observational studies show an association of albuminuria with markers of hemolysis in patients with SCD [26–29], although other studies have not shown such associations [18,25,38,39]. Chronic depletion of NO due, at least in part, to scavenging by cell-free hemoglobin from ongoing intravascular hemolysis, may contribute to the glomerulopathy in SCD. In addition, the activation of hypoxia-inducible factor 1α and the consequent increase in local endothelin-1 release, in the presence of reduced NO, leads to an increase in ROS and vasoconstriction, contributing to a cycle of chronic medullary hypoxia [40,41]. We have shown an association of sFLT-1 with markers of hemolysis [34] suggesting that sFLT-1 may be downstream of hemolysis, and increased sFLT-1 levels may further decrease the bioavailability of NO in SCD. NO deficiency has been demonstrated in individuals with chronic kidney disease, and this may lead to progression of chronic kidney disease [42]. Indeed, animal studies show that chronic inhibition of NO synthase causes systemic and glomerular hypertension, glomerular ischemia, glomerulosclerosis, tubulointerstitial injury, and proteinuria [43]. With the increasing evidence that hemolysis may play a role in the vasculopathy of SCD [44], hemolysis and NO deficiency may also contribute to the development of glomerulopathy in SCD.
Chronic treatment of transgenic sickle mice with morphine for 3–6 weeks has been reported to produce defects in renal pathology, including increased glomerular volume, mesangial expansion, mesangial cell proliferation, parietal cell metaplasia, podocyte effacement, and microvillus transformation [30]. Chronic treatment with morphine also increased the expression and activity of heme oxygenase-1, and increased albuminuria [30]. The renal effects of morphine are ameliorated by naloxone, a nonselective opioid antagonist, suggesting that opioid receptor antagonists may be effective in decreasing morphine-induced renal disease.
Clinical Implications
The normal rate of urinary albumin excretion is less than 20 mg/day (15 mcg/min), and is usually between 4 and 7 mg/day in healthy young adults. Persistent excretion of albumin between 30 and 300 mg/day (20–200 mcg/min) is called microalbuminuria, while albumin excretion above 300 mg/day (200 mcg/min) is considered overt proteinuria or macroalbuminuria. The standard urine dipstick primarily detects albumin, and is only semiquantitative. While very specific, the standard dipstick test is not sensitive to low levels of albuminuria and will not usually identify those with microalbuminuria. The gold standard for quantitative assessment of albuminuria is a 24-hr urine collection. Although proteinuria can be assessed by measuring the ratio of protein-to-creatinine (or albumin-to-creatinine ratio) from a spot first- or second-morning sample, this method has not been validated in SCD. In a study of 25 patients with albuminuria, albumin-to-creatinine ratio was reported to correlate with 12-hr urinary albumin excretion [45]. However, no correlations were observed in the percent change (from the first measurements) following treatment with angiotensin converting enzyme inhibitors at the times of second and third measurements of 12-hr urinary albumin excretion and albumin-to-creatinine ratio [45]. In another small study, the presence of microalbuminuria diagnosed by spot urine albumin-to-creatinine ratio in 19 patients was confirmed by 24-hr urine collection in only 57% of cases [46].
The prevalence of albuminuria in SCD increases with age, varying between 4.5 and 26% in patients up to 21 years [46–52], and between 26 and 68% in older patients [7,15,25,38,53,54]. Although a prospective, 25-year case-control study reported that 9 of 21 HbSS patients with nephrotic syndrome developed chronic renal failure [55], the natural history of albuminuria in SCD remains inadequately defined. A small study of 38 patients, 30 of whom had microalbuminuria and eight with macroalbuminuria, showed progression of albuminuria in two patients with microalbuminuria and two patients with macroalbuminuria after approximately 20 months of follow-up [56]. In a retrospective study of 14 patients, with estimated GFR at the time of kidney biopsy ranging from 24 to 158 mL/min per m2 and urine albumin excretion at the time of kidney biopsy ranging from 0.4 to 33 g/day, seven patients had chronic kidney disease and three patients required chronic intermittent hemodialysis due to ESRD after an average follow-up of 28 months (range, 4–79 months) [9]. Evaluation for proteinuria was only available in 12 patients, and a decrease in urinary protein was noted in 67% of patients treated with ATII receptor blockers or angiotensin converting enzyme inhibitors. In a more recent retrospective, single center study of 98 SCD patients, grade A3 albuminuria (adjusted odds ratio [OR], 5.0; 95% confidence interval [CI], 1.1–24.3; P = 0.0480) and each 1 mm Hg increase in systolic blood pressure (adjusted OR, 1.04; 95% CI, 1.0–1.07; P = 0.039) were associated with the development and progression of chronic kidney disease on multivariate analysis after 5 years of follow-up [57]. In this study, grade A3 proteinuria was defined as a urine protein-to-creatinine ratio greater than 500 mg/g, based on the 2012 KDIGO clinical practice guidelines [58].
Albuminuria is more prevalent in HbSS patients than in those with other sickling hemoglobinopathies [38] and is associated with echocardiography-derived tricuspid regurgitant jet velocity [25,54,59,60], increased systemic blood pressure and hypertension [38,15,54,61], and history of stroke [25,48]. These associated comorbidities suggest a shared vasculopathic pathophysiology with albuminuria [25–29]. Albuminuria is also reported to be associated with asymptomatic bacteruria [52], as well as a history of acute chest syndrome [46,54] and cholelithiasis [48], likely reflecting the role of hemolysis in the pathogenesis of albuminuria. Finally, a recent multicenter cross-sectional study reported an association of proteinuria, assessed by dipstick, with early mortality [62] (TableII).
Table II.
Summary of Studies of Albuminuria in Sickle Cell Disease
| Reference | Number of patients | Mean or median age/range | Type of study | Measurement | Prevalence | Comments |
|---|---|---|---|---|---|---|
| Alvarez et al. [46] | 120 | 4–20 years | Retrospective | Microalbuminuria | 15.8% | • Increased age associated with microalbuminuria. |
| • Early transfusion protective of microalbuminuria. | ||||||
| • Positive correlation with acute chest syndrome. | ||||||
| Dharnidharka et al. [47] | 102 | 2–18 years | Prospective | Microalbuminuria | 26.5% | • More common in patients older than 10years. |
| • Increasing age only variable associated with microalbuminuria. | ||||||
| McBurney et al. [49] | 142 | 21 months–20 years | Retrospective | Microalbuminuria | 19% | • Increased age and lower hemoglobin correlated with microalbuminuria. |
| McKie et al. [50] | 191 | 3–20 years | Prospective | Microalbuminuria | 19.4% | • Increased age and lower hemoglobin in patients with microalbuminuria. |
| • Four of nine patients receiving hydroxyurea demonstrated regression of microalbuminuria. | ||||||
| McPherson Yee et al. [51] | 410 | 2–21 years | Cross sectional | Microalbuminuria | 20.7% | • Increased age and lower hemoglobin in patients with microalbuminuria. |
| Aygun et al. [17] | 23 | 2.5–14 years | Prospective | Microalbuminuria | 17.4% | • All subjects treated with hydroxyrea. |
| • After 3 years of therapy, microalbuminuria resolved in two patients, persisted in two patients, and two patients developed new microalbuminuria. | ||||||
| • Treatment with hydroxyurea resulted in reduction in hyperfiltration, with associated decrease in LDH and increase in HbF levels. | ||||||
| Thompson et al. [15] | 65 | 18–23 years | Cross sectional | Albuminuria | 26.2% | • eGFR and SBP correlated positively with albumin excretion. |
| • Serum sodium and hematocrit correlated negatively with albumin excretion. | ||||||
| Bolarinwa et al. [61] | 68 | 15–60 years | Cross sectional | Albuminuria | 50.0% | • DBP associated with albuminuria. |
| • Albuminuria more common with worsening CKD stage. | ||||||
| Laurin et al. [54] | 149 | 18–71 years | Retrospective | Albuminuria | 45.0% | • Lower hemoglobin associated with albuminuria. |
| • Hydroxyurea use associated with a third lower likelihood of albuminuria. | ||||||
| Ataga et al. [25] | 73 | 39a years | Cross sectional | Albuminuria | 53.4% | • Weak correlation with age and albumin excretion. |
| • eGFR lowest in patients with macroalbuminuria. | ||||||
| • NT-proBNP, sFLT-1 higher in patients with macroalbuminuria. | ||||||
| • Higher TRV with macroalbuminuria. | ||||||
| • Association of urine albumin excretion with suspected pulmonary hypertension and history of stroke. | ||||||
| • Among HbSS and HbSβ0 patients, albuminuria associated with VCAM-1 and hypertension. | ||||||
| Guasch et al. [38] | 300 | 19–76 years | Cross sectional | Albuminuria | 58% | • Higher prevalence in HbSS (68%). |
| • Prevalence of albuminuria increased with age. | ||||||
| Iwalokun et al. [52] | 103 | 10.4b years | Cross sectional | Albuminuria | 22.3% | • Albuminuria associated with age, irreversibly sickled RBC, creatinine, packed cell volume and asymptomatic bacteruria. |
| • Irreversibly sickled RBC only independent predictor of albuminuria. | ||||||
| Asnani et al. [39] | 121 | 24.1–32.5 years | Cross sectional | Albuminuria | 33.6% | • Higher prevalence in HbSS. |
| • In HbSS, albuminuria associated with higher mean arterial pressure, higher WBC, lower hemoglobin, lower reticulocyte count, and lower serum creatinine. | ||||||
| • In HbSC, albuminuria associated with higher WBC and higher creatinine. | ||||||
| Wigfall et al. [48] | 442 | 2–21 years | Prospective | Proteinuria (urinalysis) | 4.5% | • Increased prevalence with age. |
| • Associated with stroke, acute chest syndrome, hospitalizations, and cholelithiasis. | ||||||
| Falk et al. [7] | 381 | N/A (children and adults) | Prospective | Proteinuria (urinalysis) | 26% | • In 10 patients treated with enalapril, mean reduction in proteinuria of 57% from baseline after 2 weeks. |
| Aleem [[53] | 67 | 23.8 ± 7.2 yearsb | Cross sectional | Proteinuria (24hr urine) | 40.3% | • Higher age in patients with proteinuria, but not statistically significant |
| De Castro et al. [59] | 75 | 39.3 ± 11.7 yearsb | Retrospective | Proteinuria (urinalysis) | 28% | • Proteinuria was associated with TRV ≥ 2.5 m/s. |
| • Proteinuria inversely correlated with eGFR in patients with TRV ≥ 2.5 m/s. | ||||||
| Elmariah et al. [62] | 542 | 18–84 years | Cross sectional | Proteinuria (urinalysis) | 26% | • Proteinuria and reduced renal function both associated with greater mortality. |
| Forrest et al. [60] | 85 | 6–21 years | Retrospective | Proteinuria (urinalysis) | N/A | • Elevated TRV ≥ 2.5 m/s is associated with proteinuria on longitudinal follow up. |
Median.
Mean.
CKD = Chronic kidney disease; eGFR = Estimated glomerular filtration rate; SBP = Systolic blood pressure; DBP = Diastolic blood pressure; TRV = Tricuspid regurgitant jet velocity; RBC = Red blood cells; WBC = White blood cells; HbF = Fetal hemoglobin; HTN = Hypertension; LDH = Lactate dehydrogenase; sFLT-1 = soluble fms-like tyrosine kinase-1; NT-proBNP = N-terminal pro-brain natriuretic peptide; VCAM-1 = Vascular cell adhesion molecule-1.
Treatment
Improved medical care for individuals with SCD has prolonged their survival, resulting in an increased prevalence of end-organ damage. Albuminuria occurs more commonly with increasing age and may progress to ESRD. Progression to ESRD is often preceded by increasing proteinuria, worsening anemia, and/or the appearance of hypertension [55]. There are no large, controlled studies demonstrating the efficacy of any interventions for albuminuria in SCD. However, treatment of 10 HbSS patients who had proteinuria (0.8–10.8 g/day) with the angiotensin converting enzyme inhibitor, enalapril, for 2 weeks decreased the rate of urinary protein excretion in all patients at the end of the treatment period [7]. This observation was confirmed in a 6-month study in eight HbSS patients with albuminuria [63]. In a double-blind, placebo-controlled study of 22 HbSS patients with microalbuminuria, treatment with captopril resulted in a decrease in microalbuminuria from baseline, while an increase in microalbuminuria was observed in the placebo group [64]. Based on these limited data, angiotensin converting enzyme inhibitors and/or angiotensin receptor blockers have become the “standard of care” for the treatment of albuminuria in SCD (TableIII). However, adequately powered controlled studies of these agents are required to evaluate whether they can halt the progression of SCD glomerulopathy and subsequent chronic kidney disease. Hyperkalemia and renal tubular acidosis may occur as manifestations of SCD nephropathy and the use of angiotensin converting enzyme inhibitor therapy may increase the likelihood of hyperkalemia. As such, other effective agents without this side effect are needed in patients with albuminuria. Angiotensin receptor blocking agents are increasingly used instead of angiotensin converting enzyme inhibitors because of their improved tolerability, although there are no published data in SCD to date. Two clinical trials of the angiotensin receptor blocker, losartan, are ongoing (NCT01989078, NCT01479439).
Table III.
Evaluation and Treatment of Albuminuria in Sickle Cell Disease
| • Screening for albuminuria by standard dipstick urinalysis (with serum creatinine) at least yearly. |
| • Albuminuria detected by dipstick urinalysis should be quantified by 24-hr urine collection (or spot urine for protein-to-creatinine ratioa). |
| • In patients with overt proteinuria, HIV, RPR, hepatitis B, hepatitis C, complements, and antinuclear antibodies should be measured and urine sediment evaluated, particularly for presence of RBC casts, as they may suggest other diagnoses. Serum protein electrophoresis and/or serum free light chains should also be measured in older patients. Consider a renal biopsy if acute onset of nephrotic range proteinuria. |
| • Avoid use of NSAIDs. |
| • Adequate immunization—pneumococcal and influenza. |
| • Consider use of ACE inhibitors or ARBs, if there are no contraindications. Provide counseling regarding dietary potassium intake and monitor potassium following initiation. |
| • Consider use of hydroxyurea, especially in patients with history of frequent pain episodes, acute chest syndrome, or marked anemia. |
This method has not been validated in SCD.
HIV = Human immunodeficiency virus; RPR = Rapid plasma reagin; NSAIDs = Nonsteroidal anti-inflammatory drugs; ACE inhibitors = angiotensin converting enzyme inhibitors; ARBs = Angiotensin receptor blockers.
Therapies that improve RBC survival, such as hydroxyurea, or those that improve endothelial function may be beneficial. In a retrospective study, three HbSS children with nephrotic range proteinuria on treatment with enalapril had a near-normal urine protein-creatinine ratio following the addition of hydroxyurea [65]. Further observational studies suggest that hydroxyurea may reduce albuminuria and slow the development of hyperfiltration [50,54,66]. Young age at start of chronic RBC transfusion has also been suggested to provide protection against the development of albuminuria [45], although there are no controlled studies in this setting.
As nonsteroidal anti-inflammatory agents can produce significant declines in the rates of glomerular filtration and renal blood flow in SCD [67,68], and can increase the rate of progression to ESRD, these agents should be used with caution in patients with evidence of glomerulopathy. In addition, effective treatment of hypertension has been reported to delay the progression to ESRD in SCD patients [69], and careful attention to blood pressure control is important in these patients. Despite reports of relative hypertension and its possible association with renal insufficiency as well as other complications [70–73], there are no guidelines for its treatment in patients with SCD. SCD patients can generally be treated like other adults with hypertension, although diuretics should be used with caution to avoid dehydration, which may precipitate acute painful episodes in these patients.
In SCD patients who develop ESRD, renal replacement therapies (hemodialysis and peritoneal dialysis) as well as renal transplantation are available treatment modalities. Predialysis nephrology care in patients with ESRD is associated with a lower death rate compared with those not receiving such care [74], and early referral to a nephrologist is recommended. While some reports have indicated poor allograft survival as well as a variety of disease-specific problems [75], others have found graft and patient survival rates comparable to those of other nondiabetic ESRD patients [76]. Short-term allograft results have been reported to be comparable in SCD patients and age-matched African-American kidney transplant recipients with other causes of ESRD, although there was a somewhat shorter cadaveric graft survival and a greater adjusted 3-year risk of graft loss in SCD patients beyond 1 year [77]. Furthermore, there was a trend toward improved survival when renal transplantation was compared to chronic dialysis in patients with ESRD secondary to sickle cell nephropathy.
A report based on data from the United Network for Organ Sharing registry reported patient survival at 3 years among the SCD kidney recipients of 75%, with these kidney recipients having a 7.9 times higher risk of posttransplant death compared with patients transplanted for IgA nephropathy (95% CI: 4.3–14.5) [78]. However, it has been suggested that IgA nephropathy may not be the appropriate reference condition to compare to SCD as it is uncommon in African-Americans [79,80]. When kidney transplant recipients of African descent were stratified by transplant era (1988–1999 vs. 2000–2011) using data from the Organ Procurement and Transplantation Network/United Network for Organ Sharing, patient survival at 6 years was lower in SCD patients in the earlier era compared to other patient groups (55.7 % vs. 78%, P < 0.001), with an improvement in the 6-year survival of SCD patients in the more recent era compared to other patient groups (69.8% versus 80%, P = 0.07) [81]. Although still lower than recipients with hypertensive kidney disease and glomerulonephritis, survival of SCD kidney recipients over 6 years was comparable to that of recipients with diabetes.
Renal transplantation may be complicated further by the resumption of frequent painful episodes, possibly due to an increase in whole blood viscosity that accompanies higher posttransplantation hematocrit levels [82]; posttransplantation renal allograft thrombosis and infarction [83]; and the development of sickle cell nephropathy in the donor kidney [84].
Despite these various therapeutic modalities, the prevention of sickle cell glomerulopathy and subsequent chronic kidney disease ultimately depends upon the development of an early cure for SCD. Until gene therapy fulfills its enormous promise, the only currently available curative modality for SCD is bone marrow transplantation (BMT). However, the availability of BMT remains limited. The ability to confidently identify those young patients who are at the highest risk for severe disease will make it possible to select the most appropriate candidates for BMT long before end-organ damage becomes manifest. In addition, early involvement of nephrologists in the treatment of patients with albuminuria may be beneficial.
Conclusions and Need for Future Studies
SCD is often complicated by a glomerulopathy, which commonly manifests as albuminuria. Although the natural history of albuminuria in SCD remains poorly defined, patients who exhibit albuminuria are more likely to develop ESRD. With the association of ESRD and an increased risk of mortality, early identification and treatment of albuminuria may decrease the risk of death in SCD. Further, due to its association with other SCD-related complications, albuminuria may be a useful biomarker for vasculopathic complications in SCD. The pathophysiology of albuminuria in SCD is likely multifactorial, with contributions from hyperfiltration, glomerular hypertension, ischemia-reperfusion injury, oxidative stress, decreased NO bioavailability, and endothelial dysfunction. Although most patients with albuminuria are treated with angiotensin converting enzyme inhibitors/angiotensin receptor blockers, there are no long-term data on the efficacy of these agents. With the limited data on these agents, combined with their side effect profile, other treatment agents are needed. In view of the improved patient survival following kidney transplantation, more SCD patients with ESRD should be considered for this treatment modality.
Given the high prevalence of albuminuria and its association with multiple SCD-related clinical complications, additional studies are clearly required to answer several clinically important questions in a bid to elucidate its pathophysiology, natural history, and treatment. What are the long-term renal consequences of albuminuria in SCD? Is spot urine quantification of albuminuria reliable in SCD? Can albuminuria serve as a suitable biomarker of renal and other vasculopathic complications in SCD? Will screening for microalbuminuria detect kidney disease earlier? Will treatment of microalbuminuria delay the progression to overt proteinuria and chronic kidney disease in SCD? Does treatment with opioid analgesics worsen albuminuria in SCD? Does treatment of proteinuria with angiotensin converting enzyme inhibitors/angiotensin receptor blockers lead to long-term benefit and attenuation of chronic kidney disease? Do hydroxyurea and other agents that improve RBC survival, increase NO bioavailability and improve endothelial function have a role in the treatment of SCD glomerulopathy? Only through carefully designed clinical and laboratory studies will these questions be answered.
Acknowledgments
Support for this work was also provided by an award from the North Carolina State Sickle Cell Program (KIA).
Author Contributions
KIA, VKD, and DRA wrote the paper.
References
- 1.Ataga KI, Orringer EP. Renal abnormalities in sickle cell disease. Am J Hematol. 2000;63:205–211. doi: 10.1002/(sici)1096-8652(200004)63:4<205::aid-ajh8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 2.Falk RJ, Jenette JC. Renal disease. In: Embury SH, Hebbel RP, Mohandas N, Steinberg MH, editors. Sickle Cell Disease: Basic Principles and Clinical Practice. New York: Raven Press; 1994. pp. 673–680. [Google Scholar]
- 3.Sears DA. The morbidity of sickle cell trait: A review of the literature. Am J Med. 1978;64:1021–1036. doi: 10.1016/0002-9343(78)90458-8. [DOI] [PubMed] [Google Scholar]
- 4.Kiryluk K, Jadoon A, Gupta M, Radhakrishnan J. Sickle cell trait and gross hematuria. Kidney Int. 2007;71:706–710. doi: 10.1038/sj.ki.5002060. [DOI] [PubMed] [Google Scholar]
- 5.Statius van Eps LW, Pinedo-Veels C, de Vries CH. Nature of concentrating defect in sickle cell nephropathy, microangiographic studies. Lancet. 1970;1:450–452. doi: 10.1016/s0140-6736(70)90836-6. [DOI] [PubMed] [Google Scholar]
- 6.Serjeant GR. Sickle Cell Disease. Oxford: Oxford University Press; 1992. pp. 261–281. [Google Scholar]
- 7.Falk RJ, Scheinman J, Phillips G. Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin-converting enzyme. N Engl J Med. 1992;326:910–915. doi: 10.1056/NEJM199204023261402. [DOI] [PubMed] [Google Scholar]
- 8.Bhathena DB, Sondheimer JH. The glomerulopathy of homozygous sickle hemoglobin (SS) disease: Morphology and pathogenesis. J Am Soc Nephrol. 1991;1:1241–1252. doi: 10.1681/ASN.V1111241. [DOI] [PubMed] [Google Scholar]
- 9.Maigne G, Ferlicot S, Galacteros F. Glomerular lesions in patients with sickle cell disease. Medicine (Baltimore) 2010;89:18–27. doi: 10.1097/MD.0b013e3181ca59b6. [DOI] [PubMed] [Google Scholar]
- 10.Nasr SH, Markowitz GS, Sentman RL, D'Agati VD. Sickle cell disease, nephrotic syndrome, and renal failure. Kidney Int. 2006;69:1276–1280. doi: 10.1038/sj.ki.5000234. [DOI] [PubMed] [Google Scholar]
- 11.De Paepe ME, Trudel M. The transgenic SAD mouse: A model of human sickle cell glomerulopathy. Kidney Int. 1994;46:1337–1345. doi: 10.1038/ki.1994.403. [DOI] [PubMed] [Google Scholar]
- 12.Paszty C, Brion CM, Manci E. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278:876–878. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 13.Diwan BA, Gladwin MT, Noguchi CT. Renal pathology in hemizygous sickle cell mice. Toxicol Pathol. 2002;30:254–262. doi: 10.1080/019262302753559597. [DOI] [PubMed] [Google Scholar]
- 14.Allon M, Lawson L, Eckman JR. Effects of nonsteroidal anti-inflammatory drugs on renal function in sickle cell anemia. Kidney Int. 1988;34:500–506. doi: 10.1038/ki.1988.209. [DOI] [PubMed] [Google Scholar]
- 15.Thompson J, Reid M, Hambleton I, Serjeant GR. Albuminuria and renal function in homozygous sickle cell disease: Observations from a cohort study. Arch Intern Med. 2007;167:701–708. doi: 10.1001/archinte.167.7.701. [DOI] [PubMed] [Google Scholar]
- 16.Haymann JP, Stankovic K, Levy P. Glomerular hyperfiltration in adult sickle cell anemia: A frequent hemolysis associated feature. Clin J Am Soc Nephrol. 2010;5:756–761. doi: 10.2215/CJN.08511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aygun B, Mortier NA, Smeltzer MP. Glomerular hyperfiltration and albuminuria in children with sickle cell anemia. Pediatr Nephrol. 2011;26:1285–1290. doi: 10.1007/s00467-011-1857-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King L, MooSang M, Miller M, Reid M. Prevalence and predictors of microalbuminuria in Jamaican children with sickle cell disease. Arch Dis Child. 2011;96:1135–1139. doi: 10.1136/archdischild-2011-300628. [DOI] [PubMed] [Google Scholar]
- 19.Hostetter TH, Olson JL, Rennke HG. Hyperfiltration in remnant nephrons: A potential adverse response to renal ablation. Am J Physiol. 1981;241:F85–F93. doi: 10.1152/ajprenal.1981.241.1.F85. [DOI] [PubMed] [Google Scholar]
- 20.Olson JL, Hostetter TH, Rennke HG. Altered glomerular permselectivity and progressive sclerosis following extreme ablation of renal mass. Kidney Int. 1982;22:112–126. doi: 10.1038/ki.1982.143. [DOI] [PubMed] [Google Scholar]
- 21.Nath KA, Grande JP, Haggard JJ. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am J Pathol. 2001;158:893–903. doi: 10.1016/S0002-9440(10)64037-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nath KA, Grande JP, Croatt AJ. Transgenic sickle mice are markedly sensitive to renal ischemia-reperfusion injury. Am J Pathol. 2005;166:963–972. doi: 10.1016/S0002-9440(10)62318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Juncos JP, Grande JP, Croatt AJ. Early and prominent alterations in hemodynamics, signaling, gene expression following renal ischemia in sickle cell disease. Am J Physiol. 2010;298:F892–F899. doi: 10.1152/ajprenal.00631.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy S, Konstantinidis DG, Rizvi T. Increased oxidative stress in sickle cell disease activates the renin-angiotensin-TGF-β pathway to mediate sickle nephropathy. Annual Meeting of the American Society of Hematology. 2013 et al.. Abstract # 2211. [Google Scholar]
- 25.Ataga KI, Brittain JE, Moore D. Urinary albumin excretion is associated with pulmonary hypertension in sickle cell disease: potential role of soluble fms-like tyrosis kinase-1. Eur J Haematol. 2010;85:257–263. doi: 10.1111/j.1600-0609.2010.01471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becton LJ, Kalpatthi RV, Rackoff E. Prevalence and clinical correlates of microalbuminuria in children with sickle cell disease. Pediatr Nephrol. 2010;25:1505–1511. doi: 10.1007/s00467-010-1536-8. [DOI] [PubMed] [Google Scholar]
- 27.Gurkan S, Scarponi KJ, Hotchkiss H. Lactate dehydrogenase as a predictor of kidney involvement in patients with sickle cell anemia. Pedriatr Nephrol. 2010;25:2123–2127. doi: 10.1007/s00467-010-1560-8. [DOI] [PubMed] [Google Scholar]
- 28.Maier-Redelsperger M, Levy P, Lionnet F. Strong association between a new marker of hemolysis and glomerulopathy in sickle cell anemia. Blood Cells Mol Dis. 2010;45:289–292. doi: 10.1016/j.bcmd.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Day TG, Drasar ER, Fulford T. Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica. 2012;97:201–205. doi: 10.3324/haematol.2011.050336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber ML, Vang D, Velho PE. Morphine promotes renal pathology in sickle mice. Int J Nephrol Renovasc Dis. 2012;5:109–118. doi: 10.2147/IJNRD.S33813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Jong PE, Statius van Epps LW. Sickle cell nephropathy: New insights into its pathophysiology. Kidney Int. 1985;27:711–717. doi: 10.1038/ki.1985.70. [DOI] [PubMed] [Google Scholar]
- 32.Scheinman JI. Sickle cell disease and the kidney. Nat Clin Prac. 2009;5:78–88. doi: 10.1038/ncpneph1008. [DOI] [PubMed] [Google Scholar]
- 33.Landburg PP, Elsenga H, Schnog JB. Increased serum levels of anti-angiogenic factors soluble fms-like tyrosine kinase and soluble endoglin in sickle cell disease. Acta Haematol. 2008;120:130–133. doi: 10.1159/000178143. [DOI] [PubMed] [Google Scholar]
- 34.Ataga KI, Brittain JE, Jones SK. Association of soluble fms-like tyrosine kinase-1 with pulmonary hypertension and haemolysis in sickle cell disease. Br J Haematol. 2011;152:485–491. doi: 10.1111/j.1365-2141.2010.08410.x. [DOI] [PubMed] [Google Scholar]
- 35.Fischer C, Mazzone M, Jonckx B, Carmeliet P. FLT1 and its ligand VEGFB and PlGF: Drug targets for anti-angiogenic therapy? Nat Rev Cancer. 2008;8:942–956. doi: 10.1038/nrc2524. [DOI] [PubMed] [Google Scholar]
- 36.Dimmeler S, Dernbach E, Zeiher AM. Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett. 2000;477:258–262. doi: 10.1016/s0014-5793(00)01657-4. [DOI] [PubMed] [Google Scholar]
- 37.Ahmed A. New insights into the etiology of preeclampsia: Identification of key elusive factors for the vascular complications. Thromb Res. 2011;127:S72–S75. doi: 10.1016/S0049-3848(11)70020-2. (Suppl 3): [DOI] [PubMed] [Google Scholar]
- 38.Guasch A, Navarete J, Nass K, Zayas CF. Glomerular involvement in adults with sickle cell hemoglobinopathies: Prevalence and clinical correlates of progressive renal failure. J Am Soc Nephrol. 2006;17:2228–2235. doi: 10.1681/ASN.2002010084. [DOI] [PubMed] [Google Scholar]
- 39.Asnani MR, Fraser RA, Reid ME. Higher rates of hemolysis are not associated with albuminuria in Jamaicans with sickle cell disease. PLoS One. 2011;6:e18863. doi: 10.1371/journal.pone.0018863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurbanov K, Rubinstein I, Hoffman A. Differential regulation of renal regional blood flow by endothelin-1. Am J Physiol. 1996;271:F1166–F1172. doi: 10.1152/ajprenal.1996.271.6.F1166. [DOI] [PubMed] [Google Scholar]
- 41.Heyman SN, Khamaisi M, Rosen S, Rosenberger C. Renal parenchymal hypoxia, hypoxia response and the progression of chronic kidney disease. Am J Nephrol. 2008;28:998–1006. doi: 10.1159/000146075. [DOI] [PubMed] [Google Scholar]
- 42.Baylis C. Nitric oxide deficiency in chronic kidney disease. Am J Physiol Renal Physiol. 2008;294:F1–F9. doi: 10.1152/ajprenal.00424.2007. [DOI] [PubMed] [Google Scholar]
- 43.Zatz R, Baylis C. Chronic nitric oxide inhibition model six years on. Hypertension. 1998;32:958–964. doi: 10.1161/01.hyp.32.6.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: Reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37–47. doi: 10.1016/j.blre.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lima CS, Bottini PV, Garlipp CR. Accuracy of the urinary albumin to creatinine ratio as a predictor of albuminuria in adults with sickle cell disease. J Clin Pathol. 2002;55:973–975. doi: 10.1136/jcp.55.12.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alvarez O, Montane B, Lopez G. Early blood transfusions protect against microalbuminuria in children with sickle cell disease. Pediatr Blood Cancer. 2006;47:71–76. doi: 10.1002/pbc.20645. [DOI] [PubMed] [Google Scholar]
- 47.Dharnidharka VR, Dabbagh S, Atiyeh B. Prevalence of microalbuminuria in children with sickle cell disease. Pediatr Nephrol. 1998;12:475–478. doi: 10.1007/s004670050491. [DOI] [PubMed] [Google Scholar]
- 48.Wigfall DR, Ware RE, Burchinal MR. Prevalence and clinical correlates of glomerulopathy in children with sickle cell disease. J Pediatr. 2000;136:749–753. [PubMed] [Google Scholar]
- 49.McBurney PG, Hanevold CD, Hernandez CM. Risk factors for microalbuminuria in children with sickle cell anemia. J Pediatr Hematol Oncol. 2002;24:473–477. doi: 10.1097/00043426-200208000-00013. [DOI] [PubMed] [Google Scholar]
- 50.McKie KT, Hanevold CD, Hernandez C. Prevalence, prevention, and treatment of microalbuminuria and proteinuria in children with sickle cell disease. J Pediatr Hematol Oncol. 2007;29:140–144. doi: 10.1097/MPH.0b013e3180335081. [DOI] [PubMed] [Google Scholar]
- 51.McPherson Yee M, Jabbar SF, Osunkwo I. Chronic kidney disease and albuminuria in children with sickle cell disease. Clin J Am Soc Nephrol. 2011;6:2628–2633. doi: 10.2215/CJN.01600211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iwalokun BA, Iwalokun SO, Hodonu SO. Evaluation of microalbuminuria in relation to asymptomatic bacteruria in Nigerian patients with sickle cell anemia. Saudi J Kidney Dis Transpl. 2012;23:1320–1330. doi: 10.4103/1319-2442.103589. [DOI] [PubMed] [Google Scholar]
- 53.Aleem A. Proteinuria in adult Saudi patients with sickle cell disease is not associated with identifiable risk factors. Saudi J Kidney Dis Transpl. 2010;21:903–908. [PubMed] [Google Scholar]
- 54.Laurin LP, Nachman PH, Desai PC. Hydroxyurea is associated with lower prevalence of albuminuria in adults with sickle cell disease. Nephrol Dial Transplant. 2013 doi: 10.1093/ndt/gft295. et al.. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Powars DR, Elliot-Mills DD, Chan L. Chronic renal failure in sickle cell disease: Risk factors, clinical course, and mortality. Ann Intern Med. 1991;115:614–616. doi: 10.7326/0003-4819-115-8-614. [DOI] [PubMed] [Google Scholar]
- 56.Alvarez O, Lopez-Mitnik G, Zilleruelo G. Short-term follow up of patients with sickle cell disease and albuminuria. Pediatr Blood Cancer. 2008;50:1236–1239. doi: 10.1002/pbc.21520. [DOI] [PubMed] [Google Scholar]
- 57.Gosmanova EO, Zaidi S, Wan JY, Adams-Graves PE. Prevalence and progression of chronic kidney disease in adult patients with sickle cell disease. J Investig Med. 2014 doi: 10.1097/01.JIM.0000446836.75352.72. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 58.Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int Suppl. 2013;3:1–150. [Google Scholar]
- 59.De Castro LM, Jonassaint JC, Graham FL. Pulmonary hypertension associated with sickle cell disease: clinical and laboratory endpoints and disease outcomes. Am J Hematol. 2008;83:19–25. doi: 10.1002/ajh.21058. [DOI] [PubMed] [Google Scholar]
- 60.Forrest S, Kim A, Carbonella J, Pashankar F. Proteinuria is associated with elevated tricuspid regurgitant jet velocity in children with sickle cell disease. Pediatr Blood Cancer. 2012;58:937–940. doi: 10.1002/pbc.23338. [DOI] [PubMed] [Google Scholar]
- 61.Bolarinwa RA, Akinlade KS, Kuti MA. Renal disease in adult Nigerians with sickle cell anemia: A report of prevalence, clinical features and risk factors. Saudi J Kidney Dis Transpl. 2012;23:171–175. [PubMed] [Google Scholar]
- 62.Elmariah H, Garrett ME, De Castro LM. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol. 2014;89:530–535. doi: 10.1002/ajh.23683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aoki RY, Saad ST. Enalapril reduces the albuminuria of patients with sickle cell disease. Am J Med. 1995;98:432–435. doi: 10.1016/S0002-9343(99)80341-6. [DOI] [PubMed] [Google Scholar]
- 64.Foucan L, Bourhis V, Bangou J. A randomized trial of captopril for microalbuminuria in normotensive adults with sickle cell anemia. Am J Med. 1998;104:339–342. doi: 10.1016/s0002-9343(98)00056-4. [DOI] [PubMed] [Google Scholar]
- 65.Fitzhugh CD, Wigfall DR, Ware RE. Enalapril and hydroxyurea therapy for children with sickle nephropathy. Pediatr Blood Cancer. 2005;45:982–985. doi: 10.1002/pbc.20296. [DOI] [PubMed] [Google Scholar]
- 66.Aygun B, Mortier NA, Smeltzer MP. Hydroxyurea treatment decreases glomerular hyperfiltration in children with sickle cell anemia. Am J Hematol. 2013;88:116–119. doi: 10.1002/ajh.23365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Jong PE, De Jong-Van Den Berg TW, Sewrajsingh GS. The influence of indomethacin on renal haemodynamics in sickle cell anaemia. Clin Sci. 1980;59:245–250. doi: 10.1042/cs0590245. [DOI] [PubMed] [Google Scholar]
- 68.Allon M, Lawson L, Eckman JR. Effects of nonsteroidal anti-inflammatory drugs on renal function in sickle cell anemia. Kidney Int. 1988;34:500–506. doi: 10.1038/ki.1988.209. [DOI] [PubMed] [Google Scholar]
- 69.Wong W, Elliot-Mills D, Powars D. Renal failure in sickle cell anemia. Hematol Oncol Clin North Am. 1996;10:1321–1331. doi: 10.1016/s0889-8588(05)70403-2. [DOI] [PubMed] [Google Scholar]
- 70.Rodgers GP, Walker EC, Podgor MJ. Is “relative” hypertension a risk factor for vaso-occlusive complications in sickle cell disease? Am J Med Sci. 1993;305:150–156. doi: 10.1097/00000441-199303000-00004. [DOI] [PubMed] [Google Scholar]
- 71.Gordeuk VR, Sachdev V, Taylor JG. Relative systemic hypertension in patients with sickle cell disease is associated with risk of pulmonary hypertension and renal insufficiency. Am J Hematol. 2008;83:15–18. doi: 10.1002/ajh.21016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lemonne N, Romana M, Lamarre Y. Association between relative systemic hypertension and otologic disorders in patients with sickle cell-hemoglobin C disorder. Am J Hematol. 2014;89:667. doi: 10.1002/ajh.23717. et al.. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 73.Lamarre Y, Lalanne-Mistrih ML, Romana M. Male gender, increased blood viscosity, body mass index and triglyceride levels are independently associated with systemic relative hypertension in sickle cell anemia. PLoS One. 2013;8:e66004. doi: 10.1371/journal.pone.0066004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McClellan AC, Luthi JC, Lynch JR. High one year mortality in adults with sickle cell disease and end-stage renal disease. Br J Haematol. 2012;159:360–367. doi: 10.1111/bjh.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nissenson AR, Port FK. Outcome of end-stage renal disease in patients with rare causes of renal failure. I. Inherited and metabolic disorders. Q J Med. 1989;73:1055–1062. [PubMed] [Google Scholar]
- 76.Chatterjee SN. National study in natural history of renal allografts in sickle cell disease or trait: A second report. Transplant Proc. 1987;19:33–35. [PubMed] [Google Scholar]
- 77.Ojo AO, Govaerts TC, Schmouder RL. Renal transplantation in end-stage sickle cell nephropathy. Transplantation. 1999;67:291–295. doi: 10.1097/00007890-199901270-00018. [DOI] [PubMed] [Google Scholar]
- 78.Bleyer AJ, Donaldson LA, McIntosh M. Relationship between underlying renal disease and renal transplantation outcome. Am J Kidney Dis. 2001;37:1152–1161. doi: 10.1053/ajkd.2001.24516. [DOI] [PubMed] [Google Scholar]
- 79.Nair R, Walker PD. Is IgA nephropathy the commonest primary glomerulopathy among young adults in the USA? Kidney Int. 2006;69:1455–1458. doi: 10.1038/sj.ki.5000292. [DOI] [PubMed] [Google Scholar]
- 80.Pham PT, Pham PC. The impact of mycophenolate mofetil versus azathioprine as adjunctive therapy to cyclosporine on the rates of renal allograft loss due to glomerular disease recurrence. Nephrol Dial Transplant. 2012;27:2965–2971. doi: 10.1093/ndt/gfr731. [DOI] [PubMed] [Google Scholar]
- 81.Huang E, Parke C, Mehrnia A. Improved survival among sickle cell kidney transplant recipients in the recent era. Nephrol Dial Transplant. 2013;28:1039–1046. doi: 10.1093/ndt/gfs585. [DOI] [PubMed] [Google Scholar]
- 82.Spector D, Zachary JB, Sterioff S, Millan J. Painful crises following renal transplantation in sickle cell anemia. Am J Med. 1978;64:835–839. doi: 10.1016/0002-9343(78)90524-7. [DOI] [PubMed] [Google Scholar]
- 83.Donnelly PK, Edmunds ME, O'Reilly K. Renal transplantation in sickle cell disease. Lancet. 1988;2:229. doi: 10.1016/s0140-6736(88)92344-6. [DOI] [PubMed] [Google Scholar]
- 84.Miner DJ, Jorkasky DK, Perloff LJ. Recurrent sickle cell nephropathy in a transplanted kidney. Am J Kidney Dis. 1987;10:306–313. doi: 10.1016/s0272-6386(87)80027-6. [DOI] [PubMed] [Google Scholar]