

Abstract
Rhabdomyosarcoma (RMS) is an aggressive childhood malignancy of neoplastic muscle-lineage precursors that fail to terminally differentiate into syncytial muscle. The most aggressive form of RMS, alveolar-RMS, is driven by misexpression of the PAX-FOXO1 oncoprotein, which is generated by recurrent chromosomal translocations that fuse either the PAX3 or PAX7 gene to FOXO1. The molecular underpinnings of PAX-FOXO1−mediated RMS pathogenesis remain unclear, however, and clinical outcomes poor. Here, we report a new approach to dissect RMS, exploiting a highly efficient Drosophila PAX7-FOXO1 model uniquely configured to uncover PAX-FOXO1 RMS genetic effectors in only one generation. With this system, we have performed a comprehensive deletion screen against the Drosophila autosomes and demonstrate that mutation of Mef2, a myogenesis lynchpin in both flies and mammals, dominantly suppresses PAX7-FOXO1 pathogenicity and acts as a PAX7-FOXO1 gene target. Additionally, we reveal that mutation of mastermind, a gene encoding a MEF2 transcriptional coactivator, similarly suppresses PAX7-FOXO1, further pointing toward MEF2 transcriptional activity as a PAX-FOXO1 underpinning. These studies show the utility of the PAX-FOXO1 Drosophila system as a robust one-generation (F1) RMS gene discovery platform and demonstrate how Drosophila transgenic conditional expression models can be configured for the rapid dissection of human disease.
Keywords: rhabdomyosarcoma, PAX7-FOXO1, PAX3-FOXO1, sarcoma, myogenesis
Childhood cancer differs biologically from adult neoplasia: whereas most solid adult tumors are epithelial carcinomas, solid childhood malignancies often are mesenchymal sarcomas. Soft-tissue sarcomas account for 10% of all childhood malignancies, 50% of which are skeletal muscle-lineage rhabdomyosarcomas (RMS) (Gurney et al. 1999; Scheurer et al. 2011). Despite aggressive therapies, children with high-risk RMS suffer from a 3-yr event-free survival of 20%. Treatments for high-risk RMS have not improved for three decades, underscoring the need to elucidate the molecular underpinnings of the disease.
RMS is comprised of neoplastic myoblasts that fail to exit the cell cycle and are blocked from terminally differentiating into syncytial muscle. RMS typically is divided into two clinically distinct subgroups (Huh and Skapek 2010; Wexler et al. 2011): embryonal RMS and alveolar RMS (A-RMS). Embryonal RMS is a genetically heterogeneous subtype, whereas A-RMS, which is notoriously more aggressive, is uniquely driven by the PAX-FOXO1 fusion oncoprotein.
The PAX-FOXO1 transcription factor is generated by chromosomal translocations that fuse a PAX3/7 gene (PAX3 on chromosome 2 or PAX7 on chromosome 1) to the 3′ end of the FOXO1 locus on chromosome 13 (Galili et al. 1993; Shapiro et al. 1993; Davis et al. 1994). The encoded chimera contains intact PAX3/7 DNA-binding domains fused to the FOXO1 transcriptional activation domain (Mahajan et al. 2014). Because PAX3/7 encode genetic regulators of skeletal muscle development (Lagha et al. 2008; Buckingham and Rigby 2014), it is postulated that genes regulated by PAX3/7 underlie A-RMS pathogenesis. Despite notable advances with mammalian PAX-FOXO1 RMS models (Keller et al. 2004; Naini et al. 2008; Nishijo et al. 2009; Cao et al. 2010), our understanding of RMS pathogenesis remains opaque, indicating the need for new genetic tools to dissect RMS pathobiology and uncover new molecular therapeutic targets.
As Drosophila models successfully yield critical insights into human disease, including cancer pathobiology (Gonzalez 2013), we have generated a Drosophila model to interrogate in vivo PAX-FOXO1 pathogenicity. Expression of human PAX-FOXO1 in differentiating fly muscle causes myoblast fusion defects that result in larval lethality (Galindo et al. 2006). Although tumorigenesis is not observed, in part due to quick lethality, PAX-FOXO1 cells act aggressively and infiltrate nonmuscle tissues. Although PAX7-FOXO1 RMS is less common and demonstrates better clinical outcomes than PAX3-FOXO1, PAX7-FOXO1 phenotypes exhibit better penetrance in flies due to slightly greater sequence identity between human PAX7 and Drosophila PAX3/7. Because expression of wild-type human PAX3 in flies phenocopies PAX-FOXO1, PAX3-FOXO1, PAX7-FOXO1, and wild-type PAX3/7 activity presumably overlap in vivo (Galindo et al. 2006).
PAX-FOXO1 phenotypes are susceptible to dominant genetic suppression and enhancement (Galindo et al. 2006; Avirneni-Vadlamudi et al. 2012; Crose et al. 2014). Thus, we have been exploiting this genetically tractable model to uncover new PAX-FOXO1 gene targets and cofactors. We have subsequently shown that genetic modifiers isolated from the Drosophila PAX7-FOXO1 system impact RMS oncogenesis and tumorigenesis (Avirneni-Vadlamudi et al. 2012; Crose et al. 2014). These findings establish that insights gleaned from this invertebrate model successfully uncover new RMS mechanisms, and new molecular targets for RMS therapy.
Here, we report a comprehensive deletion screen against the Drosophila autosomes to identify PAX-FOXO1 gene targets and effectors, as well as the methods used to configure the screen such that PAX-FOXO1 modifiers are quickly identifiable with only one genetic cross. We additionally report that mutation of Drosophila Myocyte Enhancer Factor-2 (D-Mef2), a critical regulator of both fly and mammalian myogenesis, dominantly suppresses PAX7-FOXO1 lethality and acts as a PAX-FOXO1 gene target. We further find that mutation of mastermind (mam), a gene encoding a MEF2 transcriptional coactivator, similarly suppresses PAX7-FOXO1, further pointing toward MEF2 transcriptional activity as a mediator of PAX-FOXO1 pathogenicity. These studies show the utility of the PAX7-FOXO1 Drosophila system as a robust one-generation (F1) RMS gene discovery platform and demonstrate how Drosophila models can be configured for rapid and effective dissection of human disease.
Materials and Methods
Genetics
In the screen for PAX7-FOXO1 genetic modifiers, the UAS-PAX7-FOXO1 and Myosin Heavy Chain-Gal4 transgenes were used, and lethality assessed, as previously described (Avirneni-Vadlamudi et al. 2012). The Gal80-containing X-chromosome is from stock #5132 from the Bloomington Drosophila Stock Center. For each experimental cross, approximately three males from the master screening stock were mated to approximately five to seven wild-type, deficiency-, or gene mutation-containing females, and at least two independent crosses performed. Crosses were reared at 23°. Multiple crosses of the master screening stock to the wild-type line w1118 were performed to generate large populations of F1 PAX7-FOXO1 male and control female siblings, from which we established a baseline percentage (22%) (SEM = 1.0%) of F1 PAX7-FOXO1 males expected upon routine outcrossing of the screening stock (Supporting Information, Table S1). Each time screening crosses were performed, we included new w1118 control crosses to insure that PAX7-FOXO1−induced semi-lethality of F1 males did not significantly differ from the established baseline. All deficiency- and mutation-containing stocks were obtained from the Bloomington Drosophila Stock Center.
The Drosophila PAX7-FOXO1 microarray raw data sets have been previously described and are publically available (Avirneni-Vadlamudi et al. 2012).
Embryo immunofluorescence
For Drosophila embryo whole-mount immunofluorescence, embryos were treated as described previously (Chen and Olson 2001), incubated in primary antibody overnight at 4° [1:1000 rabbit anti-green fluorescent protein (GFP); Molecular Probes], secondary at room temperature for 2 hr. (1:2000, Alexa-568 goat anti-rabbit; Invitrogen), and mounted in VECTASHIELD with 4′,6-diamidino-2-phenylindole. Microscopy was performed with either an LSM150-meta confocal or Zeiss Axioplan2 fluorescent microscope.
Statistics and study approval
For the genetic screen, suppressors and enhancers were identified by crosses that showed a percent-F1 male population 1 SD above or below the mean, respectively. Fold change is the % F1 males observed for each line tested divided by baseline (22%). Unpaired 2-tailed Student’s t-tests were used to calculate significance. A P value < 0.05 was considered significant.
For the microarray studies, Data represent mean ± SEM. Significance of differences was determined by unpaired 2-tailed Student’s t-test. A P value < 0.05 was considered significant. These studies did not include human tissue, and were exempt from institutional review board approval.
Results
PAX7-FOXO1 drives ectopic myogenesis in Drosophila embryos
Because PAX molecules and myogenesis show striking evolutionary conservation between Drosophila and vertebrates (Halder et al. 1995; Xue and Noll 1996; Xue et al. 2001; Daczewska et al. 2010), we generated a genetically simple and efficient Drosophila PAX7-FOXO1 transgenic platform to dissect PAX-FOXO1 pathobiology. Our approach is based on the Gal4/UAS bipartite expression system (Brand and Perrimon 1993), where transgenic human PAX7-FOXO1 is expressed from the yeast UAS enhancer/promoter by driver lines that express the Gal4 transcriptional activator in tissue-specific patterns.
Toward validating the new fly PAX-FOXO1 system, we tested whether human PAX7-FOXO1 promotes myogenesis in Drosophila. We used the daughterless-Gal4 driver, which directs ubiquitous expression of UAS-transgenes, to express PAX7-FOXO1 during embryogenesis. We then probed for expression of a GFP-tagged Myosin Heavy Chain (MHC) reporter transgene, a marker specific for myogenesis and a reporter previously used in embryonic screens to successfully identify genes involved in Drosophila somatic muscle development and patterning (Chen and Olson 2001; Chen et al. 2003). Drosophila embryos initiate native expression of MHC at embryonic stage 13—thus, we focused on embryos at stage 12 or younger for ectopic MHC expression. We observed robust expression of MHC-GFP in cells of all three germ layers, including nonmyogenic cells within the ectoderm and endoderm primordia (Figure 1), findings similar to PAX3-FOXO1 misexpression in mouse embryonic primordial cells (Scuoppo et al. 2007). These results [as well as similar results described below (MEF2 as a PAX-FOXO gene target and putative RMS effector) (Figure 4C)] show that Drosophila precursors are vulnerable to the myogenic programming properties intrinsic to the PAX-FOXO1 chimera.
Figure 1.

PAX7-FOXO1 drives myogenesis in Drosophila embryos. (A) Whole-mount wild-type and daughterless-Gal4;UAS-PAX7-FOXO1 (da>>PAX7-FOXO1) gastrulated embryos probed for expression of green fluorescent protein (GFP) from a Myosin Heavy Chain (MHC)-GFP reporter transgene. Because Drosophila embryos initiate native expression of MHC at embryonic stage 13, we focused on embryos at stage 12 or younger. Diffuse expression of MHC-GFP is only detected in the da>>PAX7-FOXO1 embryos. (B) Greater resolution images of embryo segments noted by the white bars in (A) MHC-GFP = GFP immunofluorescence from the MHC-GFP reporter; DAPI = 4′,6-diamidino-2-phenylindole nuclear staining.
Figure 4.

Isolation of the myogenesis benchmark gene D-Mef2 as a PAX7-FOXO1 suppressor and gene target. (A) Smaller, overlapping chromosomal deletions reduce the PAX7-FOXO1 deletion suppressor Df(2R)X1 to chromosomal segments 46C1-46C7, which includes D-Mef2, the master regulator of Drosophila myogenesis. (B) D-Mef2 loss-of-function mutation dominantly suppresses PAX7-FOXO1 lethality. PAX7-FOXO1-expression is semilethal. In the presence of Df(2R)X1, which deletes D-Mef2, the population of PAX7-FOXO1−positive adults is increased 2.4-fold and is a PAX7-FOXO1 suppressor. Two smaller overlapping deletions, Df(2R)BSC152 and Df(2R)BSC298, also delete D-Mef2 and suppress PAX7-FOXO1, whereas Df(2R)eve neither deletes D-Mef2 nor acts as a PAX7-FOXO1 suppressor. The D-Mef222-21 null allele (n = 193 F1 adults scored) is a strong suppressor of PAX7-FOXO1 lethality (P = 0.0018), confirming that D-Mef2 genetically interacts with PAX7-FOXO1. Of note—although the Df(2R)BSC298 deletion showed a fold change of slightly less than 1.9, the increase in PAX7-FOXO1 adults (1.8-fold) was highly significant (P = 0.0004), and in this test we considered a suppressor. (C) PAX7-FOXO1 drives D-Mef2 expression. Whole-mount wild-type and daughterless-Gal4;UAS-PAX7-FOXO1 (da>>PAX7-FOXO1) gastrulated embryos (dorsal surface upper right corner, posterior surface, lower right corner) probed for expression of yellow fluorescent protein (YFP) from a D-Mef2-YFP embryonic reporter transgene. In wild-type embryos, D-Mef2 expression is limited to differentiating myoblasts within the mesoderm. In da>>PAX7-FOXO1 embryos, D-Mef2-YFP reporter expression is seen throughout the embryo, including ectodermal and endodermal derivatives. D-Mef2 is also detectably overexpressed in myoblasts, visible in a segmentally repeating pattern. The black lines note the posterior aspect of both embryos shown in the right-most, greater resolution images. Mef2-YFP = YFP immunofluorescence from the D-Mef2-YFP reporter; DAPI = 4′,6-diamidino-2-phenylindole nuclear staining. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
A rapid, one-generation screen for PAX7-FOXO1 suppressors and enhancers
We next configured the Drosophila PAX-FOXO1 platform for unbiased forward genetic screening and RMS gene discovery. For these studies, we turned to an MHC>>PAX7-FOXO1 (MHC-Gal;UAS-PAX7-FOXO1) genetic background to hone in on PAX7-FOXO1 pathogenicity in differentiating Drosophila muscle lineage cells—a setting similar to a conditional PAX3-FOXO1 tumorigenic mouse model (Keller et al. 2004).
Because MHC>>PAX7-FOXO1 expression is lethal, a typical forward genetic screen would normally require a multigenerational scheme to bring the MHC-Gal4 driver, UAS-PAX7-FOXO1 transgene, and candidate modifiers into the same genetic background, a cumbersome and lengthy process that would need repeating for every candidate mutation-containing chromosome to be tested. To bypass this issue, we configured a stable “master” stock that would allow for candidate modifiers to be efficiently tested with a simple, one-generation (F1) scheme (Figure 2). To generate this master stock, we incorporated a transgenic X-chromosome that ubiquitously expresses the potent Gal4 physical inhibitor, Gal80. Because Gal80 antagonizes MHC>>Gal4, homozygous MHC-Gal4;UAS-PAX7-FOXO1 animals are viable and stable. Upon outcrossing of this master stock, F1 female progeny inherit the Gal4-inactivating Gal80 X-chromosome (which serve as the control cohort), whereas all F1 male siblings express PAX7-FOXO1. Additionally, we exploited that fact that Gal4 activity is partially temperature dependent to identify a rearing temperature (23°) at which PAX7-FOXO1 phenotypes are semilethal.
Figure 2.
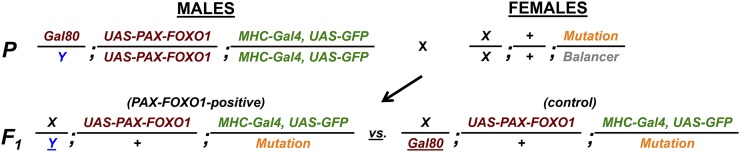
A rapid unbiased one-generation (F1) screen to uncover dominant PAX7-FOXO1 genetic modifiers. Incorporating an X-linked Gal80 transgenic chromosome allows for the MHC-Gal4;UAS-PAX7-FOXO1 screen to be performed in a single generation. Using the Gal4 inhibitor, Gal80 (carried on the X-chromosome), a viable stable stock was generated that is homozygous for UAS-PAX7-FOXO1 on the second chromosome and Myosin Heavy Chain (MHC)-Gal4 on the third chromosome, which also contains a UAS-GFP transgenic reporter. With this stock, it is possible to screen against any mutant chromosome in one generation, where the number of PAX7-FOXO1-expressing F1 males are compared to control (Gal80-positive) female siblings. Without genetic modification, PAX7-FOXO1 expression is semilethal. Genetic suppressors rescue semilethality and thus increase the number of males in the F1 population, whereas enhancers decrease the percentage of F1 males (Table 1). In the scheme shown, the mutation tested is on the third chromosome, though an equivalent scheme is used for second chromosome mutations.
Outcrossing of the screening stock to wild-type flies reared at 23° results in the PAX7-FOXO1-positive male cohort to comprise on average 22% of the F1 population (Table S1), significantly reduced from the rate of 50% that would otherwise be expected based on Mendelian ratios. When screening against this phenotype, PAX7-FOXO1 suppressors and enhancers are easily identified: suppressors and enhancers increase and decrease, respectively, the F1 percentage of males 1 SD from the mean. When compared with baseline and calculated as fold change, suppressors and enhancer show a ≥1.9-fold and ≤0.5-fold change in F1 male numbers, respectively (Table 1). A Student’s t-test is then used to confirm statistical significance for each modifier.
Table 1. Deficiency enhancers, suppressors, and nonmodifiers of PAX7-FOXO1.
| Genotype | Breakpoints | Males (P-F) | Females (Control) | Total F1 Adults | % F1 Males | Fold Change | P value | Submapped |
|---|---|---|---|---|---|---|---|---|
| Df(3L)emc-E12 | 61A;61D3 | 0 | 115 | 115 | 0% | 0.00 | 0 | No |
| Df(3L)ZN47 | 64C;65C | 0 | 53 | 53 | 0% | 0.00 | 0 | No |
| Df(3L)W10 | 75A6-7;75C1-2 | 0 | 217 | 217 | 0% | 0.00 | 0 | No |
| Df(3L)fz2 | 75F10-11;76A1-5 | 0 | 77 | 77 | 0% | 0.00 | 0 | No |
| Df(3R)crb-F89-4 | 95D7-D11;95F15 | 0 | 225 | 225 | 0% | 0.00 | 0 | No |
| Df(3R)crb87-5 | 95F7;96A17-18 | 0 | 194 | 194 | 0% | 0.00 | 0 | No |
| Df(2L)TW161 | 38A6-B1;40A4-B1 | 1 | 112 | 113 | 1% | 0.04 | 0 | No |
| Df(2R)AA21 | 57B19-C1;57E1-6 | 1 | 75 | 76 | 1% | 0.06 | 0 | Yes |
| Df(3R)p-XT103 | 85A2;85C1-2 | 3 | 189 | 192 | 2% | 0.07 | < 0.0001 | Yes |
| Df(3L)fz-GF3b | 70C2;70D4-5 | 1 | 50 | 51 | 2% | 0.09 | 0 | Yes |
| Df(2R)CX1 | 49C1-4;50C23-D2 | 2 | 88 | 90 | 2% | 0.10 | 0 | Yes |
| Df(2R)M60E | 60E6;60E11 | 5 | 140 | 145 | 3% | 0.16 | 0.02 | No |
| Df(3L)GN24 | 63F6-7;64C13-15 | 4 | 97 | 101 | 4% | 0.18 | < 0.0001 | No |
| Df(3R)23D1 | 94A3-4;94D1-4 | 3 | 71 | 74 | 4% | 0.18 | 0 | No |
| Df(2R)vg-C | 49B2;49E2 | 6 | 128 | 134 | 4% | 0.20 | 0 | No |
| Df(3R)e-R1 | 93B6-7;93D4 | 5 | 98 | 103 | 5% | 0.22 | 0.01 | No |
| Df(2R)Egfr5 | 57D2-8;58D1 | 8 | 140 | 148 | 5% | 0.25 | 0 | Yes |
| Df(3R)ea | 88E7-13;89A1 | 13 | 175 | 188 | 7% | 0.31 | 0.01 | No |
| Df(2R)BSC40 | 48E1-2;48E2-10 | 5 | 66 | 71 | 7% | 0.32 | 0.2390 | No |
| Df(2R)BSC161 | 54B2;54B17 | 5 | 66 | 71 | 7% | 0.32 | 0.07 | No |
| Df(2L)BSC32 | 32A1-2;32C5-D1 | 7 | 89 | 96 | 7% | 0.33 | 0 | No |
| Df(2L)TE35BC-24 | 35B4-6;35F1-7 | 9 | 114 | 123 | 7% | 0.33 | 0 | Yes |
| Df(2R)ED4065 | 60C8;60E8 | 30 | 332 | 362 | 8% | 0.38 | 0.01 | No |
| Df(2L)ast2 | 21E2;22B2-3 | 12 | 128 | 140 | 9% | 0.39 | 0.1480 | Yes |
| Df(2R)k10408 | 54B16,54B16 | 20 | 180 | 200 | 10% | 0.45 | 0 | No |
| Df(2R)BSC49 | 53D9-E1;54B5-10 | 12 | 112 | 124 | 10% | 0.45 | 0.01 | Yes |
| Df(3R)by10 | 85D8-12;85E7-F1 | 19 | 170 | 189 | 10% | 0.46 | 0 | Yes |
| Df(3R)D605 | 97E2;98A5 | 18 | 152 | 170 | 11% | 0.50 | 0.01 | No |
| Df(2R)M41A4 | 41A;41A | 22 | 167 | 189 | 12% | 0.53 | − | − |
| Df(2R)Kr10 | 60F1;60F5 | 11 | 80 | 91 | 12% | 0.55 | − | − |
| Df(3R)Exel6144 | 83A6;83B6 | 19 | 136 | 155 | 12% | 0.56 | − | − |
| Df(2L)drm-P2 | 23F3-4;24A1-2 | 16 | 113 | 129 | 12% | 0.56 | − | − |
| Df(3L)66C-G28 | 66B8-9;66C9-10 | 19 | 129 | 148 | 13% | 0.58 | − | − |
| Df(2L)BSC30 | 34A3;34B7-9 | 19 | 129 | 148 | 13% | 0.58 | − | − |
| Df(3L)GN34 | 63E6-9;64A8-9 | 42 | 276 | 318 | 13% | 0.60 | − | − |
| Df(3R)Antp17 | 84A5;84D9 | 18 | 106 | 124 | 15% | 0.66 | − | − |
| Df(3R)e1025-14 | 82F8-10;83A1-3 | 34 | 196 | 230 | 15% | 0.67 | − | − |
| Df(3L)Aprt-1 | 62A10-B1;62D2-5 | 14 | 80 | 94 | 15% | 0.68 | − | − |
| Df(3R)BSC140 | 96F1;96F10 | 17 | 93 | 110 | 15% | 0.70 | − | − |
| Df(3L)ri-79c | 77B-C;77F-78A | 35 | 190 | 225 | 16% | 0.71 | − | − |
| Df(2R)or-BR6 | 59B;59D8-E1 | 18 | 94 | 112 | 16% | 0.73 | − | − |
| Df(2R)H3E1 | 44D1-4;44F12 | 38 | 196 | 234 | 16% | 0.74 | − | − |
| Df(2R)BSC19 | 56F12-14;57A4 | 14 | 71 | 85 | 16% | 0.75 | − | − |
| Df(3R)L127 | 99B5-6;99F1 | 51 | 254 | 305 | 17% | 0.76 | − | − |
| Df(3R)Exel6197 | 95D8;95E5 | 22 | 109 | 131 | 17% | 0.76 | − | − |
| Df(3R)B81 | 99D3;3Rt | 41 | 197 | 238 | 17% | 0.78 | − | − |
| Df(2R)Exel7131 | 50E4;50F6 | 29 | 136 | 165 | 18% | 0.80 | − | − |
| Df(3R)Exel6202 | 96C9;96E2 | 32 | 150 | 182 | 18% | 0.80 | − | − |
| Df(3L)ME107 | 77F3;78C8-9 | 83 | 387 | 470 | 18% | 0.80 | − | − |
| Df(3L)BSC14 | 67E3-7;68A2-6 | 21 | 96 | 117 | 18% | 0.82 | − | − |
| Df(2R)en30 | 48A3-4;48C6-8 | 22 | 97 | 119 | 18% | 0.84 | − | − |
| Df(3R)Espl3 | 96F1;97B1 | 28 | 123 | 151 | 19% | 0.84 | − | − |
| Df(3R)Scr | 84A1-2;84B1-2 | 26 | 114 | 140 | 19% | 0.84 | − | − |
| Df(3R)BSC47 | 83B7-C1;83C6-D1 | 32 | 140 | 172 | 19% | 0.85 | − | − |
| Df(2R)Jp1 | 51D3-8;52F5-9 | 27 | 118 | 145 | 19% | 0.85 | − | − |
| Df(3L)ED4978 | 78D5;79A2 | 93 | 390 | 483 | 19% | 0.88 | − | − |
| Df(2R)nap9 | 42A1-2;42E6-F1 | 28 | 116 | 144 | 19% | 0.88 | − | − |
| Df(3R)WIN11 | 83E1-2;84A5 | 19 | 78 | 97 | 20% | 0.89 | − | − |
| Df(2L)BSC41 | 28A4-B1;28D3-9 | 33 | 129 | 162 | 20% | 0.93 | − | − |
| Df(3L)brm11 | 72A3;72D5 | 72 | 278 | 350 | 21% | 0.94 | − | − |
| Df(3R)IR16 | 97F1-2;98A | 46 | 176 | 222 | 21% | 0.94 | − | − |
| Df(2L)cl-h3 | 25D2-4;26B2-5 | 38 | 145 | 183 | 21% | 0.94 | − | − |
| Df(3L)Exel6087 | 62A2;62A7 | 103 | 389 | 492 | 21% | 1.0 | − | − |
| Df(2L)ed1 | 24A2;24D4 | 34 | 126 | 160 | 21% | 1.0 | − | − |
| Df(2R)robl-c | 54B17-C4;54C1-4 | 34 | 126 | 160 | 21% | 1.0 | − | − |
| Df(3L)ri-XT1 | 77E2-4;78A2-4 | 44 | 163 | 207 | 21% | 1.0 | − | − |
| w1118 (control, No Df) | N/A | 314 | 1123 | 1437 | 22% | 1.0 | − | − |
| Df(3L)81k19 | 73A3;74F | 39 | 134 | 173 | 23% | 1.0 | − | − |
| Df(3R)Exel6203 | 96E2;96E6 | 37 | 160 | 164 | 23% | 1.0 | − | − |
| Df(3R)BSC137 | 95A2-4;95A8-B1 | 43 | 141 | 184 | 23% | 1.1 | − | − |
| Df(3R)Exel9012 | 94E9;94E13 | 37 | 118 | 155 | 24% | 1.1 | − | − |
| Df(2L)XE-3801 | 27E2;28D1 | 14 | 44 | 58 | 24% | 1.1 | − | − |
| Df(3R)Exel6196 | 95C12;95D8 | 52 | 155 | 207 | 25% | 1.1 | − | − |
| Df(2L)TE29Aa-11 | 28E4-7;29B2-C1 | 39 | 113 | 152 | 26% | 1.2 | − | − |
| Df(2L)FCK-20 | 32D1;32F1-3 | 32 | 91 | 123 | 26% | 1.2 | − | − |
| Df(2R)BSC44 | 54B1-2;54B7-10 | 47 | 133 | 180 | 26% | 1.2 | − | − |
| Df(2R)Px2 | 60C5-6;60D9-10 | 39 | 109 | 148 | 26% | 1.2 | − | − |
| Df(2L)ED611 | 29B4;29C3 | 45 | 125 | 170 | 26% | 1.2 | − | − |
| Df(3L)ZP1 | 66A17-20;66C1-5 | 23 | 62 | 85 | 27% | 1.2 | − | − |
| Df(3L)vin7 | 68C8-11;69B4-5 | 47 | 126 | 173 | 27% | 1.2 | − | − |
| Df(3R)M-Kx1 | 86C1;87B1-5 | 38 | 99 | 137 | 28% | 1.3 | − | − |
| Df(3L)rdgC-co2 | 77A1;77D1 | 58 | 150 | 208 | 28% | 1.3 | − | − |
| Df(3R)3450 | 98E3;99A6-8 | 140 | 358 | 498 | 28% | 1.3 | − | − |
| Df(3L)ED4782 | 75F2;76A1 | 31 | 79 | 110 | 28% | 1.3 | − | − |
| Df(2L)Prl | 32F1-3;33F1-2 | 35 | 88 | 123 | 28% | 1.3 | − | − |
| Df(3L)eygC1 | 69A4-5;69D4-6 | 43 | 107 | 150 | 29% | 1.3 | − | − |
| Df(2R)X58-12 | 58D1-2;59A | 40 | 92 | 132 | 30% | 1.4 | − | − |
| Df(2L)b87e25 | 34B12-C1;35B10-C1 | 43 | 96 | 139 | 31% | 1.4 | − | − |
| Df(2L)dp-79b | 22A2-3;22D5-E1 | 48 | 106 | 154 | 31% | 1.4 | − | − |
| Df(2L)BSC36 | 32D1;32D4-E1 | 50 | 106 | 156 | 32% | 1.5 | − | − |
| Df(3R)ED5177 | 83B4;83B6 | 50 | 104 | 154 | 32% | 1.5 | − | − |
| Df(2R)PC4 | 55A;55F | 71 | 147 | 218 | 33% | 1.5 | − | − |
| Df(3R)Exel9014 | 95B1;95D1 | 53 | 109 | 162 | 33% | 1.5 | − | − |
| Df(3L)BSC12 | 69F6-70A1;70A1-2 | 36 | 74 | 110 | 33% | 1.5 | − | − |
| Df(2R)P34 | 55E2-4;56C1-11 | 53 | 108 | 161 | 33% | 1.5 | − | − |
| Df(2L)BSC31 | 23E5;23F4-5 | 68 | 138 | 206 | 33% | 1.5 | − | − |
| Df(2R)BSC132 | 45F6;46B12 | 43 | 86 | 129 | 33% | 1.5 | − | − |
| Df(3R)Tl-P | 97A;98A1-2 | 90 | 176 | 266 | 34% | 1.5 | − | − |
| Df(3L)pbl-X1 | 65F3;66B10 | 41 | 79 | 120 | 34% | 1.6 | − | − |
| Df(2R)BSC26 | 56C4;56D6-10 | 48 | 88 | 136 | 35% | 1.6 | − | − |
| Df(3L)st-f13 | 72C1-D1;73A3-4 | 58 | 106 | 164 | 35% | 1.6 | − | − |
| Df(2L)BSC4 | 21B7-C1;21C2-3 | 81 | 142 | 223 | 36% | 1.7 | − | − |
| Df(3L)R-G7 | 62B4-7;62D5-E5 | 51 | 89 | 140 | 36% | 1.7 | − | − |
| Df(2R)B5 | 46A;46C | 50 | 86 | 136 | 37% | 1.7 | − | − |
| Df(2R)14H10Y-53 | 54D1-2;54E5-7 | 58 | 99 | 157 | 37% | 1.7 | − | − |
| Df(2R)w45-30n | 45A6-7;45E2-3 | 83 | 140 | 223 | 37% | 1.7 | − | − |
| Df(2R)Exel7162 | 56F11;56F16 | 56 | 94 | 150 | 37% | 1.7 | − | − |
| Df(2L)BSC111 | 28F5;29B1 | 58 | 97 | 155 | 37% | 1.7 | − | − |
| Df(2R)ST1 | 42B3-5;43E15-18 | 56 | 91 | 147 | 38% | 1.7 | − | − |
| Df(2L)pr-A16 | 37B2-12;38D2-5 | 47 | 75 | 122 | 39% | 1.8 | − | − |
| Df(3L)XS533 | 76B4;77B | 42 | 67 | 109 | 39% | 1.8 | − | − |
| Df(3L)BSC8 | 74D3-75A1;75B2-5 | 53 | 84 | 137 | 39% | 1.8 | − | − |
| Df(3L)vin5 | 68A2-3;69A1-3 | 38 | 60 | 98 | 39% | 1.8 | − | − |
| Df(2R)BSC39 | 48C5-D1;48D5-E1 | 67 | 102 | 169 | 40% | 1.8 | − | − |
| Df(2R)CB21 | 48E;49A | 56 | 85 | 141 | 40% | 1.8 | − | − |
| Df(3R)3-4 | 82F3-4;82F10-11 | 59 | 87 | 146 | 40% | 1.8 | − | − |
| Df(3R)Tpl10 | 83C1-2;84B1-2 | 41 | 59 | 100 | 41% | 1.9 | 0.08 | No |
| Df(3R)BSC24 | 85B7;85D15 | 61 | 85 | 146 | 42% | 1.9 | 0 | No |
| Df(2L)JS17 | 23C1-2;23E1-2 | 90 | 125 | 215 | 42% | 1.9 | 0.02 | Yes |
| Df(2R)BSC22 | 56D7-E3;56F9-12 | 67 | 92 | 159 | 42% | 1.9 | 0 | No |
| Df(3L)BSC35 | 66F1-2;67B2-3 | 192 | 262 | 454 | 42% | 1.9 | 0.01 | No |
| Df(2R)BSC155 | 60B8;60C4 | 76 | 103 | 179 | 42% | 1.9 | 0.01 | No |
| Df(3L)BSC20 | 76A7-B1;76B4-5 | 61 | 81 | 142 | 43% | 2.0 | 0.03 | No |
| Df(3R)Exel6193 | 94D3;94E4 | 58 | 78 | 136 | 43% | 2.0 | 0.01 | No |
| Df(2L)BSC28 | 23C5-D1;23E2 | 98 | 129 | 227 | 43% | 2.0 | 0 | Yes |
| Df(3R)BSC42 | 98B1-2;98B3-5 | 129 | 169 | 298 | 43% | 2.0 | 0.01 | No |
| Df(2R)Np5 | 44F12;45DE3 | 48 | 60 | 108 | 44% | 2.0 | 0.05 | No |
| Df(3L)h-i22 | 66D10-11;66E1-2 | 51 | 63 | 114 | 45% | 2.0 | 0.05 | No |
| Df(3L)AC1 | 67A2;67D11-13 | 47 | 58 | 105 | 45% | 2.0 | 0.02 | No |
| Df(2L)BSC5 | 26B1-2;26D1-2 | 77 | 95 | 172 | 45% | 2.0 | 0.02 | Yes |
| Df(3R)Exel6195 | 95A4;95B1 | 45 | 55 | 100 | 45% | 2.0 | 0.0020 | Yes |
| Df(2R)vir130 | 59B;59D8-E1 | 59 | 72 | 131 | 45% | 2.0 | 0 | Yes |
| Df(2L)TW203 | 36E-36E3;37B10 | 46 | 55 | 101 | 46% | 2.1 | 0 | No |
| Df(2R)BSC18 | 50D1;50D2-7 | 101 | 120 | 221 | 46% | 2.1 | 0.02 | Yes |
| Df(2R)BSC29 | 45D3-4;45F2-6 | 64 | 75 | 139 | 46% | 2.1 | 0 | No |
| Df(2R)Exel7130 | 50D4;50E4 | 105 | 120 | 225 | 47% | 2.1 | 0.03 | No |
| Df(3R)mbc-R1 | 95A5-7;95D6-11 | 99 | 111 | 210 | 47% | 2.1 | 0.0050 | No |
| Df(2R)BSC3 | 48E12-F4;49A11-B6 | 79 | 88 | 167 | 47% | 2.2 | 0.0180 | No |
| Df(2R)BSC45 | 54C8-D1;54E2-7 | 98 | 109 | 207 | 47% | 2.2 | 0 | No |
| Df(2R)cn9 | 42E;44C | 59 | 64 | 123 | 48% | 2.2 | 0 | Yes |
| Df(3L)BSC10 | 69D4-5;69F5-7 | 62 | 67 | 129 | 48% | 2.2 | 0.01 | No |
| Df(3L)Scf-R6 | 66E1-6;66F1-6 | 43 | 45 | 88 | 49% | 2.2 | 0 | No |
| Df(3L)XG5 | 71C2-3;72B1-C1 | 67 | 67 | 134 | 50% | 2.3 | 0 | Yes |
| Df(3L)BSC21 | 79E5-F1;80A2-3 | 32 | 31 | 63 | 51% | 2.3 | 0.28 | No |
| Df(2L)E110 | 25F3-26A1;26D3-11 | 85 | 80 | 165 | 52% | 2.3 | 0 | Yes |
| Df(3R)mbc-30 | 95A5-7;95C10-11 | 58 | 52 | 110 | 53% | 2.4 | 0.01 | Yes |
| Df(2L)spd[j2] | 27B2-27F2 | 64 | 55 | 119 | 54% | 2.4 | 0.02 | No |
| Df(2R)X1 | 46C;47A1 | 84 | 72 | 156 | 54% | 2.4 | 0.01 | Yes |
| Df(2R)BSC11 | 50E6-F1;51E2-4 | 47 | 36 | 83 | 57% | 2.6 | < 0.0001 | Yes |
Based on Mendalian ratios, PAX7-FOXO1−positive males would be expected to represent 50% of all F1 adults. At baseline (w1118), PAX7-FOXO1 expression causes semilethality, with the percentage of F1 PAX7-FOXO1 males reduced to an average of 22% (SEM of 1.0%) (Please see Table S1.) Enhancers and suppressors decrease and increase, respectively, survival of PAX7-FOXO1 (“P-F”) F1 males 1 SD from the mean (mean = 26%) (SD = 15%). “Fold Change” = % of deficiency PAX7-FOXO1 F1 males observed divided by baseline (22%), with enhancers and suppressors showing a fold change value of ≤ 0.5 and ≥ 1.9, respectively. P values were calculated for the enhancers and suppressors. Three suppressors and three enhancers did not reach statistical significance.
We used a kit of minimally overlapping chromosomal deletions (a.k.a. “deficiencies”) (Table S1) to scan across the autosomes and identify genomic segments (or “hotspots”) that—when absent one copy—genetically modify PAX7-FOXO1 semilethality. Screening against ~95% of the Drosophila autosomes (~75% of the genome), we identified 33 suppressors and 28 enhancers (Table 1) (Figure 3), although three enhancers and three suppressors demonstrated P values above 0.05 and thus did not reach statistical significance. We next used smaller overlapping deletions to further delineate a subset of the hotspot regions, thereby significantly reducing candidate PAX7-FOXO1−interacting genes (Table 2). For the deficiency modifiers not submapped, candidate genes are provided in File S1.
Figure 3.

Distribution of the genetic lines tested in the PAX7-FOXO1 Screen. Shown is the plotted distribution of the tested deficiencies and the average baseline wild-type (w1118) control score (blue line, noted by the arrow) based on the percentage of F1 PAX7-FOXO1 males observed for each line examined. The Mean F1 male percentage for the screen was 26%, with a calculated SD of 15%. Suppressors (green) rank one SD above the mean, whereas enhancers (red) rank one SD below the mean.
Table 2. Submapping of PAX7-FOXO1-modifying Deficiencies.
| Genotype | Breakpoints | Males (P-F) | Females (Control) | Total F1 Adults | % F1 Males | Fold Change | P value | Comment |
|---|---|---|---|---|---|---|---|---|
| Df(2R)CX1 | 49C1-4;50C23-D2 | 2 | 88 | 90 | 2% | 0.10 | 0 | 1 |
| Df(2R)Exel7123 | 49D5;49E6 | 6 | 202 | 208 | 3% | 0.14 | 0 | |
| Candidate enhancers (49D5;49E6): Aats-aps, bic, CG3790, CG3814, CG13319, CG13321, CG17019, CG30487, Mdr49, NAT1, Nmda1, Psc, Sans, sug, vg | ||||||||
| Df(3R)p-XT103 | 85A2;85C1-2 | 3 | 189 | 192 | 2% | 0.07 | 0 | 1 |
| Df(3R)Exel8143 | 85A5;85B3 | 27 | 322 | 349 | 8% | 0.36 | 0 | |
| Candidate enhancers (85A5;85B3): CG8043, CG8112, CG8116, CG8136, CG8145, CG8159, CG8202, CG8223, CG8236, CG9773, CG9801, CG9837, CG9839, CG11755, CG11760, CG11762, CG11768, CG13318, Cks85A, hb, hng2, Ir85a, M1BP, mRpL19, Pif1A, ranshi, Tcp-1eta | ||||||||
| Df(3R)by10 | 85D8-12;85E7-F1 | 19 | 170 | 189 | 10% | 0.46 | 0 | 1 |
| Df(3R)Exel6153 | 85D19;85E1 | 4 | 249 | 253 | 2% | 0.09 | 0 | |
| Candidate enhancers (85D19;85E1): AP-1μ, bocksbeutel, by, CG8199, CG8273, CG8301, CG8312, CG8319, CG9386, CG9393, CG9396, CG9399, CG9427, CG16789, CG16790, Crc, Kap-a3, MBD-like, mRpL47, mura, P58IPK, Rib1, RnpS1, Vps45 | ||||||||
| Df(3L)fz-GF3b | 70C2;70D4-5 | 1 | 50 | 51 | 2% | 0.09 | 0 | 2 |
| Df(3L)Exel6122 | 70D4;70D7 | 109 | 257 | 366 | 30% | 1.4 | — | |
| Candidate enhancers (70C2-70D4): bru-3, CG43184, CG8757, CG8750, Tsp68C, Hml, CG8745, dysc, CG13737, Rgl, CG8833, Glued, Cg32137, Meics, Nxf3, ssp2, CG13738, Hsc70-1, CG17634, CG17632, CG9040, 26-29-p, CG17631, CG17359, CG8783, upSET, ptip, endos, CG6650, CG6661, Hsc70Cb, blue, CG6833, CG13484, CG32138, Pex1, breathless (FGFR), CG8100, Fbp1, Sox21a, Sox21b, Dichaete, nan, nuf, CG32141, CG7768, CG7924, CG34244, CG7906 | ||||||||
| Df(2L)E110 | 25F3-26A1;26D3-11 | 85 | 80 | 165 | 52% | 2.3 | 0 | 1 |
| Df(2L)BSC5 | 26B1-2;26D1-2 | 77 | 95 | 172 | 45% | 2.0 | 0.02 | |
| Df(2L)BSC184 | 26B1;26B3 | 85 | 104 | 188 | 45% | 2.0 | 0.01 | |
| Candidate suppressors (26B1;26B3): chickadee, eIF-4a, ifc, lid, Tsp26a, Gal, CG9098, H2.0, CG13996, CG9107, CG9109, mtm, CG9117, CG31643, ade2, mir-966, slowmo, CG34179, Cg12393 | ||||||||
| Df(2R)BSC18 | 50D1;50D2-7 | 101 | 120 | 221 | 46% | 2.1 | 0.02 | 1 |
| Df(2R)50C-36 | 50C19-23;50C21-D5 | 111 | 152 | 263 | 42% | 1.9 | 0.01 | |
| Candidate suppressors (50D1;D5): mastermind, mir-4978, CG18371, Prosap, CG42287, CG42288 | ||||||||
| Df(2R)BSC11 | 50E6-F1;51E2-4 | 47 | 36 | 83 | 57% | 2.6 | < 0.0001 | 1 |
| Df(2R)L48 | 50F6-F9;51B3 | 47 | 78 | 125 | 38%* | 1.7* | 0.04 | |
| Candidate suppressors (50F6;51B3): Shroom, CG8613, CG8617, Arc1, Arc2, Tfb1, CG34184, CG34442, CG4444, Obp50a, Obp50b, Obp50c, Obp50d, CG34185, Obp50e, CG30075, Dh44-R1, CG10104, CG17385, Sin1, CG17386, phyllopod, Oaz, Lobe, Cpsf160, Asx, Cpr51A, CG30197, tout-velu, CG30076, CG43919, LaminC | ||||||||
| Df(2L)JS17 | 23C1-2;23E1-2 | 90 | 125 | 215 | 42% | 1.9 | 0.02 | 2 |
| Df(2L)Exel7015 | 23C5;23E3 | 59 | 143 | 202 | 29% | 1.3 | — | |
| Candidate suppressors (23C1;23C5): CG8814, Prx6005, CG31950, betaggt-II, NTPase, lilliputian, Rbp9, Ts, Rrp1, gammaTub23C, CG9641, CG3165, CG9643, Chd1, Bem46, okra, CG3558, CG17265, CG17224, CG17264, alpha4GT1, CG3542, CG3605, CG17219,GABPI, CG17258, CG17259, CG17260, cnir, CG17221, CG17261 | ||||||||
| Df(2L)BSC28 | 23C5-D1;23E2 | 98 | 129 | 227 | 43% | 2.0 | 0 | 2 |
| Df(2L)Exel7015 | 23C5;23E3 | 59 | 143 | 202 | 29% | 1.3 | — | |
| Candidate suppressors (23C5;23C5): CG17219, GABPI, CG17258, CG17259, CG17260, cnir, CG17221, CG17261 | ||||||||
| Df(2R)cn9 | 42E;44C | 59 | 64 | 123 | 48% | 2.2 | 0 | 2 |
| Df(2R)Exel6053 | 43D3;43E9 | 75 | 138 | 213 | 35% | 1.6 | — | |
| Candidate suppressors (42E1;43D3): CG3358, mim, CheB42b, CheB42c, Che42a, ppk25, Cyp6u1, CG30157, vimar, CG30156, CG17002, Tsp42E-(a-r), Cg30159, CG30160, CG43646, CG43647, CG33914, lbm, pgant3, CG12831, esn, Cyp9b1, Cyp9b2, Spn43Aa, CG12828, prickle, Spn43Ab, Spn43Ad, necrotic, Cg11060, Cg33140, Cg30385, CG30384, Or43a, Ady43a, Gadd45, CG1850, Br140, Incenp, pawn, CG12164, Dscam1, costa, CG11107, Gr43a, CG1707, Eaf, CG11112, CG11113, CG43123, CG43267, mir-4977, sine oculis, CG11145, Cg11123, sPLA2, CG30503, kappaB-ras, fa2h, CG11127, p47, Aldh-III, wech, Coop, CG1620, dpa, didum, CG12763, az2, CG1603, Cg1602, Cg2144, Orc1, Drat, CG2064, mRpL52, CG12107, U2A, CG1399, CG30493, CG4096, CG34216, torso, CG19421, mir-4909, CG1942, CG1946, CG18812, CG30497, CG45093, cn, CanB2, mir-4980, CG12825, Cg12824, Gapdh1, mus205, Nop171, saxophone, Cg1550, Cg1882, cathD, CG30383, phr, phosalpha1, Cg18853, CG30382, CG12822, Atg10, Dgk, CG30377, CG30377, CG12159, Cul1, Or43b, Kdm4a, CG8791, CG30381, rnh1, drosha, CG8728, CG30380, Cg30379, CG14764, CG34430, CG34431, CG11165, CG30378, CG2906, CG2915, Sep5, Nito, CH14763, CG8726, CSN4, ACC, Nup44a, Dic3, Hey, CG11191, Odc1, Odc2, CG14762, mir-4981, Optix, CG12769, CG17977, lig, Vps28, slv, sut1, sut2, sut3, CG8713, CG8712, CG11210, Cul4, udd, Asap, Nup50, coil, Socs44a, Pbp49, CG42516, Pabp2, Obp44a, Lpin, kermit, CG8708, RagC-D, Rs1, CG30373, Gasz, Mlh1, CG14757, LRP1 | ||||||||
| Df(2R)X1 | 46C;47A1 | 84 | 72 | 156 | 54% | 2.4 | 0.01 | 1 |
| Df(2R)BSC152 | 46C1;46D7 | 88 | 124 | 212 | 42% | 1.9 | 0.01 | |
| Df(2R)BSC298 | 46B2;46C7 | 131 | 207 | 338 | 39%* | 1.8* | 0 | 1 |
| Df(2R)eve | 46C7;46C9-46C11 | 31 | 74 | 105 | 30% | 1.4 | — | 2 |
| Candidate genes (46B2;46C7): CG12744, CG1472, CG1513, CG12923, CG30008, CG30007, CG1441, FMRFa, Etf-QO, Mef2 | ||||||||
| Df(2R)vir130 | 59B;59D8-E1 | 59 | 72 | 131 | 45% | 2.0 | 0 | 2 |
| Df(2R)twi | 59C3-4;59D1-2 | 123 | 308 | 431 | 29% | 1.3 | — | |
| Candidate suppressors (59B1;59C4): CG42260, CG30270, blw, CycB, stall, CG30271, CG42284, CG30274, CG30272, CG30265, CG12490, CG9825, CG9826, CG3649, CG13531, RpL23, inaD, fd59A, CG13532, PIP5K59B, CG3501, CG3499, asrij, Gmer, MED23, CG3700, nahoda, CG30187, Nup214, CG42678, CG3788, CG3800, CG9849, CG3831, CG42694, CG32834, CG34371, CG13539, LS2, CG3092, uip3, RpL22-like, CG12782, Cg13540, ord, CG3124, CG13541, Prosbeta5R1, HP1Lcsd, CG0412, CG30416, CG9861,CG30417, CG30413, CG3502, CG9863, CG34210, CG30409, Rpi, Cg3500, CG9875, CG34423, CG34424, vir, Ice1 | ||||||||
| Df(3L)XG5 | 71C2-3;72B1-C1 | 67 | 67 | 134 | 50% | 2.3 | 0 | |
| Df(3L)brm11 | 72A3;72D5 | 72 | 278 | 350 | 21% | 1.0 | — | |
| Candidate suppressors (71C2;72A3): Best4, Best3, CG7255, Toll-6, CG33259, CG7804, Ran-like, CG12355, CG13455, CG7276, CG7275, Cg7272, CG7857, CG7841, Z600, gdl, gdl-ORF39, Eip71CD, CG13454, mex1, yellow-k, CG7945, CG33986, CG33985, CG42729, obst-H, CG42728, CG43248, CrebA, AGO2, CG7739, CG7427, dop, CG16979, mrn,CG12301, CG12304, CG7656, RhoGAP71E, CG7650, CG13449, comm3, CG7372, CG43083, CG43084, Eig71E-(a-k), CG43082, CG7304, CG7579, pgant8, CG34452, CG34451, comm2, CG42571, CG42570, comm, CG6244, CG13445, fwe, CkIIalpha-i1, DCP2, diablo, CG12713, CG18081, CG15715, CG32150, mir-263b | ||||||||
| Df(3R)mbc-30 | 95A5-7;95C10-11 | 58 | 52 | 110 | 53% | 2.4 | 0.01 | |
| Df(3R)Exel6195 | 95A4;95B1 | 45 | 55 | 100 | 45% | 2.0 | 0.02 | |
| Df(3R)Exel9014 | 95B1;95D1 | 53 | 109 | 162 | 33% | 1.5 | — | |
| Candidate suppressors (95A5;95B1): CG31145, GILT3, GILT2, eIF-3p66, CG1670, CG18754, SPE, CG10254, CG10252, prt, CG31468, CG31148, CG31413, CG31414, CG10301, CG10300, nautilus (Drosophila MyoD), CG10365 | ||||||||
| Df(2L)ast2 | 21E2;22B2-3 | 12 | 128 | 140 | 9% | 0.39 | 0.1480** | 3 |
| Df(2L)Exel6004 | 21E4;21F1 | 60 | 80 | 140 | 43% | 2.0 | 0.01 | |
| Candidate enhancers (21E2;21E4): CG2839, dachsous, Hsp60B, Eaat2, GABA-B-R3, CG12506, CG13946, CG13947, Gr21a, CG3544, Pkg21D, Nnf1b, Ddp21E2, Saf6, Pex12, CG15880, CG3867, clipper, CG3662, CG3862, dock, drongo, CG4291, kraken, CG13949, mir-375, CG13950, mir-375, aru, dbe, PNUTS, ninaA, CG15824, Lsp1beta, GluRIIC, CG4341, IA-2, Star | ||||||||
| Candidate enhancers (21F1;22B2): Tango14, CG5080, IntS14, CG14341, Plap, CG31922, CG5118, CG4887, CG4896, CG5126, Tgt, CG5001, Cg5139, CG43348, CG43349, CG5011, CG14342, CG42329, CG5397, robo3, a5, CG5440, CG33923, CG33922, Cdkc2, CG5556, CG5561, CG31924, CG5565, CG31659, NLaz, CG14346, leak, CG43401, CG43402, CG31928, CG33128, CG31926, CG31661, CG18131, CG7420, CG18132, halo, Or22a, CG44072, Or22b, haf, CG10869, CG31935, CG14352, RFeSP, chinmo, cpb, CG17660, mRpL48, frtz, Rim2, Eno, Rrp40, CG31937, CG17652, CG17646, CG17712, CG17648, Gr22f, CG17650, Gr22-(e-a), CG31933 | ||||||||
| Candidate suppressors (21E4;21F1): asteroid, Atg4a, CG4692, MtRNApol, CG14339, CG14340, Pino, CG4552, Iris, CG4577, MFS3, CG4749, Tfb4, Vsp29, capulet | ||||||||
| Df(2L)TE35BC-24 | 35B4-6;35F1-7 | 9 | 114 | 123 | 7% | 0.33 | 0 | 2 |
| Df(2L)TE35BC-7 | 35B2;35B10 | 55 | 123 | 154 | 36% | 1.6 | — | |
| Df(2L)Exel7063 | 35D2;35D4 | 46 | 64 | 110 | 42% | 1.9 | 0.03 | 3 |
| Candidate enhancers (35C1;35D2): vasa, vig, CG15270, CG15296, stc, CG4168, Sfp35C, CG43230, ZnT35C, dao, Pol32, l(2)35Cc, yuri, Cul3, UK114, CG15263, CG15260, ms(2)35Ci, CG15262, nht, esgargot, CG15258, CG44869, worniu | ||||||||
| Candidate suppressors (35D2;35D4): vasa, CG4161, snail, Tim17b2, lace, Skadu, CG15256, kek3, CG15255, Semp1, CG15254, CG15253, CG11865, Or35a, CG7631, CG18480, CG4578, CG44141, CG18477, CG18478, CG43923, CG44140, CG31780, CG1827, CG43924, CycE | ||||||||
| Df(2R)BSC49 | 53D9-E1;54B5-10 | 12 | 112 | 124 | 10% | 0.44 | 0.01 | 3 |
| Df(2R)Exel6066 | 53F8;54B6 | 100 | 144 | 244 | 41% | 1.9 | 0 | |
| Df(2R)BSC154 | 54B2;54B7 | 3 | 93 | 96 | 3% | 0.14 | 0.01 | 1 |
| Candidate enhancers (53D9;53F8): CG5522, CG15919, CG15615, CG5550, CG34459, CG34460, mir-8, Ugt37c1, IntS8, Fen1, Dek, Psi, Ef1beta, CG6426, CG6241, CG6429, CG6435, CG8910, CG6472, mir-990, inaC, Pkc53E, CG43788, CG43789, CG15614, CG43190, Vha16-4, mute, CG34191, PIG-V, CG6665, CG9010, Cbp53E, ste24c, CG30461, ste24b, ste24a, CG6796, NiPp1, CG6805, Ehbp1, CG8963, Dark, RhoGEF2, CG43327, CG43328, CG43371, CG9640, CG9642, CG9646, fat-spondin, tef, CG8950, CG6967, CG30460, Sply, CG6984 | ||||||||
| Candidate suppressors (53F8;54B2): GstS1, CG30456, CG15611, Amy-p, CG15605, Cda9, Acp54A1, CG11400, Gbp, Cg11395, CG43103, CG43107, CG17290, CG17287, CG30458, CG30457, CG10953, CG10950, CG43237, muscleblind, CG18469, CG12699, CG43272, CG43108 | ||||||||
| Candidate enhancers (54B2;54B7): Muscleblind, Sip1, CG6568, CG30101, Prosalpha5, cnk | ||||||||
| Df(2R)AA21 | 57B19-C1;57E1-6 | 1 | 75 | 76 | 1% | 0.06 | 0 | 2 |
| Df(2R)Exel6072 | 57B16;57D4 | 30 | 169 | 199 | 15% | 0.7 | — | |
| Df(2R)Exel6076 | 57E1;57F3 | 77 | 89 | 166 | 46% | 2.1 | 0.03 | 3 |
| Candidate enhancers (57D4-57E1): Rgk3, CG30391, CG30393, CG34023, MFS16, CG10505, CG30392, Sgf29, RpL29, CG9752, CG42672, CG9754, CG9485, CG33655, CG30394, dom, CG15666, CG9822, CG17974, cv-2 | ||||||||
| Candidate suppressors (57E1;57F3): Sdc, Sara, Fkbp14, TAF1c-like, MESK2, CG10494, CG30288, CG30289, EGFR, CG30286, CG30287, CG33226, CG30283, twz, CG30222, CG33225, CG10433, CG15673 | ||||||||
| Df(2R)Egfr5 | 57D2-8;58D1 | 8 | 140 | 148 | 5% | 0.25 | 0 | 3 |
| Df(2R)Exel6076 | 57E1;57F3 | 77 | 89 | 166 | 46% | 2.1 | 0.03 | |
| Candidate enhancers (57D2;57E1): CG15661, ASPP, Rgk3, CG30391, CG30393, CG34203, MFS16, CG10505, CG30392, Sgf29, RpL29, CG9752, CG42672, CG957, CG9485, CG33655, CG30394, domino, CG15666, CG9822, CG17974, cv-2 | ||||||||
| Candidate suppressors (57E1; 57F3): CG10795, EfSec, Acox57D-p, Acox57D-d, Sdc, Sara, Fkbp14, TAF1C-like, MESK2, CG10494, CG30288, CG30289, EGFR, CG30286, CG30287, CG33226, CG30283, CG10440, CG30222, CG33225, CG10433 | ||||||||
Original chromosomal deletions (a.k.a., Deficiencies, or Df) identified that genetically modify PAX7-FOXO1−induced male semilethality are noted in bold. Additional smaller Df’s tested to further delimit the critical modifying segments are shown directly below. Comments: 1) Df’s for which we were able to reduce the critical modifying chromosomal regions; 2) Df’s for which additional deletions tested showed no modification, indicating that the critical segments lie outside the smaller tested regions; 3) Df’s for which the smaller deletions showed opposite modifying behavior. Similar to Table 1, “Fold Change” = % of Deficiency PAX7-FOXO1 F1 males observed divided by the control baseline of 22%. “*” notes two smaller Df’s that, though with a fold change of slightly less than 1.9, showed a statistically significant increase in the male F1 population, and for this second-pass study we considered suppressors. “**” notes an original Df that did not reach statistical significance but was included in these submapping studies. “P-F” = PAX7-FOXO1.
MEF2 as a PAX-FOXO gene target and a putative RMS effector
We found that mutation of benchmark myogenesis genes modify PAX7-FOXO1. The D-Mef2 gene, which operates as a linchpin and critical nodal point in fly myogenesis (Lilly et al. 1995), is cytogenetically located at 46C4-46C7 on chromosome 2 and is positioned within a hotspot region (46C1-47A1) initially uncovered by the Df(2R)X1 deletion (Table 1). The hotspot region was further refined by smaller overlapping deletions to segments 46C1-46C7 (Figure 4, A and B and Table 2). Concomitantly, we found by mRNA expression profiling that D-Mef2 is overexpressed in PAX7-FOXO1 larval muscle (2.0-fold, P < 0.001, n = 3). No other gene in this region was reported as misexpressed 2.0-fold or more with statistical significance. Thus, we hypothesized that heterozygous deletion of the D-Mef2 locus might account for Df(2R)X1-mediated PAX7-FOXO1 suppression, and that D-Mef2 might act as a PAX7-FOXO1 target gene. We tested the D-Mef2−null mutation (Bour et al. 1995), D-Mef222-21, which showed that heterozygous loss of D-Mef2 suppresses PAX7-FOXO1 (2.0-fold increase in PAX7-FOXO1 F1 males) (Figure 4B). Of note, these findings do not eliminate the possibility, however, that other genes in this region might also independently interact with PAX7-FOXO1.
Because D-Mef2 mutation suppresses PAX7-FOXO1 and D-Mef2 is overexpressed by PAX7-FOXO1 in our microarray analysis, we investigated whether D-Mef2 acts as a downstream PAX-FOXO1 target. We used the daughterless-Gal4 driver to ubiquitously express PAX-FOXO1 and then probed for expression of a YFP-tagged embryonic D-Mef2 reporter (Cripps et al. 1998, 1999). We found diffuse misexpression of the D-Mef2 reporter (Figure 4C) in ectoderm and endoderm derivatives. Additionally, D-Mef2 reporter overexpression was detected in mesodermal-derived myoblasts, visible in a segmentally repeating pattern. These studies corroborate our aforementioned MHC-GFP reporter expression studies, affirming that human PAX-FOXO1 promotes myogenic fate-specification in Drosophila. These studies further show that D-Mef2 acts as a PAX7-FOXO1 downstream target gene (direct or indirect) and PAX-FOXO1 genetic effector in vivo.
We next interrogated the 50D1-50D5 hotspot suppressor, which contains only five genes (Table 2 and Figure 5A), one of which is mam. In vivo studies in mammalian models have shown that the mam ortholog Mastermind-Like 1 (Maml1) encodes a transcriptional cofactor that physically interacts with Mef2 to augment Mef2-dependent promyogenic signaling (Shen et al. 2006; Potthoff and Olson 2007). Similar to D-Mef2, mam loss-of-function mutation dominantly suppressed PAX7-FOXO1−induced lethality (Figure 5B). Of note, mam expression levels were not detectably altered in our PAX7-FOXO1 microarray studies, compatible with mam’s role as a cofactor vs. myogenesis gene target. Taken together, these Drosophila studies highlight a putative PAX-FOXO1→MEF2→RMS pathogenic axis, while also demonstrating that the one-generation (F1) genetic screen quickly uncovers dominant PAX-FOXO1 modifiers/effectors in an unbiased fashion.
Figure 5.

Mutation of mastermind, which encodes a MEF2 transcriptional cofactor, is a dominant PAX7-FOXO1 suppressor. (A) Overlapping chromosomal deletions identify a small genomic region, 50D1-50D5, as a PAX7-FOXO1−suppressing hotspot. (B) mastermind loss-of-function mutation dominantly suppresses PAX7-FOXO1 lethality. Df(2R)BSC18 was isolated in our original screen as a PAX7-FOXO1 suppressor (Table 1), which deletes mastermind (mam). The overlapping deletion, Df(2R)50C-36 also suppresses PAX7-FOXO1 (Table 2). Two well-characterized, strong loss-of-function mam alleles, mamBG02477 (n = 89 F1 adults scored) (P = 0.0044) and mam2 (n = 112 F1 adults scored) (P = 0.0043) suppress PAX7-FOXO1. Of note−although the mam2 allele showed a fold change of slightly less than 1.9, the increase in PAX7-FOXO1 adults (1.7-fold) was highly significant, and in this test we scored as a suppressor. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control.
Finally, we surveyed MEF2 expression levels in a large collection of pediatric RMS cancer cell lines, xenograft tumors, and primary tumors using the Pediatric Tumor Affymetrix Database (http://home.ccr.cancer.gov/oncology/oncogenomics/) (Khan et al. 2001). Four MEF2 orthologs (MEF2A, -B, -C, -D) are present in the mammalian genome, with -A and -C demonstrating greatest similarity to D-Mef2. Compared with normal tissues and non-RMS pediatric soft-tissue sarcomas, only MEF2C showed significant and consistent up-regulation in RMS samples (Figure 6; also shown are MYOD and MET, genes that associate with RMS), data that point toward MEF2C as influential in RMS. Similar to mam in our PAX7-FOXO1 Drosophila system, analysis of the three mammalian MASETRMIND orthologs (MAML1, -2, and -3) did not reveal a MAML overexpression pattern in these RMS data sets (Figure 6).
Figure 6.

MEF2C is overexpressed in Rhabdomyosarcoma. Shown are expression profiles for embryonal rhabdomyosarcoma (E-RMS), alveolar rhabdomyosarcoma (A-RMS), non-RMS soft-tissue sarcoma (Non-RMS STS), and Ewing sarcoma (EWS). Profiles are from cell lines (C), tumor xenografts (X), and primary human tumors (T). Three individual probes are shown for MEF2A, -B, -C (bordered in black), and -D. Probes are also shown for the three human Mastermind orthologs, MAML1, -2, and -3. Representative probes are shown for MYOD1 and MET—genes known to be up-regulated in RMS. mRNA Expression data sets are from the Pediatric Tumor Affymetrix Database (Oncogenomics; http://home.ccr.cancer.gov/oncology/oncogenomics/).
Discussion
The Drosophila PAX7-FOXO1 genetic model
Given the critical role that the PAX-FOXO1 fusion oncoprotein plays in RMS, we focused on PAX-FOXO1 as an entry-point for designing a transgenic Drosophila RMS-related model that would be amenable to forward genetic screening and RMS gene discovery. To bypass the issue of cumbersome multigenerational screening schemes that would normally be required, we incorporated a Gal80 X-linked chromosomal transgene to generate a viable screening Gal4/UAS-PAX-FOXO1 master stock that allows for the rapid identification of PAX-FOXO1 genetic modifiers in a single genetic cross.
With this platform, we have been probing for new PAX-FOXO1 pathogenesis underpinnings. Though very similar in molecular structure, PAX3-FOXO1− and PAX7-FOXO1−positive RMS demonstrate differing clinical behaviors, as PAX3-FOXO1 tumors are more common and notoriously aggressive (Kelly et al. 1997). Consequently, PAX3-FOXO1 is the PAX-FOXO1 fusion most commonly investigated in vertebrate models. In our Drosophila system, we have focused on PAX7-FOXO1, which demonstrates phenotypes that are better penetrant and experimentally tractable due to the fact that human PAX7 demonstrates slightly greater sequence identity to fly PAX3/7 than does human PAX3. Additionally, as no other animal models of PAX7-FOXO1 presently exist, the fly PAX7-FOXO1 model also conveniently serves as a complement to vertebrate PAX3-FOXO1 models.
Initially unknown was the extent to which observations from the PAX7-FOXO1 fly model would impact the clinically more aggressive PAX3-FOXO1 RMS subtype, as well as PAX-FOXO1-negative (embryonal) RMS. Notably, our previous studies have shown that genetic modifiers identified from the Drosophila system impact PAX3-FOXO1 RMS oncogenesis and tumorigenesis (Avirneni-Vadlamudi et al. 2012; Crose et al. 2014). Furthermore, unpublished studies (U. Avirneni-Vadlamudi and R. L. Galindo, unpublished data) are demonstrating that fly PAX7-FOXO1 genetic modifiers are similarly involved in Embryonal RMS. These findings provide marked validation for the applicability and value of this genetic fly system to human RMS.
Interestingly, though PAX7-FOXO1 induces expression of the late myogenic differentiation marker MHC, PAX-FOXO1 RMS myoblasts in culture and in vivo demonstrate only partial differentiation with little-to-no MHC expression. In considering this discrepancy, we first note that PAX-FOXO1 is a relatively weak driver of RMS in culture and in vivo (Keller et al. 2004; Naini et al. 2008) and requires additional/sequential genetic aberrations to induce oncogenic transformation. Thus, secondary mutations might be necessary to force the strength of RMS myoblast differentiation-arrest seen in human RMS tumors; by contrast, our PAX7-FOXO1 model differs in that the system is free of any additional background mutations. Second, previous studies have shown that expression of PAX3-FOXO1 in mouse embryonic cultured cells induces the formation of MHC-positive myocytes and myotube formation (Scuoppo et al. 2007), studies that are similar to those seen here in the Drosophila system, where the da-Gal4/UAS-PAX7-FOXO1 expression system targets undifferentiated embryonic primordia. Uncovering of the genetic/molecular sequence of RMS pathogenesis and the cell(s) origin will shed further insight into the underlying mechanisms that account for the myoblast differentiation arrest phenotypes seen in RMS in vivo.
MEF2 in myogenesis and RMS
The differentiation and fusion of myoblasts into postmitotic, syncytial muscle requires that the bHLH myogenic regulatory factors (MRFs: Myf5, Mrf4, MyoD, and Myogenin) interact with E-proteins, which drive and regulate critical aspects of myogenic fate determination (Braun and Gautel 2011). The MRFs subsequently interact with the MEF2 transcription factors that, although lacking intrinsic myogenic activity, cooperate with the MRFs to synergistically activate muscle-specific genes and the downstream myogenic terminal differentiation program (Molkentin and Olson 1996; Potthoff and Olson 2007).
Vertebrates possess four MEF2 family member genes (-A, -B, -C, -D), which demonstrate complex overlapping spatial and temporal expression patterns in embryonic and adult tissues, with greatest expression levels seen in striated muscle and brain (Potthoff and Olson 2007). Because of genetic redundancy and overlapping expression patterns of the MEF2 genes, interrogating individual MEF2 gene activity in mammals has been experimentally challenging, with loss-of-function mutation studies revealing only limited insights into MEF2 gene function in tissues in which the MEF2 genes do not overlap/compensate. Conveniently, flies possess only one Mef2 gene (D-Mef2) and have served as an excellent model system to delineate MEF2’s critical role in myogenesis (Potthoff and Olson 2007). We speculate that the lack of Mef2 redundancy in flies provided a marked experimental advantage in isolating D-Mef2 as a PAX7-FOXO1 effector. Similarly, the identification of mam was also likely facilitated by the fact that flies possess one mam gene, whereas mammals contain three mam orthologs (Saint Just Ribeiro and Wallberg 2009). Thus, we propose that the comparative lack of genetic compensation/redundancy is an attractive advantage to Drosophila as a disease model system.
Recent studies have made significant inroads toward dissecting MEF2 in myogenesis in vivo and RMS—most specifically, MEF2C and -D. Whereas global deletion of Mef2A or -D demonstrates little to no effect on embryonic myogenesis (Potthoff and Olson 2007), skeletal muscle-specific deletion of Mef2C causes neonatal lethality due to defective muscle integrity and sarcomere formation (Potthoff et al. 2007a,b). Regarding RMS, Zhang et al. (2013) have found that RMS cells lack proper expression of MEF2D, and that exogenous expression of MEF2D promotes RMS cell differentiation, diminishes oncogenesis in culture, and blocks tumorigenesis in xenograft studies. Turning to adult muscle regeneration and satellite stem cells, which is likely at least one cell of origin for human RMS, Liu et al. (2014) have now shown that Mef2A, -C, and -D are essential yet function redundantly in satellite cell differentiation. Lastly, our survey of published RMS microarrays (Khan et al. 2001), as well as PAX-FOXO1-expressing myoblast cell lines (Avirneni-Vadlamudi et al. 2012), shows a consistent pattern of MEF2C overexpression. Given the integrated and overlapping nature of the MEF2 genes, we hypothesize a potential mechanism in which overexpression of MEF2C feeds-back upon and down-regulates MEF2D, thereby preventing MEF2D from driving myoblast terminal differentiation.
We suggest that further interrogation of MEF2 in RMS will open new avenues for RMS chemotherapy, which for high-risk disease has not improved for decades. For example, since MEF2 activity is tightly governed by class IIa histone deacetylases (Haberland et al. 2007; Potthoff and Olson 2007; Nebbioso et al. 2009), histone deacetylase inhibitors are now ripe for preclinical testing as new anti-RMS agents. Additionally, we have found that the MEF2 cofactor Mastermind, which interacts with MEF2C and mediates crosstalk between Notch signals during myogenic differentiation (Shen et al. 2006; Potthoff and Olson 2007), similarly influences PAX-FOXO1 pathogenicity in flies. Interestingly, Mastermind-specific, cell-permeable peptide inhibitors have been shown to block the progression of T-cell acute lymphoblastic leukemia in mice in vivo (Moellering et al. 2009) and thus are also new agents available for RMS preclinical testing. Further characterization of MEF2 in RMS cell and mouse models will continue to refine both our understanding and the potential targeting of MEF2 activity in RMS.
In conclusion, we postulate that: 1) The Drosophila PAX7-FOXO1 model is uniquely configured for the quick uncovering of new RMS genetic effectors with one simple genetic screening cross; 2) a putative PAX-FOXO1→MEF2/MASTERMIND axis underlies A-RMS; and 3) Drosophila conditional expression models are an efficient and powerful gene discovery platform for the rapid dissection of human disease.
Supplementary Material
Acknowledgments
We are grateful to Linh Tran and Emma Simpson for excellent technical support. We thank Diego Castrillon, Corinne Linardic, and Lisa Crose for reviewing the manuscript. This work was supported by funding to R.L.G. by the Burroughs Wellcome Fund (Career Award for Medical Scientists), Alex’s Lemonade Stand Foundation (“A” Award), President’s Research Council (UTSW), Children’s Cancer Fund (Dallas, TX), American Cancer Society Research Scholars Grant (124717-RSG-13-194-01-DDC) and Cancer Prevention Research Institute of Texas (RP120685); and to K.A.G. by Alex’s Lemonade Stand Foundation (Young Investigator Award).
Footnotes
Supporting information is available online at http://www.g3journal.org/lookup/suppl/doi:10.1534/g3.114.015818/-/DC1
Communicating editor: K. S. McKim
Literature Cited
- Avirneni-Vadlamudi U., Galindo K. A., Endicott T. R., Paulson V., Cameron S., et al. , 2012. Drosophila and mammalian models uncover a role for the myoblast fusion gene TANC1 in rhabdomyosarcoma. J. Clin. Invest. 122: 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour B. A., O’Brien M. A., Lockwood W. L., Goldstein E. S., Bodmer R., et al. , 1995. Drosophila MEF2, a transcription factor that is essential for myogenesis. Genes Dev. 9: 730–741. [DOI] [PubMed] [Google Scholar]
- Brand A. H., Perrimon N., 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415. [DOI] [PubMed] [Google Scholar]
- Braun T., Gautel M., 2011. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell Biol. 12: 349–361. [DOI] [PubMed] [Google Scholar]
- Buckingham M., Rigby P. W., 2014. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev. Cell 28: 225–238. [DOI] [PubMed] [Google Scholar]
- Cao L., Yu Y., Bilke S., Walker R. L., Mayeenuddin L. H., et al. , 2010. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 70: 6497–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E. H., Olson E. N., 2001. Antisocial, an intracellular adaptor protein, is required for myoblast fusion in Drosophila. Dev. Cell 1: 705–715. [DOI] [PubMed] [Google Scholar]
- Chen E. H., Pryce B. A., Tzeng J. A., Gonzalez G. A., Olson E. N., 2003. Control of myoblast fusion by a guanine nucleotide exchange factor, loner, and its effector ARF6. Cell 114: 751–762. [DOI] [PubMed] [Google Scholar]
- Cripps R. M., Black B. L., Zhao B., Lien C. L., Schulz R. A., et al. , 1998. The myogenic regulatory gene Mef2 is a direct target for transcriptional activation by Twist during Drosophila myogenesis. Genes Dev. 12: 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripps R. M., Zhao B., Olson E. N., 1999. Transcription of the myogenic regulatory gene Mef2 in cardiac, somatic, and visceral muscle cell lineages is regulated by a Tinman-dependent core enhancer. Dev. Biol. 215: 420–430. [DOI] [PubMed] [Google Scholar]
- Crose L. E., Galindo K. A., Kephart J. G., Chen C., Fitamant J., et al. , 2014. Alveolar rhabdomyosarcoma-associated PAX3-FOXO1 promotes tumorigenesis via Hippo pathway suppression. J. Clin. Invest. 124: 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daczewska M., Picchio L., Jagla T., Figeac N., Jagla K., 2010. Muscle development and regeneration in normal and pathological conditions: learning from Drosophila. Curr. Pharm. Des. 16: 929–941. [DOI] [PubMed] [Google Scholar]
- Davis R. J., D’Cruz C. M., Lovell M. A., Biegel J. A., Barr F. G., 1994. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 54: 2869–2872. [PubMed] [Google Scholar]
- Galili N., Davis R. J., Fredericks W. J., Mukhopadhyay S., Rauscher F. J., 3rd, et al. , 1993. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 5: 230–235. [DOI] [PubMed] [Google Scholar]
- Galindo R. L., Allport J. A., Olson E. N., 2006. A Drosophila model of the rhabdomyosarcoma initiator PAX7-FKHR. Proc. Natl. Acad. Sci. USA 103: 13439–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C., 2013. Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat. Rev. Cancer 13: 172–183. [DOI] [PubMed] [Google Scholar]
- Gurney J., Young J., Roffers S., Smith M., Bunin G., 1999, pp. 111–124 in Cancer incidences and survival among children and adolescents: United States SEER Program 1975–1995, National Cancer Institute, SEER Program, edited by Ries L. National Institutes of Health, Bethesda, MD. [Google Scholar]
- Haberland M., Arnold M. A., McAnally J., Phan D., Kim Y., et al. , 2007. Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol. Cell. Biol. 27: 518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G., Callaerts P., Gehring W. J., 1995. Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science 267: 1788–1792. [DOI] [PubMed] [Google Scholar]
- Huh W. W., Skapek S. X., 2010. Childhood rhabdomyosarcoma: new insight on biology and treatment. Curr. Oncol. Rep. 12: 402–410. [DOI] [PubMed] [Google Scholar]
- Keller C., Arenkiel B. R., Coffin C. M., El-Bardeesy N., DePinho R. A., et al. , 2004. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 18: 2614–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly K. M., Womer R. B., Sorensen P. H., Xiong Q. B., Barr F. G., 1997. Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J. Clin. Oncol. 15: 1831–1836. [DOI] [PubMed] [Google Scholar]
- Khan J., Wei J. S., Ringner M., Saal L. H., Ladanyi M., et al. , 2001. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat. Med. 7: 673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagha M., Sato T., Bajard L., Daubas P., Esner M., et al. , 2008. Regulation of skeletal muscle stem cell behavior by Pax3 and Pax7. Cold Spring Harb. Symp. Quant. Biol. 73: 307–315. [DOI] [PubMed] [Google Scholar]
- Lilly B., Zhao B., Ranganayakulu G., Paterson B. M., Schulz R. A., et al. , 1995. Requirement of MADS domain transcription factor D-MEF2 for muscle formation in Drosophila. Science 267: 688–693. [DOI] [PubMed] [Google Scholar]
- Liu N., Nelson B. R., Bezprozvannaya S., Shelton J. M., Richardson J. A., et al. , 2014. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 111: 4109–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan P., Leavey P. J., Galindo R. L., 2014. PAX genes in childhood oncogenesis: developmental biology gone awry? Oncogene. 2014 Jul 21;0. doi: 10.1038/onc.2014.209. [Epub ahead of print] PMID: 25043308. [DOI] [PubMed]
- Moellering R. E., Cornejo M., Davis T. N., Del Bianco C., Aster J. C., et al. , 2009. Direct inhibition of the NOTCH transcription factor complex. Nature 462: 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin J. D., Olson E. N., 1996. Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc. Natl. Acad. Sci. USA 93: 9366–9373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naini S., Etheridge K. T., Adam S. J., Qualman S. J., Bentley R. C., et al. , 2008. Defining the cooperative genetic changes that temporally drive alveolar rhabdomyosarcoma. Cancer Res. 68: 9583–9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebbioso A., Manzo F., Miceli M., Conte M., Manente L., et al. , 2009. Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep. 10: 776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishijo K., Chen Q. R., Zhang L., McCleish A. T., Rodriguez A., et al. , 2009. Credentialing a preclinical mouse model of alveolar rhabdomyosarcoma. Cancer Res. 69: 2902–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff M. J., Olson E. N., 2007. MEF2: a central regulator of diverse developmental programs. Development 134: 4131–4140. [DOI] [PubMed] [Google Scholar]
- Potthoff M. J., Arnold M. A., McAnally J., Richardson J. A., Bassel-Duby R., et al. , 2007a Regulation of skeletal muscle sarcomere integrity and postnatal muscle function by Mef2c. Mol. Cell. Biol. 27: 8143–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff M. J., Wu H., Arnold M. A., Shelton J. M., Backs J., et al. , 2007b Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Invest. 117: 2459–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint Just Ribeiro M., Wallberg A. E., 2009. Transcriptional mechanisms by the coregulator MAML1. Curr. Protein Pept. Sci. 10: 570–576. [DOI] [PubMed] [Google Scholar]
- Scheurer M., Bondy M., Gurney J., 2011. Epidemiology of childhood cancer, pp. 2–16 in Principles and practice of pediatric oncology, edited by Pizzo P., Poplack D. Lippincott Williams & Wilkins, Philadelphia. [Google Scholar]
- Scuoppo C., Riess I., Schmitt-Ney M., Allegra P., Forni P. E., et al. , 2007. The oncogenic transcription factor PAX3-FKHR can convert fibroblasts into contractile myotubes. Exp. Cell Res. 313: 2308–2317. [DOI] [PubMed] [Google Scholar]
- Shapiro D. N., Sublett J. E., Li B., Downing J. R., Naeve C. W., 1993. Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res. 53: 5108–5112. [PubMed] [Google Scholar]
- Shen H., McElhinny A. S., Cao Y., Gao P., Liu J., et al. , 2006. The Notch coactivator, MAML1, functions as a novel coactivator for MEF2C-mediated transcription and is required for normal myogenesis. Genes Dev. 20: 675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wexler L., Meyer W., Helman L., 2011. Rhabdomyosarcoma, pp. 923–953 in Principles and practice of pediatric oncology, edited by Pizzo P., Poplack D. Lippincott Williams & Wilkins, Philadelphia. [Google Scholar]
- Xue L., Noll M., 1996. The functional conservation of proteins in evolutionary alleles and the dominant role of enhancers in evolution. EMBO J. 15: 3722–3731. [PMC free article] [PubMed] [Google Scholar]
- Xue L., Li X., Noll M., 2001. Multiple protein functions of paired in Drosophila development and their conservation in the Gooseberry and Pax3 homologs. Development 128: 395–405. [DOI] [PubMed] [Google Scholar]
- Zhang M., Truscott J., Davie J., 2013. Loss of MEF2D expression inhibits differentiation and contributes to oncogenesis in rhabdomyosarcoma cells. Mol. Cancer 12: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.