

Abstract
Pancreatic neuroendocrine tumors (PNETs) are frequent and can be non-functional (NF) in patients with multiple endocrine neoplasia type 1 (MEN1). Their identification is of clinical importance because malignant PNETs are reported to be the most common cause of death in patients with MEN1. Once the diagnosis of MEN1 is established in an individual based on clinical manifestations and/or genetic testing results, an active surveillance program is instituted for early detection and treatment of MEN1-associated disease. Ultrasonography, endoscopic ultrasonography (EUS), CT, MRI, selective arterial angiography and somatostatin receptor scintigraphy are all used for localization of tumors. Managing PNETs can be challenging and includes diagnosis, surveillance, adequate staging, and interdisciplinary, multimodal treatments to optimize patient outcome. Treatment includes surgical resection for loco-regional disease, as well as liver directed and targeted chemotherapies for advanced progressive disease. To date, the recommendation for surgical resection in NF-PNETs is based on tumor size, as a higher rate of metastases was found in patients with larger tumors. This review summarizes key concepts in managing PNETs in patients with MEN1.
Keywords: Pancreatic neuroendocrine tumors (PNETs), multiple endocrine neoplasia type 1 (MEN1), surgical management
Introduction
Multiple endocrine neoplasia type 1 (MEN1) is one of the most common familial cancer syndromes. MEN1 is an autosomal dominant hereditary syndrome caused by a germline mutation in the MEN1 tumor suppressor gene. It has a prevalence of 2-3 per 100,000 (1). MEN1 is characterized by the occurrence of tumors in the parathyroid glands, neuroendocrine tumors (NETs) in the pancreatic islets and gastrointestinal tract, the anterior pituitary, and less commonly in the thymus, lung and bronchus, and the adrenal cortex.
Although the most common manifestation of MEN1 is the development of primary hyperparathyroidism, most morbidity and mortality in patients with MEN1 results from NETs involving the gastrointestinal tract, pancreatic islets, bronchus and thymus, which have malignant potential (2,3). Thus, once a diagnosis of MEN1 is established in an individual based on clinical manifestations and/or genetic testing results, an active surveillance program is instituted for early detection and treatment of MEN1-associated disease, especially for tumor sites with malignant potential such as gastrointestinal and pancreatic NETs (4). Clinical practice guidelines have been developed for surveillance and screening for MEN1-associated tumors and include clinical, biochemical and imaging studies, which often depend on local expertise and resources that are available at each institution (4).
However, there is limited data on the most accurate methods for management of gastrointestinal and pancreatic neuroendocrine tumors (PNETs) in patients with MEN1, which account for a significant proportion of MEN1-related morbidity and mortality (2,5).
Furthermore, with the current screening protocols pancreatico-duodenal NETs are discovered very early in the disease process. Many small, less than 1 cm pancreatico-duodenal tumors are discovered in patients with MEN1, with the controversy as to surgically treat these small lesions discovered during routine follow-up and when best to intervene. Here we present a review of the current literature on the management of PNETs.
Clinical presentation and diagnosis of PNETs in patients with MEN1
The diagnosis of MEN1 is made based on (I) the presence of primary hyperparathyroidism combined with anterior pituitary tumor and/or gastrointestinal and PNETs; (II) a diagnosis of primary hyperparathyroidism combined with a diagnosis of MEN1 in at least one first-degree relative; or (III) a positive germline mutation in the MEN1 gene. All patients should undergo screening and surveillance tests for other manifestations of MEN1 as per published guidelines (Table 1) (4,6).
Table 1. Suggested biochemical and radiological screening in individuals at high risk of developing MEN1 associated PNETs.
| Pancreatic NET | Age to begin (year) | Biochemical test (plasma or serum) annually | Imaging test (time interval) |
|---|---|---|---|
| Gastrinoma | 20 | Gastrin (gastric pH) | None |
| Insulinoma | 5 | Fasting glucose, insulin | None |
| Other pancreatic NET | <10 | Chromogranin A, pancreatic polypeptide, glucagon, VIP | MRI, CT, or EUS (annually) |
MEN1, multiple endocrine neoplasia type 1; PNETs, pancreatic neuroendocrine tumors; EUS, endoscopic ultrasound; VIP, vasoactive intestinal peptide. Adapted from Thakker et al. (4).
Patients with MEN1 have follow-up with laboratory and imaging evaluations, as well as endoscopy and pH measurement to screen for primary hyperparathyroidism, kidney stones, Zollinger-Ellisson Syndrome (ZES), gastric and duodenal ulcers, bone density loss (DEXA scan), pituitary tumors, pancreatic and gastrointestinal NETs.
The incidence of PNETs in patients with MEN1 varies between 30-80% in different studies (6-9). Those tumors are classified as hormonally active or non-functional (NF) and either secrete excessive amounts of hormones (gastrin, insulin or vasoactive intestinal peptide) which are associated with specific clinical syndromes; or some tumors may not be associated with clinical symptoms (such as pancreatic polypeptide) or entirely be non-secretory (NF). Usually PNETs have an earlier onset in patients with MEN1 than in patients without MEN1 (3,10) and their hallmark is multiplicity in contrast to solitary sporadic PNETs. They are classified into three grades based on proliferation rate according to the European Neuroendocrine Tumor Society and the World Health Organization grading system (11,12).
Gastrin-secreting tumors (gastrinomas) are associated with increased gastric acid production and recurrent peptic ulceration. These tumors represent more than 50% of all neuroendocrine pancreatico-duodenal tumors in patients with MEN1 (13,14). Gastrinomas frequently appear as small (<5 mm), multiple nodular lesions arising from the duodenal mucosa. They grow slowly but can metastasize to the peripancreatic lymph nodes and the liver. They are rarely found to arise from the pancreas. The diagnosis is established by demonstrating an increased fasting serum gastrin concentration associated with increased gastric acid secretion (gastric pH). Ultrasonography, endoscopic ultrasonography (EUS), CT, MRI, selective arterial angiography and somatostatin receptor scintigraphy are all used for localization of tumors (15).
Insulinomas are the most common cause of endogenous hyperinsulinemic hypoglycemia in non-diabetic adult patients, with an incidence of 1-3 per million per year (16). They are most commonly localized in the pancreas and approximately 4-6% are associated with MEN1; 85% are solitary and 6-13% are multiple (16,17). More than 90% of insulinomas are benign. Patients present clinically with the Whipple triad: symptoms of hypoglycemia, low blood glucose (<40-50 mg/dL), and relief of symptoms after the administration of glucose (18). The gold standard for the diagnosis of insulinoma is a 48-hour supervised fast with negative levels for beta-hydroxybutyrate and sulfonylurea and using a point of discrimination for proinsulin of ≥22 pmol/L (19). Due to their small size (82% <2 cm, 47% <1 cm), insulinomas are difficult to localize (16). US, CT and MRI are widely used and EUS is positive in 70-95% of cases (20). Selective angiography with intra-arterial calcium stimulation and hepatic venous sampling for insulin levels localizes >80% of insulinomas (21,22), and with intraoperative US improves success rate of surgery.
Glucagonomas occur in fewer than 3% of patients with MEN1. The clinical manifestations are a skin rash (necrolytic migratory erythema), weight loss and anemia. Their most frequent location is the tail of the pancreas and they are often metastatic at the time of diagnosis.
Tumors secreting vasoactive intestinal peptide are rare in MEN1 and patients develop watery diarrhea, hypokalemia and achlorhydria. The diagnosis is established when elevated plasma VIP levels are associated with excess stool volume of 0.5-1 liters/day during a fast. VIPomas are also frequently located in the pancreatic tail.
The natural history of NF-PNETs in patients with MEN1 is not well established. NF-PNETs are not associated with any clinical syndrome and include cases with elevated pancreatic polypeptide or glucagon but no symptoms. With increasing sensitivities of radiological screening, especially using EUS, more small NF-PNETs are currently diagnosed (Figure 1). They are usually indolent and demonstrate slow growth, with a doubling time of 5-10 years (23). In a study using EUS for detection, up to 55% of patients with MEN1 presented with NF-PNETs (8). Estimated 10-year survival rates of 23-62% have been reported (24,25), whereas in a more recent study, this was as high as 100% (26).
Figure 1.
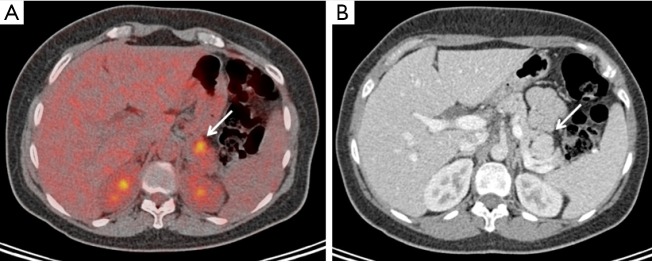
Index case of the family; a 42-year-old female with MEN1 who has a pituitary macroadenoma with acromegaly, primary hyperparathyroidism, hyperinsulinism and multiple PNETs. On imaging found to have a FDG-PET positive tail lesion (A), SUV 5.5; on pathology 2.7 cm, WHO grade 2, not staining for insulin or glucagon) with corresponding CT hyper-enhancement (B). MEN1, multiple endocrine neoplasia type 1; PNETs, pancreatic neuroendocrine tumors.
The identification of these NF-PNETs is of clinical importance because, malignant PNETs are reported to be the most common cause of death in patients with MEN1 (3,5), second, they seem to be the most common enteropancreatic NET associated with MEN1 and associated with worse prognosis than other functioning tumors such as insulinoma and gastrinoma (8). Third, the absence of clinical and biochemical abnormalities may delay diagnosis. Therefore the radiological screening for NF-PNETs in MEN1 should begin by the age of 10 years (Table 1) (4).
Management of PNETs in patients with MEN1
There is some controversy concerning the management of MEN1 associated gastrinoma. Medical management is aimed at reducing basal acid output. The goal of surgery in patients with MEN1-associated gastrinomas is to reduce the risk of distant metastases and improve survival. Their prognosis has been associated with tumor size and the presence of hepatic metastases (27). The treatment for non-metastatic gastrinoma located in the pancreas is surgical excision, because disease-related survival in patients with tumors larger than 2 cm is improved after surgery (13). However, most gastrinomas are multiple and occur within the duodenum and most centers undertake a nonsurgical management unless the gastrinomas are pancreatic and/or larger than 2 cm, in which case, surgery is recommended. Other groups recommend a more aggressive approach if the biochemical diagnosis is unequivocal and distant metastases are absent in order to prevent metastatic spread (28-30).
The situation for other secreting tumors is well defined, as most authors agree to recommend resection for insulinoma, VIPoma, glucagonoma and somatostatinoma. The main controversy, however, remains in the management of NF-PNETs, as some authors recommend an aggressive resection as soon as a tumor is identified and other authors recommend a more conservative management of small tumors (<1, <1.5 or <2 cm). The goal of treatment is to reduce morbidity and mortality due to metastatic disease while preserving as much pancreatic tissue as possible and avoiding complications of surgical intervention. This is especially true in patients with MEN1, in whom tumors are very likely to recur in the remnant pancreatic tissue (7).
Triponez et al. (31) have shown in their analysis of the French “Groupe d’ Etude des Tumeurs Endocrines” (GTE) database, that the risk to develop lymph nodes and/or distant metastasis correlates with the size of the primary tumor and that the risk of death was low for patients with MEN1 with small (<2 cm) NF-PNET. In this study, only 5 (7.7%) of 65 patients with NF-PNET <2 cm had synchronous lymph nodes or distant metastases and 2 (3.0%) of the patients who underwent surgery died of the disease. Moreover, the overall survival of patients with MEN1 and NF-PNET <2 cm was similar to the survival of patients with MEN1 who had no pancreatico-duodenal involvement (31). Because of these favorable results, they have proposed a conservative attitude for patients with MEN1 and NF-PNET <2 cm without other aggressive features. In this largest reported series on MEN1 associated NF-PNETs (n=108), distant metastases were seen in 19% of patients and disease-specific survival was 91% after a mean follow-up of 4 years (3). In other series, distant metastases were reported in about 6-22% (26,29).
However, there are limitations to these studies. There was a short follow-up time, particularly in the group of patients who were followed conservatively and there was low detection of metastases with imaging studies (EUS, CT or MRI) as compared to surgery and pathological evaluation.
Therefore, the same group performed a 10-year follow-up study: in 46 patients with NF-PNETs <2 cm without surgical treatment that were followed over 10 years, 28 displayed stable disease, 7 showed increase in size, 7 developed a functional syndrome, 1 developed a liver metastasis, 1 patient died due to metastatic NF-PNETs, 1 due to other causes, and 1 was lost to follow-up (Triponez F, Goudet P, AFCE, & GTE unpublished observations presented at WorldMEN congress, Vienna, Austria, 2014).
To date, the recommendation for surgical resection in NF-PNETs has been based on tumor size, as a higher rate of metastases was found in patients with larger tumors (3,25,31). One study reported the presence of synchronous metastases in 43% of patients with NF-PNET of more than 3 cm, in 18% of patients with tumors between 2.1-3.0 cm, and only in 4% of patients with tumors less than 1 cm had metastases (3). On the other hand, there are studies that have not confirmed this association (29,32). Bartsch et al. (29) showed no correlation between tumor size and metastatic potential, neither for gastrinomas nor for the NF-PNETs. Therefore a consensus for the indications for surgery has not yet been established.
Pancreatico-duodenal surgery can be successful in 80% of patients. Some patients however develop complications that include diabetes mellitus, steatorrhea, early and late dumping syndromes (7). Moreover, the morbidity and mortality of pancreatic surgery is significant, even when the procedure is performed in experienced centers (3,33).
There are currently several different recommendations for the management of MEN1 patients with small NF-PNETs. The Clinical Practice Guidelines for MEN1 by Thakker et al. (4) suggest to consider surgical resection for NF-PNET that are larger than 1 cm in size, like the Uppsala group (34), the Marburg group (29) and the MEN consortium in Japan (35). The National Comprehensive Cancer Center in the USA suggests a more conservative approach for tumors 1-2 cm (www.nccn.org, version 2.2014) and the GTE also suggests a conservative management of NF-PNETs <2 cm if there are no signs of aggressiveness such as rapid progression on imaging studies (31).
In conclusion, for the management of small MEN1 associated NF-PNETs, all authors currently agree that tumors <1 cm can/should be followed conservatively and all authors agree that tumors >2 cm should be resected. The management of tumors 1-2 cm of size remains a matter of debate; either resection or follow-up seems to be a safe management option for patients with MEN1.
The most important adverse prognostic factor for survival in patients with MEN1 related PNETs is the presence of liver metastases and other distant disease (2,3,25). Non surgical management of patients with advanced well-differentiated PNETs involves inhibitors of tyrosine kinase receptors (TKI) and of the mammalian target of rapamycin (mTOR), and has been reported to be effective (36,37). These two studies included mostly non-MEN1 patients, and their results showed an increased overall survival and doubling of median progression-free survival when compared to placebo.
In conclusion, PNETs are frequent and can be NF in patients with MEN1. Their identification is of clinical importance because malignant PNETs are reported to be the most common cause of death in patients with MEN1. In the longest follow-up study over 10 years, 17% of patients showed an increase in size of the lesions and 2% died of metastatic disease of NF-PNETs. To date, the recommendation for surgical resection in NF-PNETs is based on tumor size, as a higher rate of metastases was found in patients with larger tumors. Surgical resection should be performed for lesions >2 cm and is recommended for those lesions that have significant growth, such as a doubling of tumor size over a 3- to 6-month interval for tumors of any size. Further studies are needed to determine the appropriate management for tumors that are sized between 1 and 2 cm.
Acknowledgements
Disclosure: The authors declare no conflict of interest.
References
- 1.Pieterman CR, Schreinemakers JM, Koppeschaar HP, et al. Multiple endocrine neoplasia type 1 (MEN1): its manifestations and effect of genetic screening on clinical outcome. Clin Endocrinol (Oxf) 2009;70:575-81. [DOI] [PubMed] [Google Scholar]
- 2.Ito T, Igarashi H, Uehara H, et al. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: a prospective study: comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine (Baltimore) 2013;92:135-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Triponez F, Dosseh D, Goudet P, et al. Epidemiology data on 108 MEN 1 patients from the GTE with isolated nonfunctioning tumors of the pancreas. Ann Surg 2006;243:265-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97:2990-3011. [DOI] [PubMed] [Google Scholar]
- 5.Goudet P, Murat A, Binquet C, et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d'Etude des Tumeurs Endocrines) cohort study among 758 patients. World J Surg 2010;34:249-55. [DOI] [PubMed] [Google Scholar]
- 6.Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001;86:5658-71. [DOI] [PubMed] [Google Scholar]
- 7.Dralle H, Krohn SL, Karges W, et al. Surgery of resectable nonfunctioning neuroendocrine pancreatic tumors. World J Surg 2004;28:1248-60. [DOI] [PubMed] [Google Scholar]
- 8.Thomas-Marques L, Murat A, Delemer B, et al. Prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. Am J Gastroenterol 2006;101:266-73. [DOI] [PubMed] [Google Scholar]
- 9.Goudet P, Bonithon-Kopp C, Murat A, et al. Gender-related differences in MEN1 lesion occurrence and diagnosis: a cohort study of 734 cases from the Groupe d'etude des Tumeurs Endocrines. Eur J Endocrinol 2011;165:97-105. [DOI] [PubMed] [Google Scholar]
- 10.Machens A, Schaaf L, Karges W, et al. Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): a multicentre study of 258 gene carriers. Clin Endocrinol (Oxf) 2007;67:613-22. [DOI] [PubMed] [Google Scholar]
- 11.Bosman F, Carneiro F, Hruban R, et al. eds. WHO Classification of Tumors of the Digestive System. 4th ed. Lyon, France: International Agency for Research on Cancer, 2010. [Google Scholar]
- 12.Rindi G, Arnold R, Bosman FT, et al. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: Bosman FT, Carneiro F, Hruban RH, et al. eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: International Agency for Research on Cancer (IARC); 2010:13-4. [Google Scholar]
- 13.Norton JA, Jensen RT. Role of surgery in Zollinger-Ellison syndrome. J Am Coll Surg 2007;205:S34-7. [DOI] [PubMed] [Google Scholar]
- 14.Gibril F, Schumann M, Pace A, et al. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore) 2004;83:43-83. [DOI] [PubMed] [Google Scholar]
- 15.Imamura M, Komoto I, Ota S, et al. Biochemically curative surgery for gastrinoma in multiple endocrine neoplasia type 1 patients. World J Gastroenterol 2011;17:1343-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Service FJ, McMahon MM, O'Brien PC, et al. Functioning insulinoma--incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc 1991;66:711-9. [DOI] [PubMed] [Google Scholar]
- 17.Grant CS. Insulinoma. Best Pract Res Clin Gastroenterol 2005;19:783-98. [DOI] [PubMed] [Google Scholar]
- 18.Whipple AO, Frantz VK. Adenoma of islet cells with hyperinsulinism: a review. Ann Surg 1935;101:1299-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guettier JM, Lungu A, Goodling A, et al. The role of proinsulin and insulin in the diagnosis of insulinoma: a critical evaluation of the Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2013;98:4752-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLean A.Endoscopic ultrasound in the detection of pancreatic islet cell tumours. Cancer Imaging 2004;4:84-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown CK, Bartlett DL, Doppman JL, et al. Intraarterial calcium stimulation and intraoperative ultrasonography in the localization and resection of insulinomas. Surgery 1997;122:1189-93; discussion 1193-4. [DOI] [PubMed] [Google Scholar]
- 22.Guettier JM, Kam A, Chang R, et al. Localization of insulinomas to regions of the pancreas by intraarterial calcium stimulation: the NIH experience. J Clin Endocrinol Metab 2009;94:1074-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kann PH, Balakina E, Ivan D, et al. Natural course of small, asymptomatic neuroendocrine pancreatic tumours in multiple endocrine neoplasia type 1: an endoscopic ultrasound imaging study. Endocr Relat Cancer 2006;13:1195-202. [DOI] [PubMed] [Google Scholar]
- 24.Lévy-Bohbot N, Merle C, Goudet P, et al. Prevalence, characteristics and prognosis of MEN 1-associated glucagonomas, VIPomas, and somatostatinomas: study from the GTE (Groupe des Tumeurs Endocrines) registry. Gastroenterol Clin Biol 2004;28:1075-81. [DOI] [PubMed] [Google Scholar]
- 25.Kouvaraki MA, Shapiro SE, Cote GJ, et al. Management of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. World J Surg 2006;30:643-53. [DOI] [PubMed] [Google Scholar]
- 26.Lopez CL, Waldmann J, Fendrich V, et al. Long-term results of surgery for pancreatic neuroendocrine neoplasms in patients with MEN1. Langenbecks Arch Surg 2011;396:1187-96. [DOI] [PubMed] [Google Scholar]
- 27.Norton JA. Surgical treatment and prognosis of gastrinoma. Best Pract Res Clin Gastroenterol 2005;19:799-805. [DOI] [PubMed] [Google Scholar]
- 28.Bartsch DK, Langer P, Wild A, et al. Pancreaticoduodenal endocrine tumors in multiple endocrine neoplasia type 1: surgery or surveillance? Surgery 2000;128:958-66. [DOI] [PubMed] [Google Scholar]
- 29.Bartsch DK, Fendrich V, Langer P, et al. Outcome of duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Ann Surg 2005;242:757-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez CL, Falconi M, Waldmann J, et al. Partial pancreaticoduodenectomy can provide cure for duodenal gastrinoma associated with multiple endocrine neoplasia type 1. Ann Surg 2013;257:308-14. [DOI] [PubMed] [Google Scholar]
- 31.Triponez F, Goudet P, Dosseh D, et al. Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J Surg 2006;30:654-62; discussion 663-4. [DOI] [PubMed] [Google Scholar]
- 32.Lowney JK, Frisella MM, Lairmore TC, et al. Pancreatic islet cell tumor metastasis in multiple endocrine neoplasia type 1: correlation with primary tumor size. Surgery 1998;124:1043-8. [DOI] [PubMed] [Google Scholar]
- 33.Birkmeyer JD, Siewers AE, Finlayson EV, et al. Hospital volume and surgical mortality in the United States. N Engl J Med 2002;346:1128-37. [DOI] [PubMed] [Google Scholar]
- 34.Akerström G, Stålberg P, Hellman P.Surgical management of pancreatico-duodenal tumors in multiple endocrine neoplasia syndrome type 1. Clinics (Sao Paulo) 2012;67Suppl 1:173-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanazaki K, Sakurai A, Munekage M, et al. Surgery for a gastroenteropancreatic neuroendocrine tumor (GEPNET) in multiple endocrine neoplasia type 1. Surg Today 2013;43:229-36. [DOI] [PubMed] [Google Scholar]
- 36.Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011;364:514-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 2011;364:501-13. [DOI] [PubMed] [Google Scholar]