

Abstract
Although low-dose radiation (LDR) regulates a wide range of biological processes, limited information is available on the effects of LDR on the chondrocyte phenotype. Here, we found that LDR, at doses of 0.5–2 centiGray (cGy), inhibited interleukin (IL)-1β-induced chondrocyte destruction without causing side effects, such as cell death and senescence. IL-1β treatment induced an increase in the expression of α-, β-, and γ-catenin proteins in chondrocytes via Akt signaling, thereby promoting dedifferentiation through catenin-dependent suppression of Sox-9 transcription factor expression and induction of inflammation through activation of the NF-κB pathway. Notably, LDR blocked cartilage disorders by inhibiting IL-1β-induced catenin signaling and subsequent catenin-dependent suppression of the Sox-9 pathway and activation of the NF-κB pathway, without directly altering catenin expression. LDR also inhibited chondrocyte destruction through the catenin pathway induced by epidermal growth factor, phorbol 12-myristate 13-acetate, and retinoic acid. Collectively, these results identify the molecular mechanisms by which LDR suppresses pathophysiological processes and establish LDR as a potentially valuable therapeutic tool for patients with cytokine- or soluble factors-mediated cartilage disorders.
Keywords: catenin, chondrocytes, dedifferentiation, inflammation, low dose radiation
Introduction
Although even small doses of radiation have potentially harmful effects in humans, low-dose radiation (LDR), which incurs effects at the cell level, also exerts positive effects through modulation of DNA damage, longevity, and immunological responses; thus, its net effect remains a matter of controversy 1. For example, exposure of human G(0) peripheral blood lymphocytes to LDR induces mutation of the hypoxanthine-guanine phosphoribosyltransferase gene 2, and the carcinogenesis resulting from this LDR-induced DNA mutation is considered the most serious of the possible injurious effects of LDR 3. In contrast, studies have shown that LDR protects against the induction of mutations in both in vitro and in vivo models 4,5. Moreover, LDR can modify the expression of genes associated with diverse cellular functions and as such may produce beneficial effects 3. Although LDR can accelerate premature senescence in human umbilical vein endothelial cells 6, it can also extend longevity in mice 7. The potential beneficial effects of LDR, encompassed by the concept of hormesis, are exemplified by enhanced immune cell activation and increased lymphocyte transformation, which are the presumptive molecular pathways and cellular components that contribute to the anti-inflammatory effects of LDR 8.
Tests of LDR on osteoarthritis, first carried out in rabbits in the early 1930s, demonstrated improvement in joint swelling and pain but revealed no effect on degenerative changes. A subsequent study reported that LDR reduced arthritis symptoms induced by injection of granugenol in rabbit knees via inhibition of inflammatory responses 9. In addition, fractions of LDR in in vivo models of experimentally induced arthritis significantly reduced cartilage degradation, inflammatory joint swelling, and the volume of synovial fluid 10,11. To date, no clear conclusions have been drawn from published studies with regard to the molecular mechanism underlying the anti-inflammatory effect of LDR. Recent studies have established the molecular events that follow irradiation, identifying changes in the expression of immune system effectors, including cytokines, such as interleukin-1 (IL-1), IL-8, and tumor necrosis factor-α; immunoglobulin superfamily members, such as intercellular adhesion molecules and vascular cellular adhesion molecules; and enzymes, such as inducible nitric oxide synthase and heme oxygenase-1 8,12. Although there has been increasing interest in the molecular mechanisms engaged by low-intensity, pulsed ultrasound, a focused research effort is required to explore additional contributing factors involved in regulating the cellular phenotype of chondrocytes exposed to LDR.
In this work, we demonstrated that LDR, at doses of 0.5–1 centiGray (cGy), suppressed IL-1β-induced dedifferentiation and inflammation of chondrocytes, without producing any side effects. β-Catenin and γ-catenin are essential for IL-1β-induced cartilage destruction. LDR acted by suppressing signaling pathways both upstream and downstream of catenin, blocking dedifferentiation by preventing IL-1β-induced Sox-9 suppression, and inhibiting inflammation by opposing IL-1β-induced activation of NF-κB. These anti-disease effects establish LDR as a potentially valuable therapeutic tool for patients with cytokine-mediated cartilage disorders.
Materials and Methods
Cell Culture and Treatment
Primary cultured articular chondrocytes were maintained in Dulbecco's modified Eagle's medium, as described previously 13. Cells were irradiated using a 137Cs-ray source (Korea Institute of Radiological and Medical Sciences, Seoul, Korea) at a dose rate of 0.67 cGy/min. Cells were treated with 10 ng/mL IL-1β (Calbiochem, Darmstadt, Germany), 10 ng/mL epidermal growth factor (EGF) (Invitrogen, Carlsbad, CA), 10 nM phorbol 12-myristate 13-acetate (PMA) (Sigma, St. Louis, MO), and 1 μM retinoic acid (RA) (Sigma) to induce cartilage destruction-associated processes. LY294002 (Sigma), BAY 11-7082 (BAY) (Calbiochem), and Triciribine (Millipore, Billerica, MA) were used for inhibiting phosphatidylinositol 3-kinase (PI3K), NF-κB, and Akt, respectively.
Cell Proliferation and Morphology
Chondrocytes were plated on culture dishes at a density of 5 × 104 cells/cm2 and treated with radiation according to the designated experimental conditions. Cell proliferation was determined by directly counting surviving cells using a hemocytometer, and alterations in cellular morphology were observed by microscopy.
Cell Death Assay
Chondrocytes were plated on 35-mm dishes at 4 × 105 cells/plate and exposed to different doses of radiation for 48 h. Cells were incubated with propidium iodide (2.5 mg/mL) for 5 min at room temperature and analyzed with a FACScan flow cytometer (Becton Dickson, Franklin Lakes, NJ).
Senescence-Associated β-Galactosidase Staining
Detection of senescence-associated β-galactosidase (SA-β-gal) activity at pH 6 was performed as previously described 13. Briefly, radiation-treated or untreated chondrocytes were fixed with a 3.7% formaldehyde solution for 10 min. The fixative was removed by washing twice with phosphate-buffered saline, and cells were incubated with SA-β-gal staining solution (1 mg/mL of 5-bromo-4-chloro-3-indolyl β-d-galactoside [Promega, Madison, WI], 40 mM citric acid/sodium phosphate buffer pH 6.0, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, 2 mM MgCl2). Staining was evaluated after a 16-h incubation at 37 °C in a CO2-free atmosphere.
Reverse Transcription-Polymerase Chain Reaction
Reverse transcription-polymerase chain reaction (RT-PCR) was performed using primer pairs for type I collagen, type II collagen, Sox-9, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), as previously described 14. RT-PCR for COX-2 was performed at an annealing temperature of 55 °C (28 cycles) using the primer pair 5′-TCAGCCACCCAGCAAATCCT-3′ (sense) and 5′-GTCATCTGGATCTCAGCACG-3′ (antisense), which yielded a 290-bp product. Quantitative real-time PCR for COX-2 was performed in triplicate on a Chromo4 thermocycler with SYBR Premix Ex Taq (Takara Bio, Shiga, Japan). The amplification signal of the target gene was normalized to that of GAPDH.
Western Blot Analysis
Western blot analyses were performed as described previously 14 using primary antibodies against type II collagen (Chemicon, Temecula, CA); Sox-9, p53, inhibitor protein κB (I-κB), GFP, FLAG, phospho-ERK (Thr202/Tyr204) (Santa Cruz Biotechnology, Santa Cruz, CA); COX-2 (Cayman Chemical, Ann Arbor, MI); α-, β-, γ-catenin, extracellular signal-regulated kinase (ERK) (BD Transduction Laboratories, San Jose, CA); p21, GSK3β (BD PharMingen, San Diego, CA); and phospho-p53 (Ser15), phospho-p38 (Thr180/Tyr182), p38, phospho-JNK (Thr183/Tyr185), c-Jun N-terminal kinase (JNK), phospho-Akt (Ser473), Akt, phosphor-GSK3α/β (Ser21/9) (Cell Signaling Technology, Beverly, MA). β-Actin (Sigma) was used as a loading control.
Immunofluorescence Confocal Microscopy
For immunostaining, cells were incubated for 1 h with 10 µg/mL of the following primary antibodies: γH2AX (Cell Signaling Technology), α-, β-, and γ-catenin (BD Transduction Laboratories). Cells were incubated for an additional 1 h with rhodamine- or fluorescein isothiocyanate-conjugated secondary antibodies (Invitrogen), as described previously 14.
Ectopic Overexpression of Catenins
A GFP-tagged, S83A point mutant of α-catenin was generated by site-directed mutagenesis (Promega), according to the manufacturer's instructions, using wild-type α-catenin as a template 15. Constructs for nonubiquitinatable FLAG-tagged S33A β-catenin (Cat. #19286) and wild-type γ-catenin (Cat. #16827) were purchased from Addgene (Cambridge, MA). Chondrocytes were transfected with expression vectors using Lipofectamine PLUS (Invitrogen) following the manufacturer's recommended procedure.
Reporter Gene Assay
Sox-9 or NF-κB transcriptional activity was directly examined by reporter gene assay as previously described 15. NF-κB activity was also determined indirectly by analyzing the degradation of I-κB using Western blot analysis.
Immunoprecipitation
Chondrocytes were lysed by incubating in lysis buffer for 15 min as described previously 13. Samples were diluted to 500 μg of protein in 600 μL of buffer and precleared by incubating for 1 h at 4 °C with 40 μL of a 1:1 slurry of protein A-Sepharose beads (GE Healthcare, Piscataway, NJ). After a brief centrifugation step to remove precleared beads, 1 μg of antibody against β-catenin was added to each sample and incubated on a rocking platform at 4 °C overnight. Immune complexes were precipitated by incubating with 40 μL of protein A-Sepharose beads at 4°C for 2 h. Associated GFP or α-catenin in samples was examined by Western blot analysis.
Statistical Analysis
All data are expressed as means ± standard deviations. Statistical evaluations were conducted using a one-way analysis of variance. A P value < 0.05 was considered significant.
Results
LDR does not Induce the Cellular Dysfunction of Articular Chondrocytes
High-dose radiation (HDR), ranging from 3 Gy to 10 Gy, induces pathological dysregulation of chondrocytes 13. To examine whether LDR negatively affects the chondrocyte phenotype, we treated chondrocytes with radiation doses ranging from 0 to 2 cGy. None of these doses affected the protein levels of type II collagen or Sox-9, two cartilage-specific differentiation markers (Fig. 1A, top). Moreover, the transcriptional activity of Sox-9 was unchanged in LDR-treated cells compared to untreated controls (Fig. 1A, bottom). LDR also did not alter the expression of COX-2 protein, a primary mediator of cartilage inflammation, or I-κB protein, an inhibitor of the NF-κB transcription factor (Fig. 1B, top). Consistent with the absence of a change in I-κB, LDR did not induce NF-κB transcriptional activity, which is involved in COX-2 regulation (Fig. 1B, bottom). Unlike HDR (6 Gy), which induced cellular senescence of chondrocytes, as evidenced by the development of a large, flattened morphology (Fig. 1C, top) and SA-β-gal positive staining (Fig. 1C, bottom), LDR did not promote cellular senescence. Consistent with this, p53 and p21, two biochemical markers of senescence, were unchanged in chondrocytes treated with 2 cGy LDR (Fig. 1D, top), whereas p53 activation and p21 expression were markedly induced in cells exposed to 6 Gy HDR (Fig. 1D, bottom). Finally, proliferation curves for chondrocytes treated with 2 cGy LDR paralleled those of untreated control chondrocytes (Fig. 1E, left) without any induction of cell death (Fig. 1E, right). In contrast, treatment with 6 Gy HDR delayed the proliferation of chondrocytes (Fig. 1E, left). Immunofluorescence staining showed that the number of histone variant γ-H2AX foci, a marker of DNA damage, reached a peak at 30 min and then slightly declined at 3 h in chondrocytes exposed to 6 Gy HDR, whereas no foci were detected in chondrocytes exposed to 2 cGy (Fig. 1F). Taken together, these results suggest that LDR has no pathological effects on primary cultured articular chondrocytes.
Figure 1.

Effect of LDR on the chondrocyte phenotype. Chondrocytes were treated with different doses of radiation for the indicated periods in each experimental condition. Levels of differentiation- (A, top) and inflammation-associated proteins (B, top) were determined by Western blotting. Transcriptional activities of Sox-9 (A, bottom) and NF-κB (B, bottom) were determined by reporter gene assay. Data are expressed as means ± SDs (× denotes no significant difference compared with untreated controls). Changes in cell morphology were observed by light microscopy (C, top; scale bar: 1 mm), and cellular senescence was evaluated by assessing SA-β-gal-positivity (blue, C, bottom; scale bar: 1 mm). Levels of senescence-associated proteins were determined by Western blotting (D). Total cell number was quantified by counting the surviving cells using a trypan blue solution (E, left), and cell death was determined by FACS analysis (E, right). Data are expressed as means ± SDs (**P < 0.005, ***P < 0.0005, × denotes no significant difference compared with untreated controls). Phosphorylation of H2AX was determined by confocal fluorescence microscopy, and cells were identified by 4′,6-diamidino-2-phenylindole staining of nuclei (F, scale bar: 40 μm).
LDR Suppresses IL-1β-Induced Dedifferentiation and Inflammation of Articular Chondrocytes
RT-PCR analyses showed that IL-1β treatment of chondrocytes induced fibroblastic type I collagen expression and suppressed expression of type II collagen and Sox-9, a major transcriptional regulator of type II collagen (Fig. 2A, top). Sox-9 transcriptional activity was reduced by approximately 32% and 60% at 24 and 48 h after IL-1β treatment, respectively, compared to control cells (Fig. 2A, middle). IL-1β treatment reduced the level of type II collagen and Sox-9 proteins (Fig. 2A, bottom), confirming that IL-1β promotes chondrocyte dedifferentiation. Notably, LDR at doses of both 0.5 and 1 cGy markedly recovered the levels of type I collagen, type II collagen, and Sox-9 transcripts in IL-1β-treated chondrocytes (Fig. 2B, top). A promoter reporter assay showed that 0.5 and 1 cGy LDR significantly enhanced Sox-9 transcriptional activity in IL-1β-treated chondrocytes by approximately 2.3-fold and 2.9-fold, respectively, compared to cells treated with IL-1β alone (Fig. 2B, middle). The IL-1β-induced reduction in type II collagen and Sox-9 protein levels was completely abrogated by LDR (Fig. 2B, bottom), suggesting that LDR is a strong inhibitor of IL-1β-induced chondrocyte dedifferentiation. IL-1β treatment profoundly induced the expression of COX-2, a target of the NF-κB pathway and a primary mediator of cartilage inflammation, inducing approximately 9.8-fold and 7.7-fold increases in COX-2 transcript levels at 24 and 48 h, respectively, compared to control cells (Fig. 2C, top). In support of this result, I-κB protein, an inhibitor of the NF-κB transcription factor, was degraded in IL-1β-treated chondrocytes (Fig. 2C, middle). Consistent with this, reporter assays revealed approximately 8.6-fold and 6.7-fold increases in NF-κB transcriptional activity at 24 and 48 h after IL-1β treatment, respectively, compared to control cells (Fig. 2C, bottom). Interestingly, LDR markedly recovered the levels of COX-2 transcripts in IL-1β-treated chondrocytes, decreasing mRNA levels by approximately 70% and 80% at doses of 0.5 and 1 cGy, respectively, compared to cells treated with IL-1β alone (Fig. 2D, top). This phenomenon was similar to the pattern of I-κB protein recovery observed under the same experimental conditions (Fig. 2D, middle). The increase in NF-κB transcriptional activity induced by IL-β treatment was largely eliminated by 0.5 and 1 cGy LDR, which reduced this activity by approximately 76% and 83%, respectively, compared to chondrocytes treated with IL-1β alone (Fig. 2D, bottom). Collectively, these results suggest that LDR is an effective tool for inhibiting IL-1β-induced inflammation of chondrocytes.
Figure 2.
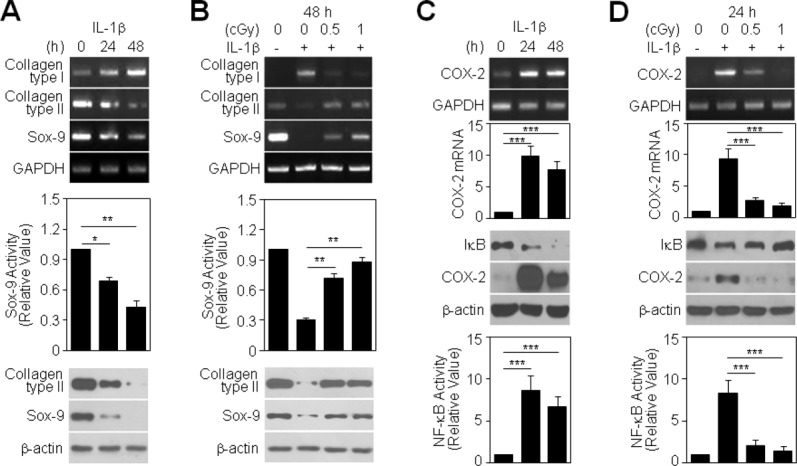
Effect of LDR on IL-1β-induced chondrocyte destruction. Chondrocytes were treated with 10 ng/mL IL-1β alone (A and C) or in combination with 0.5 or 1 cGy of LDR (B and D) for the indicated periods. Transcript levels of dedifferentiation-associated genes were determined by conventional PCR using GAPDH as a loading control (A and B, top). Sox-9 transcriptional activity was determined by reporter gene assay. Data are expressed as means ± SDs (*P < 0.05 and **P < 0.005 compared with untreated controls in A, middle; **P < 0.005 compared with cells treated with IL-1β alone in B, middle). Levels of type II collagen and Sox-9 proteins were detected by Western blotting (A and B, bottom). Levels of COX-2 transcripts were determined by conventional and quantitative real-time PCR using GAPDH as a control. Data are expressed as means ± SDs (***P < 0.0005 compared with untreated controls in C, top; ***P < 0.0005 compared with cells treated with IL-1β alone in D, top). Levels of I-κB and COX-2 proteins were detected by Western blotting (C and D, middle). NF-κB transcriptional activity was determined by reporter gene assay. Data are expressed as means ± SDs (***P < 0.0005 compared with untreated controls in C, bottom; ***P < 0.0005 compared with cells treated with IL-1β alone in D, bottom).
LDR Suppresses IL-1β-Induced PI3K/Akt Signaling in Articular Chondrocytes
To identify the mechanisms by which LDR modulates chondrocyte phenotype, we examined mitogen-activated protein kinase (MAPK) activation and Akt activation in IL-1β-treated chondrocytes before and after LDR exposure. All MAPK proteins, namely ERK, p38, and JNK, were activated by IL-1β treatment. LDR exposure did not affect IL-1β-induced activation of MAPKs (Fig. 3A), indicating that LDR acts through a MAPK signaling-independent pathway to modulate chondrocyte phenotype. Treatment of chondrocytes with IL-1β induced Akt phosphorylation and subsequently deactivated glycogen synthase kinase 3β (GSK3β), a substrate of Akt (Fig. 3B, top), whereas Triciribine, a specific inhibitor of the Akt signaling pathway, markedly attenuated the IL-1β-induced increase in Akt phosphorylation and thus reactivated GSK3β in chondrocytes (Fig. 3B, bottom). Interestingly, treatment with LY294002, another chemical inhibitor of PI3K/Akt signaling, led to recovery of type II collagen and Sox-9 expression, reduction of COX-2 expression, and inhibition of I-κB degradation in IL-1β-treated chondrocytes (Fig. 3C, top). Consistent with this, LY294002 significantly suppressed NF-κB transcriptional activity, decreasing NF-κB activity by approximately 48% and 68% at 10 and 20 μM, respectively, compared to chondrocytes treated with IL-1β alone (Fig. 3C, bottom). Notably, LY294002 treatment in IL-1β-treated chondrocytes led to downregulation of α-, β-, and γ-catenin protein levels induced by IL-1β (Fig. 3D). These results suggest that Akt activation is critically involved in IL-1β-induced chondrocyte disorders via catenin signaling. Therefore, we examined whether LDR modulates IL-1β-induced Akt activity and found that LDR at both 0.5 and 1 cGy suppressed Akt activation induced by IL-1β in chondrocytes (Fig. 3E). Taken together, our data indicate that LDR acts through blockade of the Akt signaling pathway, but not the MAPK pathway, to mitigate the IL-1β-induced destructive cellular phenotype of chondrocytes.
Figure 3.
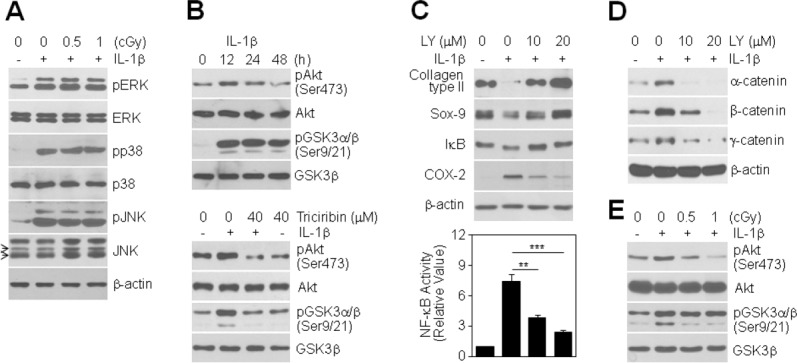
Involvement of PI3K/Akt signaling in LDR-induced inhibition of chondrocyte destruction. (A): Chondrocytes were treated with 0.5 or 1 cGy of LDR in the absence (−) or presence (+) of 10 ng/mL IL-1β for 20 min. Expression and phosphorylation status of ERK, p38, and JNK were determined by Western blotting. (B): Chondrocytes were treated with 10 ng/mL IL-1β for the indicated times (top), with or without pretreatment with 40 μM Triciribine for 1 h prior to IL-1β treatment (bottom). Expression and phosphorylation status of Akt and GSK3α/β were determined by Western blotting. (C, D): Chondrocytes were left untreated or were pretreated with 10 or 20 μM LY294002 for 1 h prior to treatment with 10 ng/mL IL-1β. After 48 h, the levels of type II collagen, Sox-9, I-κB, and COX-2 proteins were assessed by Western blotting (C, top). NF-κB transcriptional activity was determined by reporter gene assay. Data are expressed as means ± SDs (**P < 0.005 and ***P < 0.0005 compared to cells treated with IL-1β alone (C, bottom). Levels of catenin proteins were detected by Western blotting (D). (E): Chondrocytes were treated with 0.5 or 1 cGy of LDR for 12 h in the absence (−) or presence (+) of 10 ng/mL IL-1β. Expression and phosphorylation status of Akt and GSK3α/β were determined by Western blotting.
Catenin Signaling Mediates IL-1β-Induced Dedifferentiation and Inflammation of Articular Chondrocytes
We next examined downstream targets of Akt signaling, focusing on catenin proteins involved in the coordination of cell–cell adhesion and gene transcription. IL-1β treatment caused a time-dependent induction of α-, β-, and γ-catenin protein levels in chondrocytes (Fig. 4A). Ectopic overexpression of β-catenin binding-deficient mutant α-catenin (Supporting Information Fig. 1A), nonubiquitinatable mutant β-catenin, or wild-type γ-catenin in chondrocytes led to suppression of type II collagen and Sox-9 expression, induction of COX-2 expression, and degradation of I-κB protein in all experiments (Fig. 4B). Consistent with this, these maneuvers significantly suppressed Sox-9 and promoted NF-κB transcriptional activities; specifically, transfection of 3 μg of the above-mentioned α-, β-, and γ-catenin plasmids decreased Sox-9 activity by approximately 60%, 79%, and 63%, respectively, compared to the respective controls (Fig. 4C, left) and increased NF-κB activity by approximately 5.0-fold, 7.8-fold, and 6.0-fold, respectively (Fig. 4C, right). The combined effect of catenin overexpression and IL-1β treatment on chondrocyte dedifferentiation and inflammation, measured as suppression of type II collagen and Sox-9 protein expression, induction of I-κB protein degradation and promotion of COX-2 protein expression, was much greater than that produced by IL-1β alone (Fig. 4D, top). The greater expression of COX-2 under these combined treatment conditions was paralleled by a greater enhancement in NF-κB transcriptional activity; whereas IL-1β alone induced an approximately 8.5-fold increase in NF-κB activity compared to controls, when combined with overexpression of the indicated α-, β-, and γ-catenin forms, IL-1β increased NF-κB activity by approximately 12.4-fold, 15.5-fold, and 14.2-fold, respectively (Fig. 4D, bottom). These results suggest that IL-1β-induced catenin signaling has a pivotal role in promoting the destructive cellular phenotype of chondrocytes.
Figure 4.
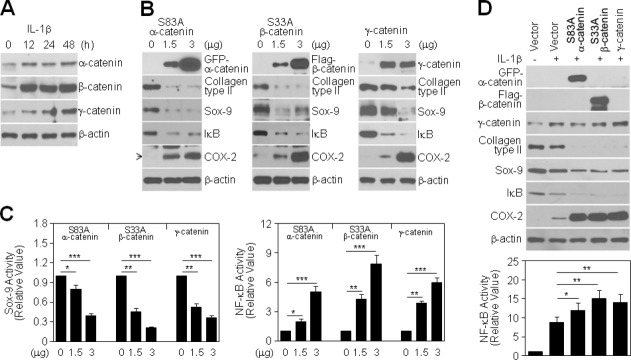
Role of IL-1β-induced catenin proteins in chondrocyte destruction. (A): Chondrocytes were treated with 10 ng/mL IL-1β for the indicated periods. Levels of catenin proteins were detected by Western blotting. (B, C): Chondrocytes were transiently transfected with GFP-tagged S83A α-catenin, FLAG-tagged S33A β-catenin, or wild-type γ-catenin, as indicated, for 48 h without (B) or with a Sox-9 (C, left) or NF-κB reporter gene (C, right). Levels of differentiation- and inflammation-associated proteins were determined by Western blotting (B), and Sox-9 or NF-κB transcriptional activity was determined by reporter gene assay. Data are expressed as means ± SDs (*P < 0.05, **P < 0.005, ***P < 0.0005 compared with untransfected controls) (C). (D): Chondrocytes were transiently transfected with 3 μg of each catenin plasmid, as indicated, for 24 h and then left untreated (−) or treated (+) with 10 ng/mL IL-1β for an additional 48 h. Levels of differentiation- and inflammation-associated proteins were determined by Western blotting (top), and NF-κB transcriptional activity 24 h after IL-1β treatment was determined by reporter gene assay. Data are expressed as means ± SDs (*P < 0.05 and **P < 0.005 compared with cells treated with IL-1β alone) (bottom).
LDR Suppresses Catenin-Induced Dedifferentiation and Inflammation of Primary Cultured Articular Chondrocytes
We next examined whether LDR, at doses of 0–2 cGy, directly modulates the expression of catenin proteins in chondrocytes. Although LDR alone did not alter the expression of catenin proteins (Fig. 5A), both 0.5 and 1 cGy LDR dramatically attenuated IL-1β-induced expression of all catenin proteins (Fig. 5B). Immunofluorescence analyses showed that treatment of chondrocytes with IL-1β significantly increased the levels of all of the tested catenin proteins in both the cytosol and nuclear regions compared to control cells. At a dose of 1 cGy, LDR reduced catenin expression in IL-1β-treated cells to basal levels (Fig. 5C), indicating that LDR inhibits the IL-1β-dependent post-translational stabilization of catenin. Finally, we examined whether LDR reverses the dedifferentiation and inflammatory response induced by overexpression of the above-mentioned α-, β-, and γ-catenin proteins in chondrocytes. Notably, LDR markedly restored the expression of type II collagen and Sox-9 proteins in catenin-overexpressing chondrocytes (Supporting Information Fig. 1B and Fig. 5D), indicating a reduced commitment to dedifferentiation. LDR also inhibited I-κB degradation and COX-2 expression under the same experimental conditions (Supporting Information Fig. 1B and Fig. 5D). Consistent with this, LDR dramatically increased in Sox-9 activity reduced by catenin proteins and decreased in NF-κB activity induced by catenin proteins in chondrocytes; at a dose of 1 cGy, LDR enhanced Sox-9 activity by approximately 2.2-fold, 2.7-fold, and 2.4-fold compared to that in cells transfected with the above-mentioned α-, β-, and γ-catenin constructs, respectively (Supporting Information Fig. 1C and Fig. 5E, left), and reduced NF-κB activity by approximately 74%, 78%, and 75%, respectively (Supporting Information Fig. 1C and Fig. 5E, right), indicating a reduced commitment to inflammation. To further confirm the role of NF-κB signaling in catenin-mediated induction of COX-2 expression, we treated catenin-overexpressing chondrocytes with the NF-κB inhibitor, BAY. Pretreatment with 5 or 10 μM BAY inhibited I-κB degradation in a dose-dependent manner and consistently reduced COX-2 expression compared to IL-1β-treated chondrocytes (Fig. 5F). Degradation of I-κB and induction of COX-2 in cells transfected with the above-mentioned α-, β-, and γ-catenin constructs were also reversed by BAY treatment (Fig. 5G), indicating that NF-κB signaling is downstream of the catenin pathway.
Figure 5.

Effect of LDR on IL-1β-induced expression of catenins and catenin-induced chondrocyte destruction. (A, B): Chondrocytes were treated with different doses of LDR for 48 h in the absence (A) or presence (B) of 10 ng/mL IL-1β. Levels of catenin proteins were determined by Western blotting. (C): Chondrocytes were left untreated or treated with 1 cGy radiation in the absence or presence of 10 ng/mL IL-1β for 48 h. Expression of each catenin was detected by confocal fluorescence microscopy (scale bar: 50 μm). (D, E): Chondrocytes were transfected with 3 μg FLAG-tagged S33A β-catenin or wild-type γ-catenin for 24 h and then exposed to different doses of LDR for an additional 48 h. Levels of differentiation- and inflammation-associated proteins were determined by Western blotting (D). Sox-9 (E, left) or NF-κB transcriptional activity (E, right) was determined by reporter gene assay. Data are expressed as means ± SDs (*P < 0.05, **P < 0.005, ***P < 0.0005 compared with cells treated with IL-1β alone) (E). (F): Cells were left untreated or were pretreated with 5 or 10 μM Bay 11-7082 for 1 h prior to treatment with 10 ng/mL IL-1β. After 48 h, the levels of I-κB and COX-2 proteins were determined by Western blotting. (G): Chondrocytes were transiently transfected with 3 μg of GFP-tagged S83A α-catenin (left), FLAG-tagged S33A β-catenin (middle), or wild-type γ-catenin (right), as indicated, for 24 h and then left untreated or treated with 5 or 10 μM Bay 11-7082 for an additional 24 h. The levels of I-κB and COX-2 proteins were determined by Western blotting.
LDR Suppresses EGF-, PMA-, and RA-Induced Dedifferentiation and Inflammation of Articular Chondrocytes
To further determine whether the inhibitory effects of LDR on chondrocyte disorders are limited to the response to IL-1β, we incubated cells with three additional agents EGF, PMA, and RA previously reported to induce dedifferentiation of chondrocytes 16. Similar to the dedifferentiation and inflammation of chondrocytes caused by IL-1β treatment, the pathophysiological events induced by treatment with EGF, PMA, or RA, exemplified by the suppression of type II collagen and Sox-9 protein expression and induction of COX-2 protein expression, were associated with increased expression of β- and γ-catenin (Fig. 6, top). Notably, LDR at doses of both 0.5 and 1 cGy dramatically attenuated EGF-, PMA-, and RA-induced expression of all catenin proteins and subsequently normalized the altered expression of type II collagen, Sox-9, and COX-2 proteins produced by treatment with these agents (Fig. 6, middle). Consistent with these phenomena, LDR markedly attenuated the EGF-, PMA-, and RA-induced increase in Akt phosphorylation and thus reactivated GSK3β in chondrocytes (Fig. 6, bottom). These results demonstrate that LDR exerts an important inhibitory effect against the loss of the chondrocyte phenotype induced by different soluble factors.
Figure 6.
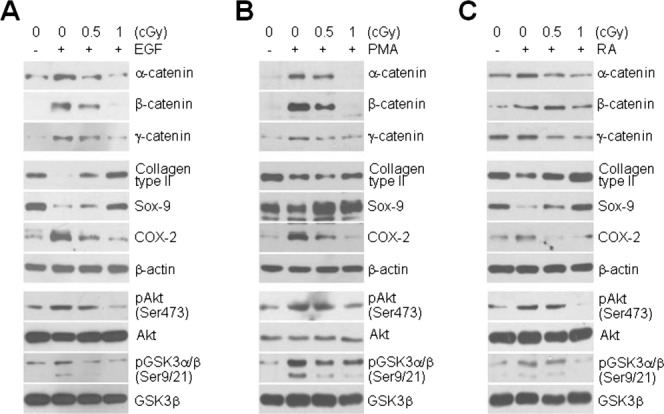
Inhibitory effect of LDR on chondrocyte destructions induced by soluble factors. Chondrocytes were incubated with (+) or without (−) 10 ng/mL EGF (A), 10 nM PMA (B), or 1 μM RA (C) for 2 h and then left untreated or exposed to 0.5 or 1 cGy of LDR for an additional 36 h. Levels of catenin proteins (top) and differentiation- and inflammation-associated proteins (middle) were determined by Western blotting. Expression and phosphorylation status of Akt and GSK3α/β were determined by Western blotting (bottom).
Discussion
HDR is not only used for cancer therapy but can also have injurious effects on normal cells, thereby causing unwanted side effects. By contrast, LDR can be largely beneficial to living organisms, as embodied by the concept of hormesis, and exemplified by improved immunological function and prolongation of lifespan 4,5,7,8, although LDR is still considered to induce slight damage at the cellular level 2,3,6. The effect of LDR (X-ray, γ-ray, and ultrasound) on health has been extensively examined in animal and human models, but almost no information is available for chondrocytes treated with γ-radiation. Our previous report has been shown that treatment of chondrocytes with 3–10 Gy radiations induced cellular senescence and dedifferentiation 13. Although 2 Gy radiation had no significant effect on both positive and negative functions in the maintenance of chondrocyte phenotype (data not shown), the present data showed that the cellular response by LDR was reversible compared to those by HDR. These indicate that the balancing between LDR and HDR is critical to decide their reversible effects to the chondrocytes. The identification and application of novel biomarkers is an unmet need in the therapeutic management of arthritis patients, prompting us to investigate catenin molecules that are significantly correlated with LDR-mediated modulation of the chondrocyte phenotype.
We previously demonstrated induction of α- and β-catenin proteins by IL-1β in chondrocytes 15. Here, we identified the γ-catenin signaling cascade as a target of IL-1β (Fig. 3). The primary role of catenin proteins is connecting cadherins to actin filaments in adhesion junctions of cells. In addition, β-catenin and γ-catenin can act as transcriptional coactivators, complexing with the lymphoid-enhancer-factor (Lef)/T cell-factor (Tcf) family of transcription factors to mediate Wnt signal transduction 17. We previously demonstrated that accumulated wild-type α-catenin inhibits the transcriptional activity of the β-catenin-Tcf/Lef complex via direct binding to β-catenin, thereby suppressing β-catenin-induced dedifferentiation of chondrocytes 15. We further confirmed this in this study using a β-catenin binding-deficient form of α-catenin containing a mutation in the β-catenin binding site (S83A). Overexpression of S83A α-catenin did not inhibit IL-1β- or β-catenin-induced phenotypic changes in chondrocytes, indicating that inhibition of β-catenin signaling by α-catenin occurs via direct binding to β-catenin. Instead, S83A α-catenin promoted chondrocyte dedifferentiation and inflammation (Fig. 3). Several binding partners of α-catenin, including vinculin, afadin, formin-1, and spectrin, are known to control actin dynamics and cytokinesis 18. These observations suggest the attractive possibility that overinteraction of mutant α-catenin with these actin cytoskeletal components is intricately involved in physiological processes and may enhance chondrocyte disorders through miscoordination of intercellular junctions. The precise role of the α-catenin complex in regulating the chondrocyte phenotype remains unclear, and its elucidation awaits further study. We previously demonstrated that acceleration of β-catenin transcriptional activity causes changes associated with chondrocyte diseases, such as dedifferentiation and inflammation 15,19. Here, we found that γ-catenin, which has a strikingly similar architecture to that of β-catenin, fulfilled some of these same functions in chondrocytes. This redundancy suggests that cells lacking either β-catenin or γ-catenin retain the ability regulate the cellular phenotype of chondrocytes. Thus, catenin proteins can be expected to provide new targets for regulation of IL-1β-induced chondrocyte destruction.
LDR alone did not directly modulate expression of catenin proteins in chondrocytes (Fig. 5A). Moreover, although treatment of chondrocytes with 26S proteasome inhibitor MG132 increased the expression of catenin proteins, LDR did not rescue MG132-enhanced proteins levels (data not shown), indicating that LDR is not involved in the induction of 26S proteasomal activity. However, LDR decreased the expression of α-, β-, and γ-catenin proteins induced by IL-1β treatment (Fig. 5B). This phenomenon is consistent with the suppression of IL-1β-induced dedifferentiation and inflammation by LDR (Fig. 2). Therefore, we assume that LDR inhibits a signaling pathway downstream of IL-1β that is involved in maintaining the posttranslational stability of catenin protein. This mechanism may include PI3K/Akt-dependent signaling, which governs increased β-catenin stability and enhanced nuclear translocation 20. Actually, one of the key actions of IL-1β is to activate Akt signaling; based on the canonical Wnt pathway, inactivation of the serine/threonine kinase GSK3 by Akt increases β-catenin accumulation and expression of Tcf/Lef target genes. Thus, we suggest that signal transduction through this cascade rather than proteasome activation is likely involved in regulating LDR-mediated catenin expression and the cellular phenotype of chondrocytes exposed to IL-1β. Although γ-catenin, like β-catenin, has multiple functions, the issue of how γ-catenin expression is regulated by IL-1β alone or in combination with LDR requires further clarification. In addition to the inhibition of downstream signal transductions of IL-1β by LDR, LDR also played an inhibitory role in β- and γ-catenin-mediated pathophysiological progression in chondrocytes, even in those not exposed to cytokines. We found that both β- and γ-catenin induced chondrocyte dedifferentiation by downregulating Sox-9 activity and promoted inflammation by upregulating NF-κB activity. Notably, these transcription factor-mediated disease mechanisms were completely reversed by LDR, although chondrocytes were maintained high catenin levels by ectopic overexpression. Thus, we suggest that LDR offers an effective therapeutic tool for osteoarthritis and rheumatism patients, and propose that γ-catenin could also serve as both a prognostic biomarker and a therapeutic target in cartilage destruction.
In conclusion, our studies demonstrate that the catenin-related cascade is an essential component of intracellular signaling mechanisms associated with cartilage pathogenesis. Notably, our findings establish that LDR governs both IL-1β signaling, which acts upstream of catenin proteins, and Sox-9 and NF-κB signaling, which act downstream of the catenin pathway in chondrocytes. Therefore, LDR may be beneficial as a new therapeutic option for controlling cartilage diseases induced by IL-1β.
Acknowledgments
This work was supported by the Nuclear Research & Development Program of the National Research Foundation and by the Nuclear Energy Technology Innovation Program of the Korea Institute of Energy Technology Evaluation Planning (No. SUBJID-0000000015133).
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- Ma S, Kong B, Liu B. Liu X. Biological effects of low-dose radiation from computed tomography scanning. Int. J. Radiat. Biol. 2013;89:326–333. doi: 10.3109/09553002.2013.756595. [DOI] [PubMed] [Google Scholar]
- Kumar PR, Mohankumar MN, Hamza VZ. Jeevanram RK. Dose-rate effect on the induction of HPRT mutants in human G0 lymphocytes exposed in vitro to gamma radiation. Radiat. Res. 2006;165:43–50. doi: 10.1667/rr-3467.1. [DOI] [PubMed] [Google Scholar]
- Dauer LT, Brooks AL, Hoel DG, Morgan WF, Stram D. Tran P. Review and evaluation of updated research on the health effects associated with low-dose ionising radiation. Radiat. Prot. Dosimetry. 2010;140:103–136. doi: 10.1093/rpd/ncq141. [DOI] [PubMed] [Google Scholar]
- Lorenz R, Deubel W, Leuner K, Gollner T, Hochhauser E, et al. Dose and dose-rate dependence of the frequency of HPRT deficient T lymphocytes in the spleen of the 137Cs gamma-irradiated mouse. Int. J. Radiat. Biol. 1994;66:319–326. doi: 10.1080/09553009414551251. [DOI] [PubMed] [Google Scholar]
- Evans HH, Nielsen M, Mencl J, Horng MF. Ricanati M. The effect of dose rate on X-radiation-induced mutant frequency and the nature of DNA lesions in mouse lymphoma L5178Y cells. Radiat. Res. 1990;122:316–325. [PubMed] [Google Scholar]
- Yentrapalli R, Azimzadeh O, Barjaktarovic Z, Sarioglu H, Wojcik A, et al. Quantitative proteomic analysis reveals induction of premature senescence in human umbilical vein endothelial cells exposed to chronic low-dose rate gamma radiation. Proteomics. 2013;13:1096–1107. doi: 10.1002/pmic.201200463. [DOI] [PubMed] [Google Scholar]
- Ina Y, Sakai K. Prolongation of life span associated with immunological modification by chronic low-dose-rate irradiation in MRL-lpr/lpr mice. Radiat. Res. 2004;161:168–173. doi: 10.1667/rr3120. [DOI] [PubMed] [Google Scholar]
- Rodel F, Keilholz L, Herrmann M, Sauer R. Hildebrandt G. Radiobiological mechanisms in inflammatory diseases of low-dose radiation therapy. Int. J. Radiat. Biol. 2007;83:357–366. doi: 10.1080/09553000701317358. [DOI] [PubMed] [Google Scholar]
- Budras KD, Hartung K. Munzer BM. [Light and electron microscopy studies of the effect of roentgen irradiation on the synovial membrane of the inflamed knee joint] Berl. Munch. Tierarztl. Wochenschr. 1986;99:148–152. [PubMed] [Google Scholar]
- Trott KR, Parker R. Seed MP. [The effect of x-rays on experimental arthritis in the rat] Strahlenther. Onkol. 1995;171:534–538. [PubMed] [Google Scholar]
- Fischer U, Kamprad F, Koch F, Ludewig E, Melzer R, et al. [The effects of low-dose Co-60 irradiation on the course of aseptic arthritis in a rabbit knee joint] Strahlenther. Onkol. 1998;174:633–639. doi: 10.1007/BF03038512. [DOI] [PubMed] [Google Scholar]
- Kracht M, Saklatvala J. Transcriptional and post-transcriptional control of gene expression in inflammation. Cytokine. 2002;20:91–106. doi: 10.1006/cyto.2002.0895. [DOI] [PubMed] [Google Scholar]
- Hong EH, Lee SJ, Kim JS, Lee KH, Um HD, et al. Ionizing radiation induces cellular senescence of articular chondrocytes via negative regulation of SIRT1 by p38 kinase. J. Biol. Chem. 2010;285:1283–1295. doi: 10.1074/jbc.M109.058628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EH, Yun HS, Kim J, Um HD, Lee KH, et al. Nicotinamide phosphoribosyltransferase is essential for interleukin-1beta-mediated dedifferentiation of articular chondrocytes via SIRT1 and extracellular signal-regulated kinase (ERK) complex signaling. J. Biol. Chem. 2011;286:28619–28631. doi: 10.1074/jbc.M111.219832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SG, Yu SS, Ryu JH, Jeon HB, Yoo YJ, et al. Regulation of beta-catenin signaling and maintenance of chondrocyte differentiation by ubiquitin-independent proteasomal degradation of alpha-catenin. J. Biol. Chem. 2005;280:12758–12765. doi: 10.1074/jbc.M413367200. [DOI] [PubMed] [Google Scholar]
- Ryu JH, Kim SJ, Kim SH, Oh CD, Hwang SG, et al. Regulation of the chondrocyte phenotype by beta-catenin. Development. 2002;129:5541–5550. doi: 10.1242/dev.129.23.5541. [DOI] [PubMed] [Google Scholar]
- Zhurinsky J, Shtutman M. Ben-Ze'ev A. Plakoglobin and beta-catenin: protein interactions, regulation and biological roles. J. Cell. Sci. 2000;113(Pt 18):3127–3139. doi: 10.1242/jcs.113.18.3127. [DOI] [PubMed] [Google Scholar]
- Kobielak A, Fuchs E. Alpha-catenin: at the junction of intercellular adhesion and actin dynamics. Nat. Rev. Mol. Cell Biol. 2004;5:614–625. doi: 10.1038/nrm1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Im DS, Kim SH, Ryu JH, Hwang SG, et al. Beta-catenin regulates expression of cyclooxygenase-2 in articular chondrocytes. Biochem. Biophys. Res. Commun. 2002;296:221–226. doi: 10.1016/s0006-291x(02)00824-0. [DOI] [PubMed] [Google Scholar]
- Sharma M, Chuang WW. Sun Z. Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta-catenin accumulation. J. Biol. Chem. 2002;277:30935–30941. doi: 10.1074/jbc.M201919200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.