

Abstract
High levels of low-density lipoprotein cholesterol (LDL-C) enhance platelet activation, whereas high levels of high-density lipoprotein cholesterol (HDL-C) exert a cardioprotective effect. However, the effects on platelet activation of high levels of LDL-C combined with low levels of HDL-C (HLC) have not yet been reported. We aimed to evaluate the platelet activation marker of HLC patients and investigate the antiplatelet effect of atorvastatin on this population. Forty-eight patients with high levels of LDL-C were enrolled. Among these, 23 had HLC and the other 25 had high levels of LDL-C combined with normal levels of HDL-C (HNC). A total of 35 normocholesterolemic (NOMC) volunteers were included as controls. Whole blood flow cytometry and platelet aggregation measurements were performed on all participants to detect the following platelet activation markers: CD62p (P-selectin), PAC-1 (GPIIb/IIIa), and maximal platelet aggregation (MPAG). A daily dose of 20 mg atorvastatin was administered to patients with high levels of LDL-C, and the above assessments were obtained at baseline and after 1 and 2 months of treatment. The expression of platelets CD62p and PAC-1 was increased in HNC patients compared to NOMC volunteers (P<0.01 and P<0.05). Furthermore, the surface expression of platelets CD62p and PAC-1 was greater among HLC patients than among HNC patients (P<0.01 and P<0.05). Although the expression of CD62p and PAC-1 decreased significantly after atorvastatin treatment, it remained higher in the HLC group than in the HNC group (P<0.05 and P=0.116). The reduction of HDL-C further increased platelet activation in patients with high levels of LDL-C. Platelet activation remained higher among HLC patients regardless of atorvastatin treatment.
Keywords: Low-density lipoprotein cholesterol, High-density lipoprotein cholesterol, Platelet activation, Statins, PAC-1, CD62p
Introduction
Platelet activation plays a key role in the pathogenesis of atherothrombosis and acute coronary syndrome (1). Several studies have demonstrated that low-density lipoprotein cholesterol (LDL-C) enhances platelet activation, leads to platelet hyperactivity, and subsequently increases the risk of arterial thrombosis (2). Hence, LDL-C is the major cause of coronary heart disease (CHD) (3). On the other hand, previous epidemiological studies found that high-density lipoprotein cholesterol (HDL-C) exerts a cardioprotective effect and reduces the risk of cardiovascular disease (4). However, inconsistent results of the HDL-C effect on platelet activation were reported in previous findings (5,6). Thus, the effect of HDL-C on platelet activation remains unclear, and the effect of high levels of LDL-C combined with low levels of HDL-C (HLC) on platelet activation in particular has not yet been reported. To clarify the relationship between them could be clinically important in the prevention and treatment of cardiovascular disease.
The 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors - statins - reduce the incidence of major coronary events in both primary and secondary prevention (7,8) owing to their antiplatelet effect (9). However, the antiplatelet effect of statins on HLC is still not fully defined.
In this study, platelet activation was analyzed by evaluating the activation markers of platelets, such as P-selectin and GPIIb/IIIa. Both of these integrins are expressed only on the surface of activated platelets. GPIIb/IIIa is a fibrinogen receptor and the binding reaction between platelets and fibrinogen leads to the formation of thrombus. Thus, the increase in GPIIb/IIIa is highly associated with acute coronary syndrome (10). Furthermore, P-selectin is an adhesion molecule, which modulates platelet-leukocyte, platelet-monocyte, and platelet-endothelium interactions (11). P-selectin also contributes to the process of stabilizing GPIIb/IIIa-fibrinogen formation, which determines the size and stability of platelet aggregates (12).
The aim of this present study was to evaluate the platelet activation markers [P-selectin, GPIIb/IIIa, and maximal platelet aggregation (MPAG)] of HLC patients and investigate the antiplatelet effect of atorvastatin on this population.
Material and Methods
Study population and protocol
Eligible for this study were patients with high levels of LDL-C [4.1-4.9 mM; (borderline high levels are 3.4-4.1 mM and very high levels are >4.9 mM, according to the classification of ATP III) (3)] and triglyceride (TG) levels less than 1.7 mM. The patients were then divided into 2 groups: the first group consisted of patients with high levels of LDL-C combined with normal levels (>1.0 mM) of HDL-C (HNC), and the second group consisted of patients with HLC (i.e., HDL-C <1.0 mM). None of these patients had been treated with lipid-lowering drugs within 2 months. Moreover, 35 normocholesterolemic (NOMC) volunteers who were matched according to age, gender, and risk factors were included as a control group. The exclusion criteria were hypertension, type 2 diabetes, treatment with antiplatelet drugs, CHD, peripheral vascular disease, hemostatic disorder, chronic inflammatory disease, thyroid disorder, nephrotic syndrome, renal insufficiency, liver disease, and mental disorder. All study participants underwent either electrocardiogram (ECG) stress testing or coronary computed tomography (CT) angiography to exclude CHD. A daily dose of 20 mg atorvastatin was administered to patients with high levels of LDL-C. Blood samples were taken from atorvastatin-treated patients at baseline and after 1 and 2 months of treatment. This study was approved by Huashan Hospital's Ethics Committee and all participants gave written, informed consent.
Blood collection
Blood was collected in the morning from the resting and fasting patients using a 21G needle without stasis. The blood was then stored in acid-citrate-dextrose (1:9) for platelet studies and in serum vacutainers for lipid profiling.
Whole blood flow cytometry
The detection of platelet surface receptors and their expression was evaluated in whole blood (13). Briefly, 30 µL citrated blood was diluted with 270 µL Tyrode buffer. Thereafter, 10 µL diluted blood was incubated with 5 µL of each of the following monoclonal antibodies: anti-GP IIb/IIIa labeled with fluorescein isothiocyanate (PAC-1 FITC; Becton Dickinson, USA), anti-P-selectin labeled with phycoerythrine (anti-CD62p PE; Becton Dickinson), and anti-cd42b labeled with allophycocyanin (anti-CD42b APC; Becton Dickinson) for 20 min at room temperature. The reaction was stopped by dosing 300 µL 1% paraformaldehyde and then analyzed on a Becton Dickinson FACSCanto Flow Cytometer (Becton Dickinson). The fluorescence of 10,000 platelets was recorded using the FACSDiva software 6.1.3 (Becton Dickinson).
Platelet preparation and measurement of platelet aggregation
The examination was performed by optical aggregometry in platelet-rich plasma (PRP) using a platelet aggregometer (model TYXN-96 I Multifunctional Smart Blood Coagulation Analyzer, Shanghai General Machinery Research Institute, China). PRP and platelet-poor plasma (PPP) were prepared by differential centrifugation of anticoagulated blood (100 g for 10 min and 1300 g for 15 min, respectively). The platelet count of PRP was adjusted to 3.0×105/µL with analogous PPP. Light transmission of PRP was adjusted to 0, and 100% for PPP served as reference. The PRP was incubated at 37°C in the aggregometer, followed by stimulation with 10 μmol/L adenosine diphosphate (ADP, Sigma-Aldrich, USA) at a constant stirring rate of 1000 rpm. The platelet aggregation curve was recorded for 5 min with MPAG as the analyzed parameter.
Lipid assay
Lipid profiles, including those of total cholesterol (TC), TG, and HDL-C (Sekisui Medical, Japan), were determined enzymatically on a Hitachi 7600 Automatic Biochemical Analyzer (Hitachi High-Technologies, Japan). In order to more accurately detect and reflect the levels of LDL-C, we used the direct method instead of Friedewald's formula to calculate the amount of LDL-C. The direct method was performed according to the manufacturer's instructions (Sekisui Medical) on the Hitachi 7600 Automatic Biochemical Analyzer.
Statistical analysis
Results are reported as means±SD. Demographic data were analyzed using one-way ANOVA, the Kruskal-Wallis test, group t-test, and Wilcoxon's test for continuous variables, and chi-square test for categorical variables. ANOVA, the Kruskal-Wallis test, group t-test, and Wilcoxon's test were applied for the assessments of lipid assay, platelet aggregation and flow cytometry between the groups. Paired t-tests and matched-pairs signed-rank tests were performed to study platelet activation and the lipid profile after atorvastatin treatment. To account for the antiplatelet effect of statins between the two distinct groups, the group t-test and Wilcoxon's test were used. Spearman's correlation coefficient was used to determine the linear relationship between the studied variables and the surface expression of platelet PAC-1 and CD62p. The statistical software package Stata 11.0 (Stata Corp. LP, USA) was used for the analysis.
Results
Characteristics of participants
A total of 48 patients with high levels of LDL-C were recruited: 25 patients in the HNC group and 23 patients in the HLC group. The NOMC group consisted of 35 healthy volunteers. The relevant biochemical and hematological results are reported in Table 1. There were no differences in age, gender, body mass index, or cardiovascular risk factors (hypertension, diabetes mellitus, smoking history) between the groups. The lipid profiles of atorvastatin-treated patients are shown in Table 2.

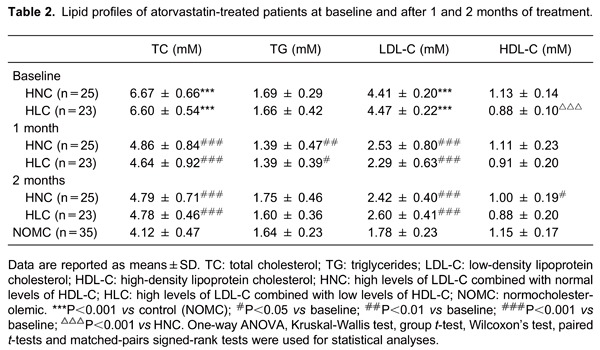
Whole blood flow cytometry
The data showed increased platelet surface expression of CD62p (P-selectin) and PAC-1 (GPIIb/IIIa) in the HNC group compared with the NOMC group (P=0.004 and P=0.014). Moreover, the expressions of platelet markers CD62p and PAC-1 were greater in the HLC group than in the HNC group (P=0.004 and P=0.026; Table 3 and Figure 1).
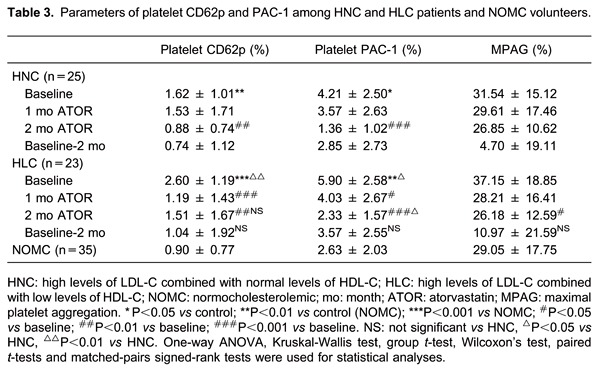
Figure 1. Results of the parameters of platelet CD62p. A, Normocholesterolemic volunteers. B, High levels of LDL-C combined with normal levels in HDL-C patients. C, High levels of LDL-C combined with low levels in HDL-C patients. LDL-C: low-density lipoprotein cholesterol; HDL-C: high-density lipoprotein cholesterol.
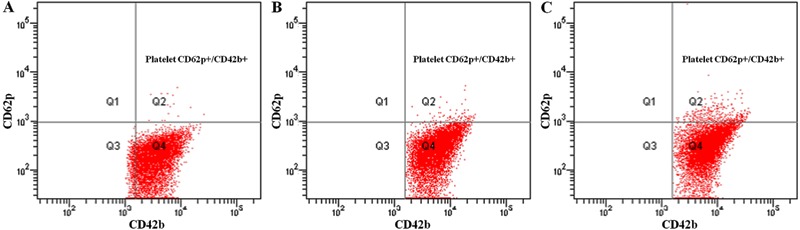
The platelet activation markers CD62p and PAC-1 decreased significantly after atorvastatin treatment (Table 3). The parameter baseline of 2 months was obtained to analyze the antiplatelet effect of atorvastatin between the HNC and HLC groups, and, as shown in Table 4, there was no correlation between HNC and HLC with that parameter (platelet CD62p, P=0.190; and platelet PAC-1, P=0.350, respectively). On the contrary, the expressions of platelet CD62p and PAC-1 remained higher in the HLC group than in the HNC group after 2 months of atorvastatin therapy (platelet PAC-1 2.33±1.57 vs 1.36±1.02%, P=0.0201, and platelet CD62p 1.51±1.67 vs 0.88±0.74%, P=0.116).
Platelet aggregation measurement
MPAG was higher in the HLC group than in the HNC and NOMC groups, although there was no significant statistical difference (P=0.222). After atorvastatin administration, MPAG decreased in both the HLC and HNC groups (P=0.023 and P=0.231, respectively). Furthermore, there was no statistical difference between the two groups with the parameter baseline 2 months (P=0.291, Table 3).
Correlation analysis
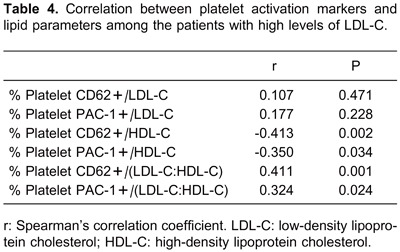
The correlation analysis of platelet activation markers and lipid parameters among the patients with high levels of LDL-C is reported in Table 4. There was no correlation between LDL-C and the expression of platelet CD62p among the patients. Although there was a linear relationship trend between LDL-C and platelet PAC-1 expression, this did not demonstrate a statistical difference (Figure 2). On the other hand, there was a significant negative linear relationship between HDL-C and platelet CD62p, and a similar relationship was found between HDL-C and platelet PAC-1.
Figure 2. Correlation analysis among the patients with high levels of LDL-C. A, Correlation between LDL-C and platelet PAC-1 (P>0.05). B, Correlation between HDL-C and platelet CD62p (P<0.05). C, Correlation between LDL-C/HDL-C and platelet PAC-1 (P<0.05). LDL-C: low-density lipoprotein cholesterol; HDL-C: high-density lipoprotein cholesterol.
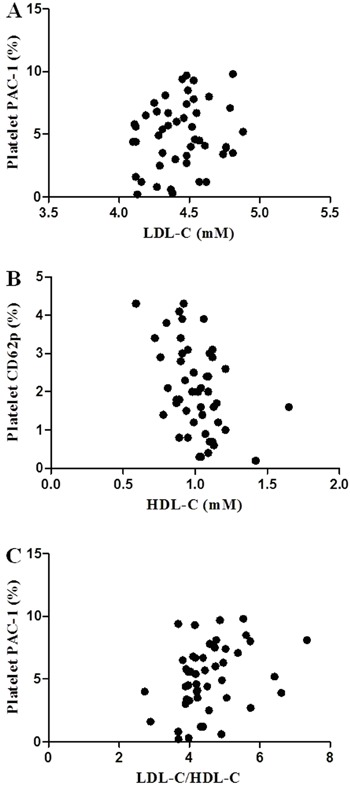
Furthermore, the data in Table 4 showed two results with positive correlations among the patients with high levels of LDL-C: 1) the correlation between LDL-C/HDL-C and platelet CD62p; and 2) the correlation between LDL-C/HDL-C and platelet PAC-1.
Discussion
Platelets play a beneficial role in maintaining hemostasis but are also involved in atherothrombotic diseases. They form platelet plugs at sites of plaque rupture via the stages of platelet adhesion, activation, and aggregation, resulting in thrombus formation (14), which is related to myocardial infarction and the no-reflow phenomenon after primary percutaneous coronary intervention (15). Our study is the first to investigate the effect of HLC on platelet activation.
In the current study, our data confirmed previous observations that high levels of LDL-C enhance platelet activation (16). Moreover, our results showed that platelet surface expression of P-selectin and GPIIb/IIIa increased to a greater extent in the HLC group than in the HNC group, indicating a further increase of platelet activation in HLC patients. In other words, the reduction of HDL-C may lead to a lack of protection from platelet activation in patients with high levels of LDL-C. In an in vitro study, the incubation of HDL-C and oxidized LDL (ox-LDL) in platelets led to inhibition of the increased number of P-selectin receptors induced by ox-LDL (17). In addition to analyzing platelet activation in patients by evaluating P-selectin receptors, we also used GPIIb/IIIa to investigate platelet activation in HLC patients via clinical research, since some studies have reported that GPIIb/IIIa is closely related to platelet aggregation as well as arterial thrombosis. Thus, our study provides further clinical evidence to clarify the relationship between plasma lipoprotein and platelet activation.
Although there was a linear relationship between LDL-C and platelet P-selectin and also between LDL-C and platelet GPIIb/IIIa in the patients with high levels of LDL-C, no statistical differences were found. On the other hand, a significant negative linear relationship was observed between HDL-C and both P-selectin and GPIIb/IIIa. This emphasized the importance of HDL-C in this population, implying the critical role that HDL-C plays in patients with high levels of LDL-C. A recent study demonstrated that low plasma levels of HDL-C in CHD patients and in healthy subjects are associated with increased platelet activation. It was found that the levels of HDL-C inversely affect platelet activation and result in an increase risk of CHD (18).
Apolipoprotein A-I (apoA-I) is the major component of HDL-C, whereas apoB is the primary apolipoprotein component of LDL-C. A previous study demonstrated that the apoB/apoA-I ratio was associated with increased carotid intima-media thickness (19). In a large standardized case-controlled study, INTERHEART unveiled the relationship between the apoB/apoA-I ratio and CHD. That study concluded that the apoB/apoA-I ratio was correlated with the risk of acute myocardial infarction, and the apoB/apoA-I ratio was considered an important predictor of CHD (20). Moreover, Assinger et al. (5) demonstrated that the balance between ox-LDL and oxidized HDL (ox-HDL) determined platelet activation in hypercholesterolemic patients. To go beyond the studies mentioned above, we introduced the ratio LDL-C/HDL-C as a parameter to evaluate platelet activation in patients with high levels of LDL-C. Surprisingly, there was a statistically significant correlation between LDL-C/HDL-C and platelet activation markers. Hence, we hypothesized that the ratio LDL-C/HDL-C could be used as a potential parameter to assess platelet activation in hypercholesterolemic patients.
The interaction between HDL-C surface LRP-8 (apoER2′) and platelet lipidated apoE resulted in increased nitric oxide production, thereby inhibiting platelet activation (21). Moreover, ox-HDL but not native HDL-C binds platelet scavenger receptor-BI (SR-BI), which inhibits platelet reactivity to ADP and other agonists by interfering with protein kinase C (PKC) activation mediated by an ox-LDL/SR-BI complex, because SR-BI is one of the important platelet receptors (22).
Numerous studies have demonstrated that statins have an antiplatelet effect via a lipid-lowering dependent mechanism or lipid-lowering independent mechanism (23,24). Recent studies found that statins and fibrates activate platelet peroxisome proliferator-activated receptors and reduce platelet aggregation in response to arachidonic acid, which is related to the downregulation of PKC in platelets (25). Other studies have shown that statins reduce thromboxane A2 (TXA2) production and thus inhibit platelet aggregation (24). Our study found that the expression of platelet P-selectin, GPIIb/IIIa, and MPAG decreased in both the HLC and the HNC groups after a 2-month treatment with atorvastatin. Such a finding may be in line with data from Labios et al. (26), which demonstrated the effect of statins on platelet activation among hypercholesterolemic patients.
Using the parameter of baseline of 2 months, we found that the antiplatelet effect of atorvastatin was similar in both the HLC and the HNC groups. Values for platelet activation markers GPIIb/IIIa and P-selectin remained higher in the HLC group than in the HNC group after atorvastatin treatment. This may be attributed to the absent effect of atorvastatin on HDL-C, which further leads to a deficiency in the antiplatelet effect that could be compensated by HDL-C. Thus, medical providers should take notice of this situation. Antiplatelet therapy or HDL-elevating treatment might be considered for such patients in clinical practice.
Generally low numbers of patients were included in this study owing to the strictness of the inclusion and exclusion criteria. Thus, further multicenter studies with larger samples need to be carried out in order to define the assumption. In this study, we focused on phenomenon-based investigations, and were unable to interpret the microscopic changes between HDL-C and platelet activation because of a lack of a mechanism study.
In conclusion, LDL-C levels do not lead to any difference in platelet activation in patients with high levels of LDL-C; however, HDL-C levels lead to the following difference in platelet activation: a reduction in HDL-C levels increases platelet activation. Furthermore, the balance between LDL-C and HDL-C may determine the platelet activation of hypercholesterolemic patients. On the other hand, platelet activation remains higher among patients in the HLC group regardless of atorvastatin treatment.
Acknowledgments
We thank Sun Wei, Joan Wong Ka Ghee, Ma Wei Zhe, Xu Xiao for their kind advice and support during this study. Research supported by Shanghai Municipal Bureau Foundation.
Footnotes
First published online.
References
- 1.Fuster V, Badimon L, Badimon JJ, Chesebro JH. The pathogenesis of coronary artery disease and the acute coronary syndromes (2) N Engl J Med. 1992;326:310–318. doi: 10.1056/NEJM199201303260506. [DOI] [PubMed] [Google Scholar]
- 2.Nagy B, Jr, Jin J, Ashby B, Reilly MP, Kunapuli SP. Contribution of the P2Y12 receptor-mediated pathway to platelet hyperreactivity in hypercholesterolemia. J Thromb Haemost. 2011;9:810–819. doi: 10.1111/j.1538-7836.2011.04217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- 4.Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA. 1986;256:2835–2838. doi: 10.1001/jama.1986.03380200073024. [DOI] [PubMed] [Google Scholar]
- 5.Assinger A, Schmid W, Eder S, Schmid D, Koller E, Volf I. Oxidation by hypochlorite converts protective HDL into a potent platelet agonist. FEBS Lett. 2008;582:778–784. doi: 10.1016/j.febslet.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Robertson JO, Li W, Silverstein RL, Topol EJ, Smith JD. Deficiency of LRP8 in mice is associated with altered platelet function and prolonged time for in vivo thrombosis. Thromb Res. 2009;123:644–652. doi: 10.1016/j.thromres.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naci H, Brugts JJ, Fleurence R, Tsoi B, Toor H, Ades AE. Comparative benefits of statins in the primary and secondary prevention of major coronary events and all-cause mortality: a network meta-analysis of placebo-controlled and active-comparator trials. Eur J Prev Cardiol. 2013;20:641–657. doi: 10.1177/2047487313480435. [DOI] [PubMed] [Google Scholar]
- 8.Athyros VG, Katsiki N, Tziomalos K, Gossios TD, Theocharidou E, Gkaliagkousi E, et al. Statins and cardiovascular outcomes in elderly and younger patients with coronary artery disease: a post hoc analysis of the GREACE study. Arch Med Sci. 2013;9:418–426. doi: 10.5114/aoms.2013.35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pignatelli P, Carnevale R, Pastori D, Cangemi R, Napoleone L, Bartimoccia S, et al. Immediate antioxidant and antiplatelet effect of atorvastatin via inhibition of Nox2. Circulation. 2012;126:92–103. doi: 10.1161/CIRCULATIONAHA.112.095554. [DOI] [PubMed] [Google Scholar]
- 10.Arnaout MA, Goodman SL, Xiong JP. Structure and mechanics of integrin-based cell adhesion. Curr Opin Cell Biol. 2007;19:495–507. doi: 10.1016/j.ceb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blann AD, Nadar SK, Lip GY. The adhesion molecule P-selectin and cardiovascular disease. Eur Heart J. 2003;24:2166–2179. doi: 10.1016/j.ehj.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 12.Merten M, Thiagarajan P. P-selectin expression on platelets determines size and stability of platelet aggregates. Circulation. 2000;102:1931–1936. doi: 10.1161/01.CIR.102.16.1931. [DOI] [PubMed] [Google Scholar]
- 13.Gorchakova O, von Beckerath N, Gawaz M, Mocz A, Joost A, Schomig A, et al. Antiplatelet effects of a 600 mg loading dose of clopidogrel are not attenuated in patients receiving atorvastatin or simvastatin for at least 4 weeks prior to coronary artery stenting. Eur Heart J. 2004;25:1898–1902. doi: 10.1016/j.ehj.2003.10.039. [DOI] [PubMed] [Google Scholar]
- 14.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–666. doi: 10.1016/S0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 15.Zhao B, Li J, Luo X, Zhou Q, Chen H, Shi H. The role of von Willebrand factor and ADAMTS13 in the no-reflow phenomenon: after primary percutaneous coronary intervention. Tex Heart Inst J. 2011;38:516–522. [PMC free article] [PubMed] [Google Scholar]
- 16.Gocmen AY, Burgucu D, Gumuslu S. Effect of resveratrol on platelet activation in hypercholesterolemic rats: CD40-CD40L system as a potential target. Appl Physiol Nutr Metab. 2011;36:323–330. doi: 10.1139/h11-022. [DOI] [PubMed] [Google Scholar]
- 17.Sener A, Enc E, Ozsavci D, Vanizor-Kural B, Yanikkaya-Demirel G, Oba R, et al. Exogenous L-arginine and HDL can alter LDL and ox-LDL-mediated platelet activation: using platelet P-selectin receptor numbers. Clin Appl Thromb Hemost. 2011;17: doi: 10.1177/1076029610385223. [DOI] [PubMed] [Google Scholar]
- 18.Vazzana N, Ganci A, Cefalu AB, Lattanzio S, Noto D, Santoro N, et al. Enhanced lipid peroxidation and platelet activation as potential contributors to increased cardiovascular risk in the low-HDL phenotype. J Am Heart Assoc. 2013;2: doi: 10.1161/JAHA.113.000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang F, Yang Z, Xu B, Bi Y, Xu M, Xu Y, et al. Both serum apolipoprotein B and the apolipoprotein B/apolipoprotein A-I ratio are associated with carotid intima-media thickness. PLoS One. 2013;8: doi: 10.1371/journal.pone.0054628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McQueen MJ, Hawken S, Wang X, Ounpuu S, Sniderman A, Probstfield J, et al. Lipids, lipoproteins, and apolipoproteins as risk markers of myocardial infarction in 52 countries (the INTERHEART study): a case-control study. Lancet. 2008;372:224–233. doi: 10.1016/S0140-6736(08)61076-4. [DOI] [PubMed] [Google Scholar]
- 21.Mehta JL, Chen LY. Reversal by high-density lipoprotein of the effect of oxidized low-density lipoprotein on nitric oxide synthase protein expression in human platelets. J Lab Clin Med. 1996;127:287–295. doi: 10.1016/S0022-2143(96)90097-9. [DOI] [PubMed] [Google Scholar]
- 22.Valiyaveettil M, Kar N, Ashraf MZ, Byzova TV, Febbraio M, Podrez EA. Oxidized high-density lipoprotein inhibits platelet activation and aggregation via scavenger receptor BI. Blood. 2008;111:1962–1971. doi: 10.1182/blood-2007-08-107813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–1719. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 24.Ma LP, Nie DN, Hsu SX, Yin SM, Xu LZ, Nunes JV. Inhibition of platelet aggregation and expression of alpha granule membrane protein 140 and thromboxane B2 with pravastatin therapy for hypercholesterolemia. J Assoc Acad Minor Phys. 2002;13:23–26. [PubMed] [Google Scholar]
- 25.Ali FY, Armstrong PC, Dhanji AR, Tucker AT, Paul-Clark MJ, Mitchell JA, et al. Antiplatelet actions of statins and fibrates are mediated by PPARs. Arterioscler Thromb Vasc Biol. 2009;29:706–711. doi: 10.1161/ATVBAHA.108.183160. [DOI] [PubMed] [Google Scholar]
- 26.Labios M, Martinez M, Gabriel F, Guiral V, Martinez E, Aznar J. Effect of atorvastatin upon platelet activation in hypercholesterolemia, evaluated by flow cytometry. Thromb Res. 2005;115:263–270. doi: 10.1016/j.thromres.2004.08.016. [DOI] [PubMed] [Google Scholar]