

Significance
MITF, a master regulator of melanocytes and a major melanoma oncogene amplified in 30-40% of melanomas, determines proliferative or invasive phenotypes. Previously unrecognized as a driver of lysosomal biogenesis, we found that MITF expression correlates with many lysosomal genes and generates late endosomes that are not functional in proteolysis. This accumulation of incomplete organelles expands the late endosomal compartment, enhancing Wnt signaling by entrapping the Wnt machinery in multivesicular bodies. Wnt signaling can stabilize many proteins besides β-catenin. Our study identifies MITF as an oncogenic protein stabilized by Wnt, and describes three novel glycogen synthase kinase 3-regulated phosphorylation sites in this oncogene. This study deepens our knowledge on proliferative stages of melanoma: MITF, multivesicular bodies, and Wnt may form a feedback loop that drives proliferation.
Keywords: MITF, Wnt-STOP, lysosome, melanoma, multivesicular body
Abstract
Canonical Wnt signaling plays an important role in development and disease, regulating transcription of target genes and stabilizing many proteins phosphorylated by glycogen synthase kinase 3 (GSK3). We observed that the MiT family of transcription factors, which includes the melanoma oncogene MITF (micropthalmia-associated transcription factor) and the lysosomal master regulator TFEB, had the highest phylogenetic conservation of three consecutive putative GSK3 phosphorylation sites in animal proteomes. This finding prompted us to examine the relationship between MITF, endolysosomal biogenesis, and Wnt signaling. Here we report that MITF expression levels correlated with the expression of a large subset of lysosomal genes in melanoma cell lines. MITF expression in the tetracycline-inducible C32 melanoma model caused a marked increase in vesicular structures, and increased expression of late endosomal proteins, such as Rab7, LAMP1, and CD63. These late endosomes were not functional lysosomes as they were less active in proteolysis, yet were able to concentrate Axin1, phospho-LRP6, phospho-β-catenin, and GSK3 in the presence of Wnt ligands. This relocalization significantly enhanced Wnt signaling by increasing the number of multivesicular bodies into which the Wnt signalosome/destruction complex becomes localized upon Wnt signaling. We also show that the MITF protein was stabilized by Wnt signaling, through the novel C-terminal GSK3 phosphorylations identified here. MITF stabilization caused an increase in multivesicular body biosynthesis, which in turn increased Wnt signaling, generating a positive-feedback loop that may function during the proliferative stages of melanoma. The results underscore the importance of misregulated endolysosomal biogenesis in Wnt signaling and cancer.
Wnt signaling is required for tissue differentiation, growth, and homeostasis (1, 2). Misregulation of Wnt signals can result in abnormal development and disease, most notably cancer (3). The canonical Wnt pathway influences transcription through the stabilization of β-catenin, a transcriptional activator. In the absence of Wnt ligands, β-catenin is rapidly turned over by a destruction complex composed of adenomatous polyposis coli, Axin1, casein kinase 1α, and glycogen synthase kinase 3 (GSK3) (4). During Wnt signaling, the destruction complex is inhibited, allowing newly synthesized β-catenin to accumulate in the nucleus and regulate transcription of Wnt target genes (5). However, Wnt signaling stabilizes many other cellular proteins in addition to β-catenin (6, 7). Wnt achieves this stabilization of proteins by inhibiting GSK3, a kinase that generates phosphodegrons in β-catenin and many other proteins. These phosphodegrons are then recognized by E3 ubiquitin ligases and polyubiquitinated, targeting proteins for proteosomal degradation (7). Upon Wnt binding, the Wnt receptor low-density lipoprotein receptor-related protein 6 (LRP6) recruits the β-catenin destruction complex, including Axin1, GSK3, and phospho-β-catenin (p-β-catenin), which are endocytosed as “Wnt/LRP6 signalosomes” (8) and translocated from the cytosol into multivesicular bodies (MVBs) (6, 9). Wnt signal transduction requires an intact endosomal sorting complexes required for transport (ESCRT) machinery (6), which is required for the formation of the intraluminal vesicles of late endosomes (10). In this way, Wnt signaling causes GSK3 and Axin1 to become sequestered from their potential cytosolic substrates inside membrane-bounded organelles (6, 9). This mechanism, which results in the stabilization of many proteins by decreasing the polyubiquitination triggered by the generation of GSK3-induced phosphodegrons, has been recently designated “Wnt-dependent STabilization Of Proteins” or Wnt/STOP (7).
We became interested in MITF (micropthalmia-associated transcription factor) when a bioinformatic screen of the human proteome for putative GSK3 targets containing three or more consecutive phosphorylation sites followed by a priming site revealed that a remarkable 20% of the human proteome contained such sites (6). Among these sites, the highest score for phylogenetically conserved putative GSK3 phosphorylation motifs (www.hhmi.ucla.edu/derobertis/EDR_MS/GSK3%20Proteome/Table_1-full_table.xls) belonged to a group of basic helix–loop–helix leucine zipper transcription factors called the MiT family (11). The four members of this family (MITF, TFEB, TFE3, and TFEC) possess three putative GSK3 sites followed by a priming site close to the carboxyl terminus. MiT genes behave as oncogenes (12, 13). In the case of MITF and TFEB, the priming sites were shown to be phosphorylated by RSK1/p90 and PKCβ, respectively (14, 15), but it was not known whether the new GSK3 sites were phosphorylated.
MITF stands for micropthalmia-associated transcription factor, because mutations in the mitf gene give rise to smaller eyes (16) as a result of defects in the development and function of the retinal pigment epithelium (17, 18). MITF is also a melanocyte master regulator gene and a melanoma oncogene (18, 19). A point mutation in MITF that inhibits sumoylation and leads to increased activity predisposes to familial melanoma, indicating that MITF is indeed a melanoma oncogene (20). In addition, the MITF gene is amplified in 30–40% of melanomas (19). MITF is expressed in many tissues and is subject to alternative splicing and differential promoter use, giving rise to multiple tissue-specific isoforms. MITF-M, also known as variant 4, is an isoform with an N-terminal truncation that is specifically expressed in melanocytes and melanomas (18, 21).
Another important member of the MiT family is TFEB, which is the master regulator of lysosome biogenesis (22). TFEB binds to a specific DNA sequence known as the “coordinated lysosomal expression and regulation” element (CLEAR element) in the promoter region of many lysosomal genes (22). Regulation of lysosome biogenesis is controlled by the mammalian target of rapamycin (mTOR) pathway through phosphorylations of TFEB that retain this transcription factor in the cytoplasm (23, 24), coupling lysosomal biogenesis to nutritional sensors. TFE3 has also been shown to act in a similar manner, promoting autophagy and lysosomal biogenesis (25). Although MITF has not, to our knowledge, been recognized as participating in lysosomal biogenesis (22, 25), it is strongly expressed in cell types with high levels of lysosome-related organelles, such as melanocytes, osteoclasts, mast cells, and retinal pigment epithelium (18).
In the present study, we report that MITF mRNA expression levels significantly correlated with the expression of lysosomal gene transcripts in a large panel of melanoma cell lines. MITF up-regulated many, but not all, lysosomal genes in an inducible MITF melanoma model, and activated transcription of a CLEAR element synthetic promoter. MITF not only induced lysosomal gene transcripts, but also protein markers of late endosomal trafficking and acidic organelles. However, this marked increase in late endosomes failed to increase overall lysosomal degradation of endocytosed BSA. Previous work from our laboratory had shown that expansion of late endolysosomal structures [via chloroquine (CQ) treatment or presenilin knockdown] enhanced Wnt signaling by increasing relocalization of GSK3 into MVBs (26). Induction of MITF expression in a melanoma model, in addition to increasing late endosomal vesicles, also increased Wnt signaling in an ESCRT-dependent manner. In the presence of Wnt, the MITF-induced vesicular structures contained Axin1, GSK3, p-β-catenin, and phospho-LRP6 (pLRP6). Wnt prolonged the half-life of MITF protein, and enhanced MITF activity in cultured cells and in Xenopus embryo explants. A custom-made antiphospho antibody confirmed that the novel C-terminal sites on MITF were indeed phosphorylated by GSK3. The results suggest a positive regulatory loop by which MITF expands MVBs/late endosomes, resulting in increased Wnt signaling which in turn stabilizes MITF by decreasing its GSK3 phosphorylations.
Results
MITF Has Three Consecutive Putative GSK3 Sites.
The best-characterized members of the MiT family of helix–loop–helix leucine zipper transcription factors are MITF, the melanocyte master regulator and melanoma oncogene (18), and TFEB, the master coordinator of lysosomes and cellular clearance pathways (22, 27). Although posttranslational modifications had been extensively studied in this family, a bioinformatics screen discovered three previously unrecognized putative GSK3 phosphorylation sites at their C terminus (Fig. 1A) (6). The high degree of conservation of these serines during evolution (Fig. 1B) suggested that these sites might be important for protein function. GSK3 prefers prephosphorylated “primed” substrates, phosphorylating Ser or Thr at position −4 (S/TXXXS/T[PO3]) (28). Importantly, in the case of MITF and TFEB the serine residues that could serve as the priming phosphate for GSK3 had been previously demonstrated to be phosphorylated in vivo (14, 15). This finding prompted us to investigate whether MITF was regulated by Wnt, and whether MITF, like TFEB, also had a role in endolysosomal biogenesis.
Fig. 1.

MITF mRNA correlates with lysosomal gene expression in melanoma cell lines. (A) MITF and the lysosomal master gene regulator TFEB have three putative C-terminal GSK3 phosphorylation sites, with a previously validated priming site. (B) The C-terminal GSK3 sites on MITF have been highly conserved throughout evolution, including in the oriental fruit fly Bactrocera dorsalis. (C) Heat map obtained from a RNA microarray panel of 51 melanoma cell lines, which cluster into two distinct groups, one with high MITF and the other with low MITF expression, when queried for a panel of 89 lysosomal genes. The group with high MITF expression (which includes all cell lines with MITF genomic amplifications) up-regulates many, but not all, lysosomal genes (dashed line). (D and E) GSEA of an expression dataset consisting of a panel of 83 additional, different, melanoma lines confirms that MITF, but not TFEB, significantly correlates with the lysosomal gene set in melanoma. Microarray data for melanoma cell lines was obtained from Hoek et al. (30). Genes were ranked by their correlation (cor) with MITF or TFEB (red to green = high to low correlation). The positions of lyosomal genes (22) among over 12,000 genes per cell line are marked as vertical lines (GS). Enrichment of the lysosomal gene set at the top of the ranked lists was assessed with a permutation based Kolmogorov–Smirnoff nonparametric rank test. A significant correlation for MITF, but not for TFEB, was found. (F) Transfection of MITF activated a CLEAR element-Firefly luciferase reporter (22), in transient transfections of HEK 293T cells. Renilla luciferase driven by the CMV promoter was used for normalization purposes. ***P < 0.001.
MITF Expression Levels Correlate with Many Lysosomal Genes in Melanomas.
Although MITF and TFEB share extensive sequence homology, MITF had not been previously linked to lysosomal biogenesis (22). We investigated whether melanomas in which the MITF gene was amplified had increased transcription of lysosomal genes. We first analyzed a panel of RNA microarray data for 51 melanoma cell lines generated at the University of California, Los Angeles. Cell lines were allowed to sort out according to their levels of expression of a set of 89 lysosomal genes (22) using the Rosetta Resolver gene-expression data analysis system. As shown in Fig. 1C, the melanoma lines clustered into two distinct groups: one with high MITF expression and the other with low MITF expression. The group with high MITF expression (indicated in red in the vertical axis of Fig. 1C) was composed mainly of melanoma cell lines harboring genomic MITF amplifications. Many, but not all, lysosomal genes were up-regulated in the lines expressing high levels of MITF. When the same samples were specifically queried for the expression of a subset of 63 lysosomal genes that contain a CLEAR response element within their promoter region (22), the melanoma lines also strongly clustered into two groups, one with high MITF mRNA expression and DNA amplification, and another with low MITF (Fig. S1).
These results suggested a positive correlation between MITF levels and lysosomal gene expression. To confirm this finding, we performed gene set enrichment analysis (GSEA) (29) on the lysosomal gene set using publicly available microarray expression profile datasets for melanoma (Philadelphia, Zurich, and Mannheim datasets) (30), encompassing a different group of 83 melanoma lines (i.e., in addition to the 51 lines described above). GSEA revealed that MITF transcripts significantly correlated with the expression of the lysosomal gene set in melanomas (P < 0.001) (Fig. 1D). Surprisingly, TFEB, the master regulator of lysosomes, did not correlate with the lysosomal gene set (Fig. 1E). This finding indicated that the up-regulation of lysosomal genes observed in melanomas with high levels of MITF is not mediated by a secondary increase in TFEB expression levels.
To test whether MITF could directly activate the CLEAR element, we cotransfected HEK 293T cells with a CLEAR element luciferase reporter (22) and MITF. Cotransfection with TFEB was used as a positive control. We found that MITF, like TFEB, could significantly activate the synthetic CLEAR element reporter (Fig. 1F). We conclude from these experiments that MITF expression can drive the transcription of many, but not all, CLEAR element lysosomal genes in melanomas.
MITF Expands the Late Endolysosomal Compartment.
To further test the effect of MITF on lysosomal biogenesis, we took advantage of the C32 melanoma cell line model (20), which has undetectable levels of endogenous MITF, but contains a tetracycline-inducible MITF-M (Fig. 2A). After tetracycline (Tet) treatment, MITF was strongly expressed, as detected by an anti-MITF monoclonal antibody (Fig. 2B). In response to MITF induction, C32 cells underwent a phenotypic switch displaying a striking increase in large vesicular structures visible by differential interference contrast light microscopy (Fig. 2 C–D′, arrows). MITF induction also increased the lysosomal membrane marker lysosomal-associated membrane protein 1 (LAMP1) as detected by immunofluorescence (Fig. 2 E and F), and this increase could be quantified by flow cytometry (Fig. 2G). In addition, CD63, a tetraspanin protein that marks the intraluminal vesicles of MVBs, was also enriched in MITF induced-vesicles (Fig. 2 H–J). These two endolysosomal markers are themselves lysosomal genes whose promoters contain CLEAR elements, and were found to be highly correlated with MITF in the heat map of Fig. 1C.
Fig. 2.

Tet-inducible MITF expression increases late endolysosomal vesicles in the C32 melanoma cell line. (A) Schematic diagram of the C32 Tet-inducible MITF melanoma cell line (20). (B) Strong induction of MITF after Tet treatment was detected in C32 cells by Western blot with anti-MITF antibody. (C–D′) Increase in the number of vesicular structures upon MITF induction observed by differential interference contrast light microscopy. Note vesicular structures seen at high power (arrows). (E and F) MITF induction increases immunostaining of the late endosomal marker LAMP1. (G) Quantification by flow cytometry of the increase in LAMP1 levels upon MITF induction. (H and I) MITF induction increases immunostaining of the MVB marker CD63. (J) Quantification by flow cytometry of the increase in CD63 upon MITF induction. (K) MITF induction increased the transcripts of many lysosomal genes containing CLEAR elements in C32 melanoma cells, as validated by RT-qPCR. MITF induction in C32 melanoma cells up-regulated transcripts for the CLEAR element lysosomal genes NAGLU, CLCN7, PSAP, CTSD, CTSA, NEU1, GLA, MCOLN1, GBA, and SCPEP1. Error bars indicate the SEM from three independent experiments.
Using quantitative RT-PCR (RT-qPCR), we validated that expression of multiple lysosomal mRNAs increased by MITF induction in C32 cells. These included α-N-acetylglucosaminidase (NAGLU), chloride channel voltage sensitive 7 (CLCN7), prosaposin (PSAP), cathepsin D (CTSD), cathepsin A (CTSA), sialidase 1 (NEU1), α-galactosidase (GLA), mucolipin 1 (MCOLN1), β-glucocerebrosidase (GBA), and serine carboxypeptidase 1 (SCPEP1) (Fig. 2K). As a negative control, hypoxanthine phosphoribosyltransferase 1 (HPRT1) was shown not to change upon MITF induction in C32 melanoma cells. The ability of MITF to up-regulate lysosomal genes was not confined to melanoma cells, as HEK 293T cells transiently transfected with MITF also had increased transcripts for CTSA, MCOLN1, PSAP, GNS, SCPEP1, NEU1, and GLA (Fig. S2). TFEB, which contains a CLEAR element in its own promoter as part of a positive-feedback mechanism (31), was also induced by MITF (Fig. S2), but was not correlated with lysosomal gene expression in the GSEA analysis of melanoma lines (Fig. 1E). F-box protein 11 (FBX011), used in this case as a negative control, was not induced by MITF in HEK 293T cells (Fig. S2).
In addition, the vesicles induced by MITF were strongly enriched in the late endosomal marker Rab7 (Fig. S3). Although this small GTPase is not itself considered a CLEAR network lysosomal gene, Rab7 has been recently shown to play a key role in the regulation of melanoma proliferation by exploiting a lineage-specific endolysosomal pathway wiring (32).
TFEB elicits a coordinated response to nutritional and homeostatic cellular demands by regulating the synthesis of lysosomes (33). By using LysoTracker dye, a specific marker of acidic organelle compartments, we found that MITF induction in C32 cells increased the number of acidic organelles detected by fluorescence microscopy and flow cytometry (Fig. 3 A–C).
Fig. 3.

MITF expands acidic organelle compartments but does not increase lysosomal activity. (A and B) Acidic organelles visualized by treatment of living cells with LysoTracker dye. (C) Quantification by flow cytometry of the increase in acidic organelles observed upon MITF induction. (D) Schematic representation of the BSA-DQ reagent used for detecting lysosomal proteolytic activity. BSA-DQ added to the culture medium is endocytosed, but only fluoresces when cleaved by proteases inside lysosomes. (E) MITF induction decreases lysosomal activity, as quantified by flow cytometry.
We next tested whether these MITF-induced vesicles were functional lysosomes. To this end, we used the dequenched BSA (BSA-DQ) reagent (Fig. 3D). BSA-DQ is normally self-quenched, as a result of heavy labeling by BODIPY dyes. When added to the culture medium, BSA-DQ is incorporated into the liquid-phase cellular endosomal compartment by nonreceptor-mediated endocytosis. Upon fusion with endolysosomes, BSA-DQ is digested into smaller fragments, relieving the self-quenching and causing a fluorescent signal (Fig. 3D). Although MITF induction greatly expanded the endolysosomal system, increased acidic organelles, and up-regulated the transcription of many lysosomal markers, we were surprised to find that lysosomal activity was not increased, but instead moderately decreased (Fig. 3E). This result is likely because of the inability of overexpressed MITF to induce the complete repertoire of lysosomal genes, as reflected in the heat map in Fig. 1C. We conclude that MITF expression in the C32 melanoma model expands late endosome/MVB vesicles but not the number of functional lysosomes.
MITF Enhances Wnt Signaling.
The increase in late endosome/MVB structures was reminiscent of the effects of CQ or presenilin depletion. We had shown in a previous study that in these conditions Wnt signaling was enhanced through the expansion of the MVB compartment and subsequent increased sequestration of GSK3 (26). This finding prompted us to test whether the responsiveness to the Wnt pathway, which is critical in melanoma (34), was affected by MITF expression.
The Xenopus system provides an efficient way of analyzing Wnt signaling. We found that MITF mRNA microinjections significantly potentiated Wnt8 signaling in animal cap ectodermal explants (Fig. 4A), using a SuperTopFlash-Luciferase Wnt reporter as the assay (Materials and Methods). In accordance to the GSK3 sequestration model (6, 9), this MITF-driven enhancement of Wnt activity required the ESCRT machinery components hepatocyte growth factor-regulated tyrosine kinase substrate (HRS, also known as Vps27) and vacuolar protein sorting protein 4 (Vps4) (Fig. 4A). In addition, MITF mRNA microinjection expanded the expression domain of chordin, a downstream target of Wnt in the developing Xenopus gastrula, when coinjected with suboptimal amounts of Wnt8 mRNA (Fig. 4 B–E, arrows). These results showed that MITF expression enhanced the Wnt signaling response in the Xenopus system, and that this required MVB formation.
Fig. 4.
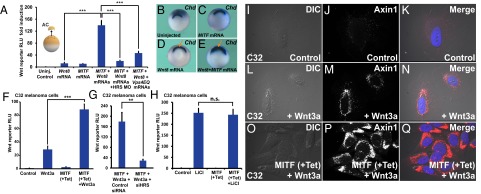
MITF enhances Wnt signaling in Xenopus and in melanoma cells in an ESCRT-dependent manner, and causes increased MVB localization of the destruction complex component Axin1. (A) MITF enhances Wnt signaling in Xenopus ectodermal explants. This enhancement is ESCRT-dependent, as it is blocked by HRS/Vps27 morpholino or a dominant-negative form of the Vps4 ATPase (HRS MO and VPS4EQ) (***P < 0.001). (B–E) MITF cooperates with a low dose of Wnt8 mRNA, expanding the Spemann organizer (arrows), the region that expresses chordin mRNA in Xenopus whole-mount in situ hybridization. (F) MITF induction increases Wnt signaling in the C32 MITF-inducible melanoma cell line. The Wnt BAR firefly luciferase reporter and EF1α-driven Renilla luciferase were permanently introduced with lentivectors into the C32 cell line. (G) HRS/Vps27 knockdown by HRS siRNA decreased Wnt signaling in Tet-induced C32 cells (**P < 0.01). (H) MITF induction did not affect Lithium chloride-induced β-catenin signaling. (I–Q) Immunostaining for Axin1, the key scaffold of the β-catenin destruction complex. Note that Wnt signaling relocalizes Axin1 to vesicular structures, and that this effect is strongly enhanced by MITF induction with Tet. For relocalization of other Wnt components (pLRP6, GSK3, and p-β-catenin) after MITF induction and Wnt3a protein treatment, see Figs. S6–S8.
To study the effect of MITF on Wnt signaling specifically in a melanoma background, we generated permanent lines of MITF-inducible C32 cells transduced with the Wnt/β-batenin-activated reporter (BAR) firefly luciferase reporter together with a Renilla luciferase driven by an EF1α promoter for normalization purposes (Materials and Methods). Using this cell line, we found that MITF induction with Tet significantly enhanced the response to the addition of Wnt3a protein (Fig. 4F, brackets). HRS/Vps27 was required for Wnt signaling in this system as well, because HRS siRNA inhibited Wnt signaling (Fig. 4G). LiCl is a direct inhibitor of GSK3 that stabilizes β-catenin and this effect is expected to be independent of endolysosomal organelles. In agreement with this prediction, MITF had no significant effect on LiCl-induced Wnt reporter activity (Fig. 4H). As mentioned above, CQ treatment enhances Wnt signaling by accumulating late endosome/MVB structures (26). In C32 melanoma cells, CQ also enhanced Wnt3a signaling, and the extent of its effect was comparable to that of MITF induction by Tet (Fig. S4). Taken together, these results indicate that MITF enhances Wnt signaling, both in early embryos and in a melanoma setting, in an ESCRT-dependent manner.
The Destruction Complex Localizes to MITF-Induced Vesicles During Wnt Signaling.
To test whether MITF induction affected the relocalization of the β-catenin destruction complex to endolysosomes/MVBs during Wnt signaling (6, 9), we first examined the localization of endogenous Axin1, the crucial scaffold protein of this complex, using a monoclonal antibody (35). In C32 cells, Wnt3a treatment relocalized Axin1 to vesicles (Fig. 4 I–N), and this vesicular localization was greatly enhanced in cells expressing MITF after Tet treatment (Fig. 4 O–Q), which caused an enlargement of the MVB compartment.
MITF-induced vesicles strongly colocalized with endogenous pLRP6, specifically when C32 cells were treated with Wnt3a (Fig. S5, arrows). This finding indicated that the activated Wnt receptor complex relocalized into MITF-induced vesicles. In addition, we found that RFP-GSK3 colocalized with CD63-containing vesicles upon Wnt treatment, and that the overlap was particularly prominent in C32 melanoma cells expressing MITF (Fig. S6 A–I; note yellow arrows in Fig. S6I′). We also observed that p-β-catenin, a component of the destruction complex (35) that accumulates in MVBs following Wnt signaling (6), colocalized with the MVB marker CD63 in MITF-expressing C32 cells in the presence of Wnt3a (Fig. S7). The results indicated that the destruction complex components Axin1, GSK3, and p-β-catenin are translocated to CD63+ endolysosome/MVBs induced by MITF in the presence of Wnt. The enhanced sequestration of GSK3 and Axin1 provides a cell biological explanation for how MITF induction increased Wnt signaling (Fig. 4F).
MITF Protein Is Stabilized by Wnt.
Wnt signaling has been shown to stabilize many proteins via GSK3-regulated polyubiquitination and degradation (6). This Wnt/STOP is emerging as an important branch of canonical Wnt signaling (7). MITF was previously known to be phosphorylated at S409 by p90/RSK1 (14), providing a possible priming for the three previously unrecognized carboxyl-terminal (S405, S401, S397) putative GSK3 sites identified here (Fig. 1A). The priming phosphorylation at S409, in conjunction with another phosphorylation at S73, had been shown to trigger proteasomal degradation of MITF (14).
To investigate whether Wnt stabilized MITF protein, we treated C32 melanoma cells (with or without previous induction of MITF expression) with Wnt3a protein (Fig. 5 A–D). Cycloheximide (CHX) was also added to inhibit new protein synthesis. After 5 h, C32 cells were fixed and immunostained using an anti-MITF monoclonal antibody. Nuclear MITF staining was observed in cells that had been previously induced with Tet. Upon Wnt3a treatment, MITF protein was significantly stabilized (Fig. 5, compare C and D). Nuclear MITF was quantified and found to be stabilized nearly twofold by Wnt3a (Fig. 5E).
Fig. 5.

MITF protein stabilization by Wnt via novel C-terminal GSK3 phosphorylation sites. (A–D) MITF immunostainings in C32 cells using a 40× objective. Wnt3a protein treatment for 5 h stabilized MITF protein in the presence of CHX. (E) Quantification of MITF staining per nucleus from a previous experiment (***P < 0.001). (F) MITF protein levels (normalized to total Erk1/2) from three independent Western blot experiments upon treatment of M308 melanoma cells with Wnt3a; Wnt prolongs the half-life of endogenous MITF protein. (G) RT-qPCR of the MITF target gene MART1 in Tet-induced C32 cells treated with CHX and Wnt3a (**P < 0.01). (H) RT-qPCR for the MITF target gene x-tyrosinase obtained from Xenopus laevis embryos microinjected with Wnt8, MITF, or MITF + Wnt8 mRNAs. Wnt8 markedly increased MITF activity (**P < 0.01). (I) Diagram depicting MITF wild-type (MITF-WT) and a MITF GSK3 phosphorylation-resistant mutant (MITF-GM). (J) Western blot of Xenopus laevis embryos injected with equal amounts of mRNA for MITF-WT or MITF-GM and blotted for MITF. GAPDH was used as a loading control. (K) Quantification of Western blots from three independent Xenopus experiments showing that MITF-GM is more stable than MITF-WT (**P < 0.01). (L) Diagram of a pMITFGSK3 antibody raised against the indicated peptide corresponding to the C-terminal region of MITF with two phosphorylations. (M) pMITFGSK3 antiserum mirrors the total MITF immunostaining pattern detected with an anti-MITF mAb in Tet-induced C32 cells. This indicates that the phospho-antiserum is specific for MITF. (N) Western blot of HEK 293T cells transiently transfected with MITF-WT or MITF-GM and blotted for pMITFGSK3, total MITF, and GAPDH as a loading control. Note that MITF-GM is not recognized by the phospho-specific MITF antibody. (O) Western blot of HEK 293T cells transiently transfected with MITF-WT treated with or without BIO, a specific GSK3 inhibitor. Note that pMITFGSK3, but not total MITF (anti-MITF mAb), is inhibited by BIO.
To assess the effect of Wnt3a on endogenous MITF protein, we used the M308 melanoma cell line that expresses MITF from its endogenous promoter at detectable levels. M308 melanoma cells were treated with CHX and control medium, or CHX and Wnt3a, and harvested for Western blots at different time points. It was found that Wnt3a treatment prolonged the endogenous MITF half-life by 45% (Fig. 5F). In accordance with this, Wnt3a plus CHX treatment also increased transcript levels of the MITF target gene melanoma antigen recognized by T cells 1 (MART1) (36) in C32 cells induced to express MITF (Fig. 5G), measured by RT-qPCR. In addition, MITF mRNA was significantly more active in promoting the expression of endogenous x-tyrosinase mRNA when coinjected together with Wnt8 mRNA in Xenopus laevis embryos (Fig. 5H). Tyrosinase is an MITF target gene that is not normally expressed at the early gastrula stage. Importantly, in early embryos, microinjection of Wnt8 mRNA alone does not increase x-tyrosinase transcripts, suggesting that the strong increase in x-tyrosinase transcripts by MITF in combination with Wnt is mediated by the stabilization and increased activity of the microinjected MITF (Fig. 5H). Induction of MITF, as well as Wnt3a treatment, increased cellular proliferation in C32 melanoma cells (Fig. S8).
To test whether the novel putative GSK3 sites on MITF played a role in the stability of MITF, we mutated these serine residues into alanine in the human MITF-M (Fig. 5I). The resulting MITF GSK3 mutant (MITF-GM) protein was more stable than wild-type MITF (MITF-WT) protein when the same amount of synthetic mRNA was microinjected into Xenopus laevis embryos and analyzed by Western blot (Fig. 5J). Because equal amounts of mRNA were injected, the effect of the mutations should be posttranscriptional. Quantification of three independent experiments confirmed that MITF-GM was more stable than MITF-WT (Fig. 5K). These results indicate that MITF stability and activity are regulated by these novel GSK3 sites.
A Phospho-Specific MITF Antibody Demonstrates That the C-Terminal GSK3 Sites Are Phosphorylated.
To determine whether MITF was phosphorylated at the C-terminal GSK3 sites predicted by the bioinformatics screen, we raised a phospho-specific rabbit antibody directed against the first two GSK3 phosphorylation sites (S401 and S405) adjacent to the S409 priming site (Fig. 5L). This anti-pMITFGSK3 antibody reproduced the nuclear localization of the anti-MITF monoclonal antibody in melanoma C32 cells upon Tet-driven MITF induction (Fig. 5M), indicating that this reagent is specific for MITF and that phosphorylation takes place. Unfortunately, purification in affinity columns failed, but the crude antiserum was effective at 1:5,000 dilutions. The pMITFGSK3 antibody was phospho-specific, as it did not cross react with the phosphorylation-resistant mutant MITF-GM, while the MITF-WT band was strongly stained in Western blots from transiently transfected HEK 293T cells (Fig. 5N).
We next tested whether the novel phosphorylation sites in MITF were phosphorylated by GSK3. HEK 293T cells were transiently transfected with MITF-WT and treated the next day with the GSK3 inhibitor BIO for 5 h. GSK3 inhibition led to the stabilization of total MITF and disappearance of the pMITFGSK3 band in Western blots (Fig. 5O), indicating that the C-terminal phosphorylations of MITF were indeed mediated by GSK3. In Tet-stimulated C32 melanoma cells, MITF was also recognized by pMITFGSK3, and this band disappeared upon GSK3 inhibition with BIO (Fig. S9), suggesting that MITF was phosphorylated at the novel C-terminal GSK3 sites in a melanoma background as well. Taken together, these results indicate that the MITF protein contains three previously unnoticed C-terminal GSK3 phosphorylation sites that regulate its stability.
Discussion
MITF is a melanoma oncogene that is amplified in 30–40% of melanomas (13). Other members of the MiT family are also disregulated in cancer, for example TFEB in pediatric renal carcinomas and TFE3 in alveolar soft-part sarcomas (12, 13). In this study we uncovered a positive regulatory loop in which Wnt signaling stabilizes MITF independently of de novo protein synthesis. We describe three previously undetected GSK3 phosphorylation sites that are highly conserved in MITF, TFEB, TFE3, and TFEC, and show that these sites in MITF mediate protein stabilization when Wnt inhibits GSK3. We found that high levels of MITF in melanoma cell lines correlated with the expression of a large subset of lysosomal genes. Late endolysosomal structures were greatly expanded when MITF was expressed in the Tet-inducible C32 melanoma cell line. However, these late endolysosomes/MVBs did not form proteolytically active lysosomes in assays using BSA-DQ added to the culture medium. The expansion of late endosomes resulted in increased sensitivity to Wnt signaling through an ESCRT-dependent mechanism. The endosomal/MVB vesicles induced by MITF colocalized with pLRP6, Axin1, GSK3, and p-β-catenin in the presence of Wnt, explaining the increased Wnt signaling by the sequestration of the destruction complex in vesicular organelles (6, 9, 26).
The model in Fig. 6 proposes that a positive-feedback signaling loop drives the proliferative stages of melanoma. Increased MITF would cause an expansion of late endolysosomal vesicles that potentiates Wnt signaling. In turn, Wnt signaling stabilizes MITF by inhibiting GSK3 and reducing the C-terminal GSK3 phosphorylations of MITF. The proliferative stages of melanoma are associated with a peak in canonical Wnt signaling and also increased MITF activity (37). Vesicular trafficking misregulation is emerging as a fundamental hallmark of melanomas and perhaps of other cancers (32, 38). Our model helps explain how a lineage-addiction oncogene could cause perturbations in the endolysosomal pathway and increase cellular responses to Wnt signaling (Fig. 6).
Fig. 6.
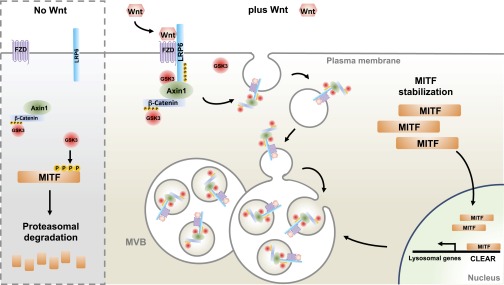
Model portraying a positive feedback loop involving MITF, MVBs, and Wnt signaling in proliferative stages of melanoma. Without Wnt signaling, GSK3 phosphorylates MITF on novel C-terminal phosphorylation sites, targeting MITF for proteasomal degradation. Upon Wnt signaling, destruction complex components are sequestered into MVBs, inhibiting GSK3 and stabilizing MITF. In turn, MITF induces late endolysosomes that further sequester destruction complex components upon Wnt signaling, enhancing overall Wnt responsiveness. This positive-feedback loop is proposed to function in the proliferative stages of melanoma, in which MITF and Wnt signaling peak.
MITF and Lysosomal Biogenesis.
TFEB is the master regulator of lysosomal biogenesis (22), capable of orchestrating cellular clearance pathways by promoting transcription of a set of genes downstream of the CLEAR element (33). Recently, it has been shown that TFE3 is capable of a similar response (25). Although MITF, TFEB, and TFE3 share high-sequence homology, MITF had not been previously recognized as being capable of triggering lysosomal biogenesis (22, 25). However, the idea that MITF may promote a lysosomal gene response is not entirely surprising. One of the MITF E-box consensus DNA binding sites (5′-CACGTG-3′) (39) is contained within the TFEB consensus binding site (CLEAR element) (5′-GTCACGTGAC-3′) (22). In addition, melanomas were recently shown to have a strong enrichment of the lysosome Gene Ontology (GO:0005764) gene set in comparison with other cancers (32).
MITF, the master regulator of melanocytes, regulates the expression of melanosomal genes (40). Melanosomes are commonly termed lysosome-related organelles. Although it is tempting to speculate that the correlation between MITF and lysosomal genes described here derives from the relationship between melanosomes and lysosomes, melanosomes are distinct from conventional endosomes and lysosomes, and represent a distinct lineage of organelles (41, 42). Proteomic analyses have revealed that melanosomes contain a unique proteomic profile and share only a few proteins with lysosomes (6 in the case of premelanosomes and 12 in the case of mature melanosomes), (43, 44). Our analyses revealed that MITF correlates with, and is capable of up-regulating, many genes that are exclusively lysosomal and not considered melanosomal. The likely explanation is that MITF, when amplified at the genomic or overexpressed at the transcriptional level in melanomas, may bind promiscuously to TFEB binding sites (CLEAR elements) and drive expression of a large subset of lysosomal genes.
The observation that MITF-M, but not TFEB, significantly correlated with lysosomal genes in melanoma may be a reflection of their distinct regulations. The activity of TFEB is controlled by mTORC1 (23, 24). In nutrient-rich conditions, TFEB localizes to the lysosomal outer membrane through binding to Rag GTPases (45) via its amino terminal domain. Active mTORC1 on the lysosomal surface phosphorylates TFEB and causes its retention in the cytoplasm by 14-3-3 proteins (23, 24). Only when mTOR is inhibited, for example during starvation or lysosomal stress, is unphosphorlylated TFEB free to enter the nucleus (23, 24). Therefore, TFEB-mediated lysosomal biogenesis depends more on nutritional status and cellular localization than on absolute TFEB levels. The MITF-A and MITF-D isoforms are similarly regulated through their 30 N-terminal amino acids required for binding to Rag GTPases at the lysosome surface (23, 45).
However, the MITF-M isoform, which is the one expressed in melanomas, lacks this N-terminal domain required for lysosomal localization and mTOR phosphorylation. Consequently, MITF-M is a constitutively nuclear protein (45). We propose that, when overexpressed, MITF-M binds promiscuously to CLEAR element lysosomal genes without being restrained by mTOR signaling. This would be sufficient to drive partial endolysosomal biogenesis, resulting in inactive lysosomes and enhanced Wnt signaling.
Alterations in lysosomal content, distribution, and volume have been associated with cancer (46). Recently, it has been shown how melanomas use a lineage-specific Rab7-mediated wiring of the endolysosomal pathway to promote proliferation (32). This finding is consistent with the very strong increase in Rab7+ vesicles we found upon MITF induction in C32 cells. It appears that endolysosomal regulation is particularly important in melanoma (32). Perhaps disregulated endolysosomal pathways are a feature of malignancies associated with other MiT family oncogenes as well.
MITF Enhances Wnt Signaling.
Although MITF induction increased expression of lysosomal genes in melanoma, it did not increase lysosomal activity assessed by the cleavage of BSA-DQ. This result was reminiscent of the effects of CQ treatment or presenilin mutations in which accumulation of late endolysosomal structures enhances Wnt signaling through an increase in the sequestration of the Wnt receptor/β-catenin destruction complex (26).
Because MITF induction in C32 melanoma cells caused an expansion of MVBs and late endolysosomal/MVB vesicles marked by CD63, LAMP1, and Rab7, we investigated whether MITF expression could affect Wnt signaling and found that Wnt signaling was enhanced by MITF both in Xenopus embryos and in an MITF-inducible melanoma cell line. This increase in Wnt responsiveness required the ESCRT machinery necessary for intraluminal vesicle formation in MVBs. Axin1, GSK3, p-β-catenin, and pLRP6 colocalized with MITF-induced vesicles upon Wnt signaling.
Although Wnt is one of the main signaling pathways commonly perturbed in metastatic melanomas (47), its role in oncogenesis is paradoxical. Canonical Wnt signaling and β-catenin activation seem to be a key step in the initiation of melanoma (34). However, β-catenin has also been shown to suppress invasion, and loss of β-catenin predicts poor rates of survival in patients (48–50). Additionally, BRAF signaling, a hallmark of most melanomas, has been reported to inhibit Wnt signaling in melanoma cells (51). In some melanoma lines, Wnt signaling synergizes with BRAF inhibitors in decreasing tumor growth and increasing apoptosis (51). This finding has caused re-evaluation of the oncogenic nature of Wnt signaling in these cancers (52). Given the importance of the endolysosomal pathway in Wnt signaling (6, 9) and Rab7 wiring in melanomas (32), it is tempting to speculate that the sequestration of the destruction complex components, which confers canonical Wnt signaling, is particularly high in melanomas.
MITF Protein Stabilization by Wnt/GSK3.
Wnt, through GSK3 inhibition, stabilizes many proteins in addition to β-catenin (6, 7, 53). Wnt-dependent stabilization of proteins (Wnt/STOP) peaks during G2/M, preventing protein degradation in preparation for cellular division (8). The possibility that Wnt signaling could increase the stability of the MiT family of transcription factors, which behave as oncogenes (12, 13), is potentially significant. We found that Wnt signaling indeed stabilized MITF protein levels in melanoma cell lines. It is well documented that Wnt signaling also participates (through Lef1 and other transcription factors) in driving transcriptional expression of MITF (54–56). We now find that Wnt can also regulate MITF at the protein degradation level, independently of transcription.
MITF is clearly a driver of melanoma, because a recurrent mutation in MITF predisposes to familial melanoma. This mutant, MITF E318K, has impaired sumoylation and increased activity (20). The same mutation also predisposes for renal cell carcinoma (57). Ectopic MITF expression, in combination with activated BRAF, can transform human primary melanocytes (19). We found that Wnt signaling strongly potentiated MITF transcriptional activity, as measured by an increase in MART1 transcripts. Importantly, this experiment was carried out in the presence of CHX to prevent de novo synthesis of MITF. During Wnt signaling, only the newly synthesized β-catenin is competent for signaling (35). By preventing protein synthesis, we also ruled out any effect of β-catenin accumulation on the observed increase in MITF activity, indicating that the enhanced activity is caused by increased stability, activity, or both. We conclude that Wnt signaling, by inhibiting GSK3, could enhance the stability of MITF and contribute to its oncogenic effects (Fig. 6).
The C-Terminal GSK3 Phosphorylation Sites of MITF.
MITF is highly regulated at the transcriptional, posttranscriptional, and posttranslational level (58). MITF-M protein has been shown to be phosphorylated at S73, via ERK1/2, and at S409, via p90/RSK1, in response to tyrosine kinase KIT receptor activation (18). MITF has also been proposed to be phosphorylated at S298 by GSK3, and this residue is often mutated in Waardenburg syndrome (59).
Here are presented three novel GSK3 phosphorylation sites (S397, S401, and S405) conserved in the C terminus of MITF. When these residues were mutated into alanines, the MITF protein became more stable, suggesting a role for GSK3 in targeting MITF for proteasomal degradation. In agreement with this proposal, the priming phosphorylation at S409 has been shown to participate in MITF proteasomal targeting (14). Using a custom antiphospho MITFGSK3 antibody, we confirmed that the new sites are indeed phosphorylated by GSK3 in vivo. Phosphopeptides corresponding to the GSK3 phosphorylations have been recorded by proteomic discovery-mode mass spectrometry (see Phosphosite.org), but their regulation has not been previously analyzed in the scientific literature.
MITF, MVBs, Wnt, and Melanoma Proliferation.
As in the case of Wnt signaling, the role of MITF in melanoma is paradoxical, promoting transcription of genes with antagonistic behaviors (60). As a melanocyte master regulator, MITF drives cells toward differentiation, and high MITF levels have antiproliferative effects (61). However, MITF has been also described as a lineage addiction—or lineage survival—oncogene required for melanoma survival and proliferation (18, 19, 62). The levels of MITF protein are critical in melanomas, as they determine cellular phenotype: low levels cause G1 arrest, stem cell-like properties, and often confer invasive behavior (63). Intermediate MITF levels endow cells with increased proliferative capacity. However, higher levels drive cells toward differentiation and G1 arrest. Thus, MITF is considered a molecular rheostat (39, 63–65).
Melanomas, in terms of their expression profiles and clinical behavior, can be subdivided into two phenotypes: proliferative and invasive. Among the genes up-regulated in the proliferative melanoma state are many MITF targets, as well as many Wnt target genes (37). This finding contrasts to the invasive state, in which MITF and Wnt targets are down-regulated (37). These different cellular phenotypes coexist within a heterogeneous tumor and are determined by MITF levels (37). However, melanoma cells have the capability to switch phenotypes by modifying MITF levels. Recently, this phenotype switch has been elegantly exploited as part of an effective antimelanoma therapy strategy (66).
The results presented here suggest that in the proliferative stages of melanoma increased MITF levels may potentiate Wnt signaling by expanding the endolysosomal compartment, and that Wnt in turn stabilizes MITF by preventing GSK3-mediated proteasomal degradation.
Materials and Methods
Analysis of Gene Expression by Microarray.
Melanoma cell lines were cultured in RPMI medium (Life Technologies) supplemented with penicillin/streptomycin and FBS. All 51 cell line cultures used for arrays were harvested at 50–70% confluency, centrifuged, quick frozen, and characterized by mtDNA sequence before use. For microarray analysis of gene-expression, the RNA was isolated using the Qiagen RNeasy protocol and quantitated using a NanoDrop Spectrophotometer (Agilent Technologies). Next, 825 ng of high-quality total RNA with RNA integrity number greater than 8.0 was labeled with cyanine 5-CTP or cyanine 3-CTP using the Low RNA Input Fluorescent Linear Amplification Kit (Agilent Technologies) and purified on RNeasy Mini columns (Qiagen). Labeled RNA was then hybridized to Agilent Human 44K expression arrays that include 44,000 probes and compared with a labeled mixed-reference sample consisting of a pool of equal amounts of RNA from each of 47 melanoma cell lines. Analysis of the microarray data were done using Rosetta Resolver (v7.2.2.0) system with P values equal or less than 0.01.
GSEA.
The correlation between MITF or TFEB and the set of lysosomal genes was analyzed by the GSEA approach (29). The set of 89 lysosomal genes was from Sardiello et al. (22). Normalized gene expression data for melanoma cell lines was obtained from the Hoek et al. dataset (30), which consists of three independent subsets—Zurich, Mannheim, and Philadelphia—containing among them 83 additional melanoma lines. Each subset was scaled and the Pearson correlation between MITF or TFEB and all genes was calculated. MITF was represented as a single probe and for TFEB the average scaled expression vector of two probes was used. Each gene was collapsed to its probe with the highest absolute correlation. Genes were ranked by their correlation with MITF or TFEB and the three subsets combined using average gene ranks. The combined ranked lists were analyzed for enrichment of the lysosomal gene set. Statistical significance was assessed with a permutation based Kolmogorov–Smirnoff nonparametric rank test (1,000 permutations) (29).
Cell Culture.
For MITF induction in the C32 MITF Tet-inducible melanoma line, cells were grown in medium containing Tet 1 μg/mL (Sigma) for 4 d to allow MITF protein to be synthesized. Recombinant murine Wnt3a protein (PeproTech) was added to C32 cells at 80 ng/mL for 5–6 h. CQ was dissolved in water and added to C32 cells at 100 μM (Sigma). For GSK3 inhibition, cells were treated with BIO (or meBIO as a control) (67) at 5 μM for 5 h or LiCl at 30 mM for 8 h. For inhibition of protein synthesis, CHX (Sigma) was dissolved in ethanol and used at a final concentration of 20 mg/mL (6).
Immunostainings.
For immunostainings, round coverslips were placed in 12-well plates and extensively washed with ethanol, Dulbecco’s PBS (DPBS, Gibco), and culture medium. C32 melanoma cells, previously grown with or without Tet for 4 d, were seeded into these 12-well plates, and 24 h later treated with Wnt3a. After 6 h of Wnt3a treatment, cells were fixed with fresh 4% (wt/vol) paraformaldehyde (Sigma #P6148) (PFA) in DPBS for 20 min, washed for 15–20 min three times in freshly prepared DPBS, permeabilized by treatment with 0.2% (vol/vol) Triton X-100 in DPBS for 10 min, washed for 15–20 min three times in DPBS and treated with 0.5% (wt/vol) SDS for 5 min for antigen retrieval. Wells were then washed for 15–20 min three times in DPBS and blocked with 5% (vol/vol) goat serum plus 0.5% (wt/vol) BSA in DPBS for at least 2 h (blocking solution). Cells were then incubated with primary antibodies, diluted 1:4 in blocking solution, overnight at 4 °C. The following day, wells were washed for 15–20 min three times DPBS, and secondary antibodies (diluted in 1:4 blocking solution) applied for 2 h at room temperature. After washing in DPBS, coverslips were removed and mounted in ProLong Gold antifade reagent with DAPI (Life Technologies) to stain cell nuclei. A Zeiss Imager Z.1 microscope with Apotome was used to analyze and photograph immunofluorescence, using a 63× objective unless otherwise stated. To quantify immunostainings for MITF protein in C32 cells, staining of individual nuclei was measured with ImagJ software (NIH). Because uninduced C32 cells lack MITF staining, the perimeter of the nucleus was determined by DAPI staining. The MITF protein levels of 120–150 individual nuclei were measured for each condition. Primary antibodies used for immunostaining in this study were: anti-MITF 1:250 (DAKO #M3621), anti–p-β-catenin 1:300 (S33/37/T41) (Cell Signaling Technologies #9561), anti-Axin1 clone A5 1:300 (Millipore #05–1579), anti–pLRP6 1:300 (S1490) (Cell Signaling Technologies #2568S), anti-CD63 1:400 (BD Pharmingen #556019), anti-Rab7 1:400 (D95F2) (Cell Signaling Technologies #9367S), and anti-LAMP1 1:400 (DSHB #H4A3). For generation of the custom pMITFGSK3 antibody, a synthetic peptide [Ac-C(Ahx)PGA(pS)KTS(pS)RRS-amide] was used to immunize a rabbit (Covance). Although affinity purification failed, the remaining high-titer antiserum is effective at a concentration of 1:250 for detecting pMITFGSK3 in immunostainings of C32 melanoma cells. Secondary antibodies were Alexa Fluor 488-conjugated AffiniPure Donkey anti-Rabbit IgG and Cy3 conjugated AffiniPure Donkey anti-Mouse IgG (Jackson Immuno Research Laboratories).
BSA-DQ Lysosomal Activity Assay and Lysotracker on Living Cells.
For studying lysosomal proteolysis on endocytosed BSA, C32 cells previously grown with or without Tet, were plated in six-well plates. Upon 60% confluency, cells were treated with 5 μg/mL of BSA-DQ (DQ Red BSA, Molecular Probes, #D-12051) diluted in prewarmed medium and incubated at 37 °C for 6 h. Cells were then briefly washed twice with prewarmed DPBS, trypsinized, fixed in suspension with 4% (wt/vol) PFA for 15 min at room temperature, and washed extensively with DPBS at room temperature. Fluorescence of cleaved BSA-DQ was analyzed by flow cytometry. For visualizing LysoTracker stainings by microscopy, C32 cells were previously grown with or without Tet were plated in 12-well plates containing coverslips as described above for immunostainings. Upon 60% confluency, cells were treated with Lysotracker (LysoTracker Red DND-99, Invitrogen, #L7528), diluted 1:1,000 in prewarmed medium, incubated for 1 min at 37 °C, and washed with prewarmed DPBS. Cells were fixed with 4% PFA for 15 min at room temperature in the dark, and washed extensively with DPBS. Coverslips were then mounted with ProLong Gold antifade reagent with DAPI (Life Technologies) and visualized with Zeiss Imager Z.1 microscope with Apotome. For quantitatively analyzing Lysotracker stainings, C32 cells were previously grown with or without Tet were plated in six-well plates and, upon 60% confluency, cells were treated with Lysotracker as described above in prewarmed medium and incubated for 1 min at 37 °C, and washed extensively with prewarmed DPBS. Cells were then trypsinized, fixed in suspension with 4% PFA for 15 min at room temperature in the dark, washed several times with DPBS, and analyzed by flow cytometry.
DNA Constructs.
Human MITF-M (GB# NM000248) was cloned into the pCS2 vector. This MITF-M construct was used to generate the MITF GSK3 mutant (MITF-GM) by PCR-based site-directed mutagenesis (QuikChange II Site-Directed Mutagenesis, Stratagene). RFP-GSK3 was from ref. 6. The CLEAR element Luciferase construct was from ref. 22, and was transfected at 0.6 µg per 12-well plate, together with 0.2 µg of SV40-Renilla luciferase construct for normalization (6).
Statistical Analyses.
Results from three or more independent experiments are presented as the mean ± SEM. Excel (Microsoft) was used for statistical analyses, applying the two-tailed t test as appropriate. Significant differences of means are indicated as **P < 0.01 and ***P < 0.001. For Xenopus embryo assays, Western blot analyses, siRNA in tissue culture, lentiviral transductions, luciferase assays, RT-qPCR analysis, and additional cell culture information, see SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Dr. Glen Boyle (Brisbane, Australia) for providing the C32 inducible micropthalmia-associated transcription fact melanoma cell line; Andrea Ballabio for coordinated lysosomal expression and eegulation element luciferase reporter; and Roel Nusse for β-catenin activated reporter firefly and Renilla luciferase lentiviral constructs. Lentivirus production was by University of California, Los Angeles (UCLA) Vectorcore, supported by CURE/P30DK04130; flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center Flow Cytometry Core Facility supported by NIH awards CA-16042 and AI-28697; D.P. was supported by an International Fulbright Science and Technology Award of the State Department and by a UCLA Dissertation Year Fellowship; and L.R. was supported by the V Foundation-Gil Nickel Family Endowed Fellowship in Melanoma Research. This work was made possible by support from NIH HD 21502-26, the Norman Sprague Molecular Oncology Endowment, and the Howard Hughes Medical Institute, of which E.M.D.R. is an Investigator.
Footnotes
The authors declare no conflict of interest.
Data deposition: The microarray expression data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE64497).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424576112/-/DCSupplemental.
References
- 1.MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009;10(7):468–477. doi: 10.1038/nrm2717. [DOI] [PubMed] [Google Scholar]
- 3.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 4.Cadigan KM, Peifer M. Wnt signaling from development to disease: Insights from model systems. Cold Spring Harb Perspect Biol. 2009;1(2):a002881. doi: 10.1101/cshperspect.a002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peifer M, Sweeton D, Casey M, Wieschaus E. Wingless signal and Zeste-white 3 kinase trigger opposing changes in the intracellular distribution of Armadillo. Development. 1994;120(2):369–380. doi: 10.1242/dev.120.2.369. [DOI] [PubMed] [Google Scholar]
- 6.Taelman VF, et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143(7):1136–1148. doi: 10.1016/j.cell.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acebron SP, Karaulanov E, Berger BS, Huang Y-L, Niehrs C. Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol Cell. 2014;54(4):663–674. doi: 10.1016/j.molcel.2014.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Bilić J, et al. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science. 2007;316(5831):1619–1622. doi: 10.1126/science.1137065. [DOI] [PubMed] [Google Scholar]
- 9.Vinyoles M, et al. Multivesicular GSK3 sequestration upon Wnt signaling is controlled by p120-catenin/cadherin interaction with LRP5/6. Mol Cell. 2014;53(3):444–457. doi: 10.1016/j.molcel.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 10.Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464(7290):864–869. doi: 10.1038/nature08849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hemesath TJ, et al. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994;8(22):2770–2780. doi: 10.1101/gad.8.22.2770. [DOI] [PubMed] [Google Scholar]
- 12.Davis IJ, Fisher DE. MiT transcription factor associated malignancies in man. Cell Cycle. 2007;6(14):1724–1729. doi: 10.4161/cc.6.14.4484. [DOI] [PubMed] [Google Scholar]
- 13.Haq R, Fisher DE. Biology and clinical relevance of the micropthalmia family of transcription factors in human cancer. J Clin Oncol. 2011;29(25):3474–3482. doi: 10.1200/JCO.2010.32.6223. [DOI] [PubMed] [Google Scholar]
- 14.Wu M, et al. c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev. 2000;14(3):301–312. [PMC free article] [PubMed] [Google Scholar]
- 15.Ferron M, et al. A RANKL-PKCβ-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev. 2013;27(8):955–969. doi: 10.1101/gad.213827.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Packer SO. The eye and skeletal effects of two mutant alleles at the microphthalmia locus of Mus musculus. J Exp Zool. 1967;165(1):21–45. doi: 10.1002/jez.1401650103. [DOI] [PubMed] [Google Scholar]
- 17.Hodgkinson CA, et al. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell. 1993;74(2):395–404. doi: 10.1016/0092-8674(93)90429-t. [DOI] [PubMed] [Google Scholar]
- 18.Steingrímsson E, Copeland NG, Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet. 2004;38(1):365–411. doi: 10.1146/annurev.genet.38.072902.092717. [DOI] [PubMed] [Google Scholar]
- 19.Garraway LA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436(7047):117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 20.Yokoyama S, et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. 2011;480(7375):99–103. doi: 10.1038/nature10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuse N, Yasumoto K, Suzuki H, Takahashi K, Shibahara S. Identification of a melanocyte-type promoter of the microphthalmia-associated transcription factor gene. Biochem Biophys Res Commun. 1996;219(3):702–707. doi: 10.1006/bbrc.1996.0298. [DOI] [PubMed] [Google Scholar]
- 22.Sardiello M, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 23.Roczniak-Ferguson A, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5(228):ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Settembre C, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31(5):1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martina JA, et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014;7(309):ra9. doi: 10.1126/scisignal.2004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobrowolski R, et al. Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Reports. 2012;2(5):1316–1328. doi: 10.1016/j.celrep.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Settembre C, Ballabio A. TFEB regulates autophagy: An integrated coordination of cellular degradation and recycling processes. Autophagy. 2011;7(11):1379–1381. doi: 10.4161/auto.7.11.17166. [DOI] [PubMed] [Google Scholar]
- 28.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2(10):769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian A, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoek KS, et al. Novel MITF targets identified using a two-step DNA microarray strategy. Pigment Cell Melanoma Res. 2008;21(6):665–676. doi: 10.1111/j.1755-148X.2008.00505.x. [DOI] [PubMed] [Google Scholar]
- 31.Settembre C, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15(6):647–658. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alonso-Curbelo D, et al. RAB7 controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell. 2014;26(1):61–76. doi: 10.1016/j.ccr.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 33.Palmieri M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20(19):3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 34.O’Connell MP, Weeraratna AT. Hear the Wnt Ror: How melanoma cells adjust to changes in Wnt. Pigment Cell Melanoma Res. 2009;22(6):724–739. doi: 10.1111/j.1755-148X.2009.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li VSW, et al. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 2012;149(6):1245–1256. doi: 10.1016/j.cell.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Du J, et al. MLANA/MART1 and SILV/PMEL17/GP100 are transcriptionally regulated by MITF in melanocytes and melanoma. Am J Pathol. 2003;163(1):333–343. doi: 10.1016/S0002-9440(10)63657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoek KS, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008;68(3):650–656. doi: 10.1158/0008-5472.CAN-07-2491. [DOI] [PubMed] [Google Scholar]
- 38.Ibarrola-Villava M, et al. Genes involved in the WNT and vesicular trafficking pathways are associated with melanoma predisposition. Int J Cancer. 2014 doi: 10.1002/ijc.29257. [DOI] [PubMed] [Google Scholar]
- 39.Strub T, et al. Essential role of microphthalmia transcription factor for DNA replication, mitosis and genomic stability in melanoma. Oncogene. 2011;30(20):2319–2332. doi: 10.1038/onc.2010.612. [DOI] [PubMed] [Google Scholar]
- 40.Levy C, Khaled M, Fisher DE. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12(9):406–414. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 41.Raposo G, Tenza D, Murphy DM, Berson JF, Marks MS. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J Cell Biol. 2001;152(4):809–824. doi: 10.1083/jcb.152.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raposo G, Marks MS. Melanosomes—Dark organelles enlighten endosomal membrane transport. Nat Rev Mol Cell Biol. 2007;8(10):786–797. doi: 10.1038/nrm2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basrur V, et al. Proteomic analysis of early melanosomes: Identification of novel melanosomal proteins. J Proteome Res. 2003;2(1):69–79. doi: 10.1021/pr025562r. [DOI] [PubMed] [Google Scholar]
- 44.Chi A, et al. Proteomic and bioinformatic characterization of the biogenesis and function of melanosomes. J Proteome Res. 2006;5(11):3135–3144. doi: 10.1021/pr060363j. [DOI] [PubMed] [Google Scholar]
- 45.Martina JA, Diab HI, Li H, Puertollano R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell Mol Life Sci. 2014;71(13):2483–2497. doi: 10.1007/s00018-014-1565-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kallunki T, Olsen OD, Jäättelä M. Cancer-associated lysosomal changes: Friends or foes? Oncogene. 2013;32(16):1995–2004. doi: 10.1038/onc.2012.292. [DOI] [PubMed] [Google Scholar]
- 47.Valsesia A, et al. Network-guided analysis of genes with altered somatic copy number and gene expression reveals pathways commonly perturbed in metastatic melanoma. PLoS ONE. 2011;6(4):e18369. doi: 10.1371/journal.pone.0018369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arozarena I, et al. In melanoma, beta-catenin is a suppressor of invasion. Oncogene. 2011;30(45):4531–4543. doi: 10.1038/onc.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chien AJ, et al. Activated Wnt/β-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci USA. 2009;106(4):1193–1198. doi: 10.1073/pnas.0811902106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kageshita T, et al. Loss of β-catenin expression associated with disease progression in malignant melanoma. Br J Dermatol. 2001;145(2):210–216. doi: 10.1046/j.1365-2133.2001.04336.x. [DOI] [PubMed] [Google Scholar]
- 51.Biechele TL, et al. Wnt/β-catenin signaling and AXIN1 regulate apoptosis triggered by inhibition of the mutant kinase BRAFV600E in human melanoma. Sci Signal. 2012;5(206):ra3. doi: 10.1126/scisignal.2002274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lucero OM, Dawson DW, Moon RT, Chien AJ. A re-evaluation of the “oncogenic” nature of Wnt/β-catenin signaling in melanoma and other cancers. Curr Oncol Rep. 2010;12(5):314–318. doi: 10.1007/s11912-010-0114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim N-G, Xu C, Gumbiner BM. Identification of targets of the Wnt pathway destruction complex in addition to β-catenin. Proc Natl Acad Sci USA. 2009;106(13):5165–5170. doi: 10.1073/pnas.0810185106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dorsky RI, Raible DW, Moon RT. Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev. 2000;14(2):158–162. [PMC free article] [PubMed] [Google Scholar]
- 55.Takeda K, et al. Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. J Biol Chem. 2000;275(19):14013–14016. doi: 10.1074/jbc.c000113200. [DOI] [PubMed] [Google Scholar]
- 56.Hsiao JJ, Fisher DE. The roles of microphthalmia-associated transcription factor and pigmentation in melanoma. Arch Biochem Biophys. 2014;563:28–34. doi: 10.1016/j.abb.2014.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bertolotto C, et al. French Familial Melanoma Study Group A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011;480(7375):94–98. doi: 10.1038/nature10539. [DOI] [PubMed] [Google Scholar]
- 58.Arnheiter H, et al. MITF—A matter of life and death for developing melanocytes. In: Hearing VJ, Leong SPL, editors. From Melanocytes to Melanoma: The Progression to Malignancy. Humana; Totowa, NJ: 2007. pp. 27–49. [Google Scholar]
- 59.Takeda K, et al. Ser298 of MITF, a mutation site in Waardenburg syndrome type 2, is a phosphorylation site with functional significance. Hum Mol Genet. 2000;9(1):125–132. doi: 10.1093/hmg/9.1.125. [DOI] [PubMed] [Google Scholar]
- 60.Palmieri G, et al. Main roads to melanoma. J Transl Med. 2009;7(1):86. doi: 10.1186/1479-5876-7-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wellbrock C, Marais R. Elevated expression of MITF counteracts B-RAF-stimulated melanocyte and melanoma cell proliferation. J Cell Biol. 2005;170(5):703–708. doi: 10.1083/jcb.200505059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 63.Carreira S, et al. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20(24):3426–3439. doi: 10.1101/gad.406406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoek KS, Goding CR. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010;23(6):746–759. doi: 10.1111/j.1755-148X.2010.00757.x. [DOI] [PubMed] [Google Scholar]
- 65.Cheli Y, et al. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene. 2011;30(20):2307–2318. doi: 10.1038/onc.2010.598. [DOI] [PubMed] [Google Scholar]
- 66.Sáez-Ayala M, et al. Directed phenotype switching as an effective antimelanoma strategy. Cancer Cell. 2013;24(1):105–119. doi: 10.1016/j.ccr.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 67.Meijer L, et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10(12):1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.