

Significance
In the endoplasmic reticulum (ER), N-glycans on glycoproteins play important roles in dictating the folding status of proteins by a sophisticated N-glycan–dependent protein quality control machinery. In this study we identified the dysregulation of ER-associated degradation (ERAD) in cells that were defective in the cytosolic deglycosylating enzyme, Ngly1. ERAD dysregulation was caused by an unexpected deglycosylating activity of endo-β-N-acetylglucosaminidase, another cytosolic deglycosylation enzyme, and this action resulted in the intracellular formation of protein aggregates. Our results clearly point to the critical role of N-glycans even in cytosolic events of the ERAD process by controlling the conformation/solubility of proteins. This study may also provide a potential mechanism for explaining the pathology of a human genetic disorder caused by mutations in the NGLY1 gene.
Keywords: PNGase (Ngly1), ENGase, protein aggregates, glycoprotein, ERAD
Abstract
The cytoplasmic peptide:N-glycanase (PNGase; Ngly1 in mice) is a deglycosylating enzyme involved in the endoplasmic reticulum (ER)-associated degradation (ERAD) process. The precise role of Ngly1 in the ERAD process, however, remains unclear in mammals. The findings reported herein, using mouse embryonic fibroblast (MEF) cells, that the ablation of Ngly1 causes dysregulation of the ERAD process. Interestingly, not only delayed degradation but also the deglycosylation of a misfolded glycoprotein was observed in Ngly1−/− MEF cells. The unconventional deglycosylation reaction was found to be catalyzed by the cytosolic endo-β-N-acetylglucosaminidase (ENGase), generating aggregation-prone N-GlcNAc proteins. The ERAD dysregulation in cells lacking Ngly1 was restored by the additional knockout of ENGase gene. Thus, our study underscores the functional importance of Ngly1 in the ERAD process and provides a potential mechanism underlying the phenotypic consequences of a newly emerging genetic disorder caused by mutation of the human NGLY1 gene.
Endoplasmic reticulum (ER)-associated degradation (ERAD) constitutes one of the quality control mechanisms for newly synthesized proteins in the ER. The ERAD process involves a series of events, including aberrant domain recognition, ubiquitination, translocation from the ER to the cytosol, and degradation by proteasomes. Numerous lines of evidence point to the existence of an ERAD system dedicated to N-linked glycoproteins; in this system, specific N-glycan structures dictate the folding status of client glycoproteins (1, 2). Once glycoproteins in the ER lumen are targeted for degradation, they are retrotranslocated into the cytosol, where the 26S proteasome plays a central role in their degradation (3). During the degradation process, N-glycans are removed by the action of the cytoplasmic peptide:N-glycanase (PNGase) (4–6).
Activity of the cytoplasmic PNGase was first described in mammalian cells (7, 8), and the gene encoding cytoplasmic PNGase (PNG1 in yeast; Ngly1/NGLY1 in mice/human) is widely distributed throughout eukaryotes (9). The functional importance of cytoplasmic PNGase in the ERAD process is evident in yeast (10–13). On the other hand, the suppression of Ngly1 gene expression by siRNA in mammalian cells resulted in a reduced deglycosylation of T-cell receptor α subunit (TCRα) or MHC class I heavy chain, whereas no significant delay in their degradation was observed (14, 15). Moreover, Z-VAD-fmk, a pan-caspase inhibitor, was shown to inhibit cytoplasmic PNGase activity in vivo, but it did not impede the degradation of MHC class I heavy chain (16). Consequently, the functional importance of the cytoplasmic PNGase remains elusive in mammalian cells.
PNGase-mediated deglycosylation generates free oligosaccharides in the cytosol (17). Recent evidence suggests that a nonlysosomal degradation pathway exists for these cytosolic free glycans (17). This degradation process involves cytosolic endo-β-N-acetylglucosaminidase (ENGase) (18, 19). Although the ENGase is believed to be involved in the catabolism of cytosolic free oligosaccharides, recent evidence shows that it can deglycosylate glycoproteins in vivo to generate N-GlcNAc–bearing proteins in Arabidopsis thaliana (20), raising the possibility that this enzyme may also act as a deglycosylation enzyme for misfolded glycoproteins in the cytosol (21, 22) (Fig. 1A).
Fig. 1.
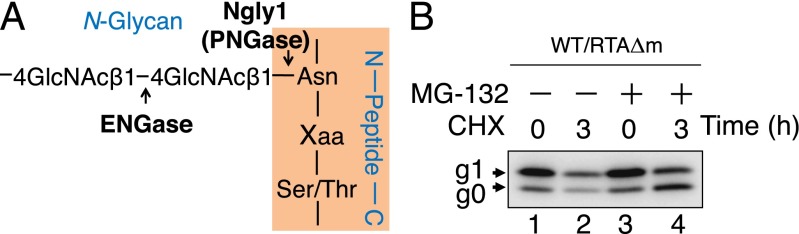
RTAΔm serves as an ERAD substrate in MEF cells. (A) Schematic representation of the ENGase and Ngly1 (PNGase) cleavage sites on N-linked glycans. (B) Effect of cycloheximide and MG-132 on the stability of RTAΔm. Each lane contains a cell extract from ∼5 × 104 cells. Cell extracts were subjected to Western blot analysis, and RTAΔm was detected using anti-V5 antibody.
Recently, patients harboring mutations on the NGLY1 gene, an ortholog of the cytoplasmic PNGase in mammalian cells (23), have been described (24, 25). Although this observation emphasizes the functional importance of this protein in mammalian cells, mechanistic insight into the phenotypic consequences of these patients remains unclarified. In this study, we established an ERAD model substrate, RTAΔm, and demonstrated that the delay in its degradation was evident in Ngly1−/− mouse embryonic fibroblast (MEF) cells. Interestingly, the delay was canceled by additional gene knockout of ENGase. The degradation of RTAΔm in double-knockout cells remains proteasome-dependent, clearly indicating that the presence of an N-glycan on RTAΔm did not affect the efficiency of proteasomal degradation. Moreover, the occurrence of N-GlcNAc–modified RTAΔm in Ngly1−/− MEF cells was identified by MS analysis, demonstrating that the ENGase-mediated deglycosylation of an ERAD substrate can indeed occur in vivo. Interestingly, the N-GlcNAc protein was shown to form stable, mild detergent-insoluble protein aggregates. We propose the critical function of N-glycans, maintaining the conformation/solubility of the cognate proteins destined for proteasomal degradation in the cytosol. In addition, our results provide not only insight into the pathology of NGLY1 deficiency, a newly found human genetic disorder, but also a therapeutic strategy for the disease by inhibiting ENGase activity.
Results
Establishment of RTAΔm as an Ngly1-Dependent Model Glycoprotein ERAD Substrate in Mammalian Cells.
To provide insight into the functional significance of Ngly1 in the glycoprotein ERAD processes in mammalian cells, we aimed to establish a model ERAD substrate that could be degraded in an Ngly1-dependent fashion. Because we have previously established the ricin A chain nontoxic mutant (RTAΔ) as the first Png1 (yeast PNGase)-dependent ERAD substrate in yeast (10, 11), we examined whether this protein could also be degraded by a similar mechanism in MEF cells. To target RTAΔ to the N-glycosylation pathway, an ER-targeting signal peptide and an ER retention signal was introduced to the N and C terminus of RTAΔ, respectively (Fig. S1A). RTAΔ has two potential N-glycosylation sites at N10 and N236 (Fig. S1A), with the N10 site almost exclusively occupied by N-glycan in yeast (10). To avoid any potential deglycosylation by Ngly1 or ENGase, RTAΔ was expressed in Ngly1−/−Engase−/− double knockout (DKO) MEF cells and was analyzed by Western blotting. As shown in Fig. S1B, three bands corresponding to diglycosylated (g2), monoglycosylated (g1), and nonglycosylated (g0) RTAΔ were detected. To clarify which of the potential glycosylation sites were used, mutants for each N-glycosylation site were generated (N10Q and N236Q mutant; Fig. S1A). As shown in Fig. S1B, essentially no N-glycosylation was observed for the N10Q mutant, whereas N236Q is expressed mainly as a monoglycosylated form. As expected, N10Q/N236Q mutation resulted in the absence of glycosylated forms (Fig. S1B). These results clearly suggest that N10 of RTAΔ is the main N-glycosylation site in mammalian cells, which is consistent with the case in yeast. For the following experiments, we used the monoglycosylatable RTAΔN236Q mutant (RTAΔm) to simplify the N-glycosylation pattern.
When protein translation was inhibited for 3 h by a cycloheximide treatment, the amount of RTAΔm in WT MEF cells was clearly decreased (Fig. 1B, lanes 1 and 2). Moreover, the degradation of RTAΔm was inhibited by addition of MG-132, a proteasome inhibitor (Fig. 1B, compare lanes 2 and 4). Furthermore, proteasome inhibition resulted in an increase in the g0 form of RTAΔm, indicating the deglycosylation of RTAΔm (Fig. 1B, compare lanes 3 and 4). The deglycosylation of glycoprotein ERAD substrates is often observed upon treatment of mammalian cells with proteasome inhibitors (26–32). Taken together, these results show that, as in the case of yeast (10, 11), RTAΔm is degraded by proteasomal activity and therefore can serve as an ERAD model substrate in mammalian cells.
g0-RTAΔm Is Generated and Stabilized in Ngly1−/− MEF Cells.
Next, we expressed RTAΔm in MEF cells derived from WT, Engase−/−, Ngly1−/−, and DKO mice and carried out Western blotting analysis to compare the relative ratios of the g0/g1 forms of RTAΔm. In DKO MEF cells only ∼10% of RTAΔm was detected as a g0 form (Fig. 2A, lane 4). Because DKO MEF cells do not have deglycosylating enzymes in the cytoplasm, the g0 form RTAΔm observed in DKO MEF cells must represent the nonglycosylated form. Interestingly, when RTAΔm was expressed in Ngly1−/− MEF cells, the proportion of the g0 form (Fig. 2B, column 3, 35% ± 9%) was significantly higher than in DKO MEF cells (Fig. 2B, column 4, 10% ± 3%). Furthermore, we noted that the g0 proportion in Ngly1−/− MEF cells was comparable to that in WT (Fig. 2B, column 1, 26% ± 10%) and Engase−/− (Fig. 2B, column 2, 29% ± 10%) MEF cells. These data strongly suggest that the deglycosylation of RTAΔm can occur independently of Ngly1 activity (Fig. 2A; quantitative data in Fig. 2B).
Fig. 2.

Ngly1−/− caused delayed degradation of RTAΔm. (A) Western blot analysis of RTAΔm expressed in WT, Engase−/−, Ngly1−/−, and DKO MEF cells. Cell extracts from 5 × 104 cells (WT), 3.3 × 104 cells (Engase−/−), 1.5 × 104 cells (Ngly1−/−), and 2 × 104 cells (DKO) in each lane were analyzed after transfection with pEF/RTAΔN236Q/V5-myc/ER. (B) Quantification of the g0 form of RTAΔm in the four MEF cells. The data and error bars represent the average ± SD from seven independent samples. *P < 0.05; **P < 0.01 by Student’s t test. (C) Cycloheximide chase assay of RTAΔm in different MEF cells. Each lane contains ∼5 × 104 cells, and RTAΔ expression was detected using anti-V5 antibody after separation by SDS/PAGE. (D) (Top) Quantification of RTAΔm (g1 plus g0 form). The data and error bars represent the average ± SD from at least three independent experiments. The amount of RTAΔm at the zero time point for each cell type was set to 1. (Middle and Bottom) Relative quantitation of g1 (Middle) and g0 (Bottom) form of RTAΔm in WT, Ngly1−/−, and Engase−/− cells. The amount at the zero time point for each cell type was set to 1. (E) Effect of MG-132 on the stability of RTAΔm in WT, Ngly1−/−, and Engase−/− and DKO MEF cells. Each lane contains a cell extract from ∼5 × 104 cells.
To examine the stability of RTAΔm in MEF cells, RTAΔm was expressed in MEF cells and a cycloheximide-decay experiment was carried out. Our results clearly showed that RTAΔm in Ngly1−/− MEF cells was significantly stabilized (t1/2 >3 h) compared with WT (t1/2 = 1.4 ± 0.7 h), Engase−/− (t1/2 = 1.0 ± 0.2 h), or DKO MEF cells (t1/2 = 1.4 ± 0.5 h) (Fig. 2 C and D). The cytoplasmic PNGase-dependent ERAD of RTA-derived protein was consistent with observations in Saccharomyces cerevisiae (10, 11). However, it was noted that the g0 form of RTAΔm was substantially accumulated in Ngly1−/− MEF cells, which is in sharp contrast to S. cerevisiae png1Δ cells, where the g1 form of RTAΔ is predominantly accumulated (10, 11). Although the degradation of RTAΔm in DKO MEF cells was as efficient as in WT MEF cells, its degradation remains proteasome dependent (Fig. 2E). Moreover, inhibiting lysosomal degradation did not cause a delay in the degradation of RTAΔm (Fig. S2 A and B). These results suggest that the presence of N-glycans on RTAΔm did not result in impairment in its proteasomal degradation.
In sharp contrast to the case with RTAΔm, nonglycosylated RTAΔm, which is also stabilized by MG-132 treatment and therefore can be regarded as another ERAD/proteasome substrate, was shown to be degraded in Ngly1−/− MEF cells as efficiently as in WT MEF cells (Fig. S2 C and D). This result indicates that the proteasome activity itself is not likely to be impaired in Ngly1−/− MEF cells. Possible secondary effects due to a defect of Ngly1 to influence the experimental results, however, cannot be excluded at this point.
ENGase Generates N-GlcNAc–Modified RTAΔm, Which Forms Radioimmunoprecipitation Assay Buffer-Insoluble Aggregates in MEF Cells Lacking Ngly1.
Because S. cerevisiae does not contain cytoplasmic ENGase (18, 33, 34), we assume that in Ngly1−/− MEF cells cytoplasmic ENGase generates the g0 form of RTAΔm and that the remaining GlcNAc modification on RTAΔm results in its stability. To evaluate whether cytoplasmic ENGase is directly involved in deglycosylation, we coexpressed ENGase and RTAΔm in DKO MEF cells. As shown in Fig. 3A, the amount of the g0 form of RTAΔm was significantly higher in DKO MEF cells expressing ENGase, compared with control (GFP-expression; Fig. 3A, compare lanes 1 and 2). Interestingly, the major fractions containing g0 RTAΔm formed by ENGase expression were not solubilized in mild detergent [radioimmunoprecipitation assay (RIPA) buffer], indicating the formation of a RIPA-insoluble aggregate (Fig. 3A, lane 6). In sharp contrast, the majority of the g1 form was observed in the RIPA soluble fraction (Fig. 3A, lanes 3 and 4). These results support our contention that ENGase is involved in the deglycosylation of RTAΔm and can generate an aggregation-prone g0 form of RTAΔm.
Fig. 3.

Identification of the N-GlcNAc modification on RTAΔm. (A) Coexpression of RTAΔm with ENGase results in formation of the RIPA-insoluble g0 form of RTAΔm. ENGase and RTAΔm were coexpressed in DKO MEF cells. RTAΔm and GFP coexpressing cells were used as a transfection control. Cell extracts were obtained by resuspending cells in SDS/PAGE sample buffer (lanes 1 and 2; Total), or RIPA buffer (lanes 3 and 4, RIPA-soluble); the RIFs were solubilized using SDS/PAGE sample buffer (lanes 5 and 6, RIPA-insoluble). RTAΔm (Top) was visualized using anti-V5 antibody, whereas expression of ENGase (Middle) was confirmed using anti-FLAG antibody, and GFP (Bottom) was confirmed using anti-GFP antibody. (B) Silver staining pattern of immunopurified RTAΔm in Ngly1−/− MEF cells. Lane 1, immunoprecipitated sample; lane 2, immunoprecipitated sample followed by Endo-H (Roche) treatment. *Antibody heavy chain (upper) and light chain (lower). (C) MS/MS spectrum of 1,354.68, corresponding to a doubly charged tryptic peptide ion containing HexNAc. The mass vale of y17-y16 implies that Asn10 was modified by a HexNAc.
To further confirm the reaction product of ENGase, we characterized N-GlcNAc–modified RTAΔm using mass spectrometry. Ngly1−/− MEF cells stably expressing RTAΔm were treated with MG-132 for 3 h to maximize the yield of RTAΔm. Proteins were solubilized in SDS/PAGE sample buffer, and RTAΔm was immunopurified from the lysate. As shown in Fig. 3B, both the g0 and g1 forms of RTAΔm were identified, and the position of g0 on SDS/PAGE was identical to the authentic Endo H-treated sample (lane 2). The g0 band of the untreated sample (lane 1) was subjected to nanoflow liquid chromatography–electrospray ionization mass spectrometry. It was found that the peptide fragment corresponding to the N-GlcNAc–modified tryptic peptide (QYPIINFTTAGATVQSYTNFIR + HexNAc; Fig. S3A shows the complete RTAΔm sequence) was detected at approximately 25.3 min in both the g0 band of the sample and the Endo H-treated authentic GlcNAc-RTAΔm sample (Fig. S3B, Upper). The exact distribution pattern of isotopic ions for the peptide (Fig. S3B, Upper) was also very similar to the control (Fig. S3B; Lower). Furthermore, the parent ion equivalent to the N-GlcNAc peptide was subjected to MS/MS analysis, and attachment of a HexNAc residue on the N10 residue was confirmed (Fig. 3C). Taken together, we can safely conclude that formation of N-GlcNAc–modified RTAΔm by the action of ENGase can be, at least to some extent, attributed to the occurrence of the g0 form of RTAΔm in Ngly1−/− MEF cells.
Reactivity of ENGase Toward Misfolded Glycoproteins Is Distinct Among the Possible Substrates.
It was noted that both g0 and g1 forms of RTAΔm were shown to be stabilized in Ngly1−/− MEF cells (Fig. 2D). No significant increase in the g0 form during the chase period, however, was observed for Ngly1−/− MEF cells, even in the presence of MG-132, suggesting that ENGase-mediated deglycosylation is not as efficient as Ngly1-mediated deglycosylation (Fig. S4 A and B). In sharp contrast, an increase in the g0 form during the 3-h chase was evident upon MG-132 incubation in WT MEF cells (Fig. 2E and Fig. S4 A and B).
To obtain deeper insights into the generality of the ENGase-mediated deglycosylation of ERAD substrates, the deglycosylation status of other glycoprotein ERAD model substrates, such as the TCRα or the null Hong Kong variant of α1-antitrypsin (NHK), was examined. As shown in Fig. S5 A and B, in both cases the formation of a deglycosylated protein was evident, especially in the presence of the proteasomal inhibitor MG-132 (Fig. S5A, lane 4; Fig. S5B, lane 8 at the position of the red arrows) in WT MEF cells, but with less efficiency compared with the case of RTAΔm (Fig. 2 A and B). Deglycosylated bands were not observed for DKO MEF cells, clearly indicating that they were most likely formed by the action of Ngly1 and/or ENGase. Interestingly, a protein band corresponding to deglycosylated TCRα was observed for Engase−/− and WT MEF cells, whereas it was not observed for Ngly1−/− MEF cells, suggesting that Ngly1 is the predominant deglycosylating enzyme for TCRα. On the other hand, the presence of deglycosylated NHK was clearly observed in Ngly1−/− and WT MEF cells, whereas it was somewhat less obvious in Engase−/− MEF cells (Fig. S5B). In the presence of MG-132, the ratio of deglycosylated/total proteins was found to be similar between WT, Ngly1−/−, and Engase−/− MEF cells, implying that both enzymes act on NHK with similar efficiency (Fig. S5C). Collectively, these results imply that the susceptibility of misfolded glycoproteins toward cytoplasmic PNGase/ENGase is distinct among the possible substrates.
Higher Levels of Cytosolic N-Glycoproteins Were Observed in Ngly1−/−Engase−/− DKO MEF Cells.
Although the deglycosylation of RTAΔm by ENGase occurs in Ngly1−/− MEF cells, it is not clear whether such deglycosylation could also proceed in endogenous substrates. Accordingly, cytosolic glycoproteins were detected by lectin staining using Con A in different MEF cells in the presence of MG-132. Although little difference in Con A staining among WT, Ngly1−/−, and Engase−/− MEF cells was found, significantly higher levels of Con A-stained bands were observed for the cytosolic fraction of DKO MEF cells (Fig. S6). Most of the Con A-stained bands disappeared in the case of PNGase F-treated samples, suggesting that the Con A-positive signals are derived from N-glycoproteins. Collectively, these findings indicate that ENGase-mediated deglycosylation may occur on ERAD substrates more frequently than originally thought.
Formation of RIPA-Insoluble RTAΔm Is More Pronounced in MEF Cells Expressing ENGase.
Although accumulation of the g0 form RTAΔm in the RIPA-insoluble fraction (RIF) was observed in ENGase-expressing DKO MEF cells, it was not certain whether this phenomenon is specifically caused by ENGase in cells lacking Ngly1. We therefore performed RIPA extraction of RTAΔm from the WT, Engase−/−, Ngly1−/−, and DKO MEF cell lines. As shown in Fig. 4A, the RIPA-insoluble g0 form was observed in Ngly1−/− MEF cells, as well as in WT and Engase−/− MEF cells. However, the proportion of the g0 form of RTAΔm in RIF was significantly higher in Ngly1−/− MEF cells (91% ± 7%) compared with other cells (Fig. 4B). Moreover, the proportion in WT MEF cells (69% ± 5%) was also found to be significantly higher compared with Engase−/− MEF cells (29% ± 10%) (Fig. 4B). These results strongly suggest that the formation of the RIPA-insoluble aggregate is more prominent in cells expressing cytoplasmic ENGase.
Fig. 4.

Non- or de-glycosylated RTAΔm are mainly recovered from RIFs. (A) Separation of RIPA-soluble and RIPA-insoluble RTAΔm in WT, Engase−/−, Ngly1−/−, and DKO MEF cell lines. Cells expressing RTAΔm were extracted with SDS/PAGE sample buffer (lane 1, Total), RIPA buffer (lane 2, RIPA soluble), and the RIPA-insoluble fraction was solubilized with SDS/PAGE sample buffer (lane 3, RIPA insoluble). For each sample, an extract equivalent to ∼5 × 104 cells was loaded. (B) Quantitation of RIPA-insoluble g0 in WT, Engase−/−, and Ngly1−/− MEF cells. Data represent average ± SD of at least three independent samples. *P < 0.05; **P < 0.01 by Student’s t test.
RIPA-Insoluble Aggregates Are Stabilized in Ngly1−/− MEF Cells.
Because RTAΔm is stabilized specifically in Ngly1−/− MEF cells during cycloheximide-decay analysis (Fig. 2 C and D), we speculated that in Ngly1−/− MEF cells, the RIPA-insoluble RTAΔm aggregate is sequestered from proteasomal degradation. To address this issue, a cycloheximide-decay experiment of RTAΔm was performed using WT, Ngly1−/−, and Engase−/− MEF cells, with RIPA-soluble and -insoluble fractions separated at 0, 1.5, and 3 h. As shown in Fig. 5 A and B, the accumulation of g0 RTAΔm in RIF of Ngly1−/− MEF cells was evident, compared with Engase−/− or WT MEF cells. This result implies that deglycosylation by ENGase may change the soluble glycosylated RTAΔm into an aggregated g0 form. In contrast, we did not find an increase in the level of the RIPA-insoluble g0 form in WT or Engase−/− MEF cells (Fig. 5 A and B). These results indicate that the RIPA-insoluble g0 form of RTAΔm formed in Ngly1−/− MEF cells is highly stable, whereas other g0 forms (i.e., nonglycosylated or PNGase-deglycosylated RTAΔm) did not seem to be stabilized.
Fig. 5.

RIPA-insoluble proteins are stabilized in Ngly1−/− MEF cells. (A) Cycloheximide chase assay of RIPA soluble/insoluble RTAΔm in WT, Ngly1−/−, and Engase−/− MEF cells. Cells expressing RTAΔm were chased using cycloheximide for the indicated time intervals. The RIPA soluble (lanes 1–3) and RIPA-insoluble extracts (lanes 4–6) were assessed. For each sample, extracts equivalent to 5 × 104 cells were loaded. (B) Time course analysis of g1 and g0 levels in WT, Ngly1−/−, and Engase−/− MEF cells. Data represent average ± SD of three independent samples.
Discussion
Since the identification of the gene encoding the cytoplasmic PNGase (9), several reports have demonstrated that this enzyme is indeed involved in the glycoprotein ERAD process (10, 12–15, 26–33, 35). More recently, the in vivo deglycosylation has been detected by a very sophisticated assay system, using a model ERAD substrate that exhibits fluorescence in a deglycosylation-dependent manner (36). It was also reported that the EDEM1 protein, a key component of the glycoprotein ERAD process, was stabilized upon the inhibition of cytosolic PNGase, implying that the EDEM1 may be the endogenous substrate for the PNGase (37). No critical evidence, however, has been provided in terms of the biological significance of PNGase-mediated deglycosylation during the ERAD process in mammalian cells, because inhibition of PNGase activity did not cause the impairment of deglycosylation of EDEM1. In this study we show in mammalian cells that Ngly1 is important for preventing ERAD substrates from forming mild detergent-insoluble aggregates. Using a model ERAD substrate, RTAΔm, ENGase was shown to act on misfolded glycoproteins in MEF cells lacking Ngly1, resulting in the generation of aggregation-prone N-GlcNAc–modified proteins. Moreover, aggregates containing N-GlcNAc–modified proteins were found to be significantly stabilized in Ngly1−/− MEF cells. Our results clearly indicate the functional importance of Ngly1 in the ERAD process and further indicate a detrimental effect of ENGase, especially in the absence of Ngly1, on proper glycoprotein ERAD (Fig. 6). It should also be noted that, according to our current findings, to unequivocally confirm the action of Ngly1 on ERAD substrates in vivo, detecting deglycosylation by SDS/PAGE is not sufficient, and the introduction of negative charge(s) into the core peptide (i.e., the conversion of glycosylated Asn to Asp) should be independently confirmed, using methods such as isoelectoric focusing (15, 26).
Fig. 6.

Schematic representation of ENGase-mediated formation of N-GlcNAc proteins in Ngly1−/− cells. (Left) In Ngly1+ cells, misfolded proteins like RTAΔm are deglycosylated predominantly by Ngly1 and are degraded efficiently by the proteasome. (Right) In the absence of Ngly1, ENGase acts on some portion of unfolded glycoproteins and makes N-GlcNAc proteins. Too much occurrence of N-GlcNAc proteins somehow causes detrimental effects on cells, possibly by an intrinsic toxicity of protein aggregates and/or impairment of intracellular signaling pathways involving O-GlcNAc.
To date, the direct action of cytoplasmic ENGase on glycoproteins has not been unequivocally demonstrated in mammalian cells. However, there is experimental evidence showing the intracellular occurrence of N-GlcNAc proteins (i.e., potential cytoplasmic ENGase reaction products) in murine synapses (38–40). It was also clearly shown that at least some of the N-GlcNAc proteins are formed by the cytoplasmic ENGase activity in plants (20). These results indicate that formation of N-GlcNAc proteins by ENGase may not be a rare event in cells. We therefore speculate that the action of ENGase may only be detrimental to a subset of glycoprotein ERAD substrates, especially in cells lacking Ngly1, thereby causing the formation of stable N-GlcNAc proteins.
A recent study has shown that the innermost GlcNAc on N-glycans may contribute to glycoprotein stability by forming stabilizing interactions between the GlcNAc and carrier proteins (41). Therefore, one can speculate that N-GlcNAc proteins may somehow affect the tertiary structure surrounding the glycosylation region and may lead to inefficient degradation by the proteasome. On the other hand, our study clearly showed that the tendency to be recovered in an RIF is a general feature of nonglycosylated or deglycosylated RTAΔm, compared with the N-glycosylated form (Fig. 4A). This phenomenon may reflect the fact that, at least for RTAΔm, attachment of a hydrophilic N-glycan may be critical in maintaining the solubility of the carrier protein. Consistent with this hypothesis, it is of interest to note that RTAΔm degradation in Ngly1−/−Engase−/− DKO MEF cells is as efficient as in WT, whereas its degradation remains to be proteasome dependent. These observations further support the idea that, at least for RTAΔm, the efficiency of proteasomal degradation seems to be normal for N-glycan–containing proteins (which probably retain their solubility), even in the absence of Ngly1.
In Engase−/− MEF cells, it was found that minimal aggregate formation is observed for the g0 form of RTAΔm (most likely the product of Ngly1). These results imply that, under physiological conditions, Ngly1-mediated deglycosylation and proteasomal degradation may be tightly coupled, so that the deglycosylated proteins undergo proteolytic degradation without forming aggregates. However, once this regulation is lost in Ngly1−/− MEF cells, detrimental aggregates may form. It should also be noted that, except in Ngly1−/− MEF cells, RIPA-insoluble RTAΔm does not seem to accumulate over time during cycloheximide-decay experiments, implying that cells can somehow manage the aggregation-prone g0 forms, except the N-GlcNAc RTAΔm formed by the action of ENGase. The proportion of the RIPA-insoluble RTAΔm g0 form seems to be considerably higher in WT MEF cells in comparison with Engase−/− MEF cells (Fig. 4 A and B), suggesting that ENGase-catalyzed deglycosylation may occur in WT MEF cells. However, this does not cause a delay in RTAΔm degradation (Fig. 2 C and D), indicating that ENGase activity did not affect the overall efficiency of glycoprotein ERAD in WT MEF cells. It is therefore conceivable that the effect of ENGase toward glycoprotein ERAD substrates is somehow more pronounced in the absence of Ngly1.
Although Png1 mutants in budding yeast (9) or in plants (42) did not show any significant phenotypic consequences, more recent studies suggests that PNGase orthologs may play important physiological roles in other eukaryotes (43–46). Recent exome analysis studies have identified human patients with mutations in the NGLY1 gene (24, 25). These patients exhibited multiple symptoms that include developmental delay, multifocal epilepsy, involuntary movement, abnormal liver function, and absent tears. Currently it is unclear how this observation correlates with our current finding (i.e., compromised degradation of RTAΔm caused by the RIPA-insoluble aggregates in Ngly1−/− MEF cells). However, if the formation/aggregation of N-GlcNAc proteins could somehow be related to the various symptoms in NGLY1 patients, it is tempting to speculate that inhibition of ENGase activity may serve as a therapeutic target for patients carrying mutations in the NGLY1 gene.
Materials and Methods
Preparation of MEFs.
Details on the generation of Ngly1−/− and Engase−/− knockout mice are described in Fig. S7. MEF cells were established from fetuses of the Ngly1−/+Engase−/+ mice with a C57BL/6 congenic background (>96% isogenic for all MEF cells used), for which experimental details are described in SI Materials and Methods and Table S1.
Cell Cultures.
MEF cells were cultured in Dulbecco’s modified Eagle’s medium (Nacalai Tesque Co.) supplemented with 10% FBS and antibiotics (100 U/mL penicillin G, 100 ng/mL streptomycin; Nacalai Tesque Co.) at 37 °C in humidified air containing 5% (vol/vol) CO2.
Plasmid Transfection.
Detailed methods for plasmid construction are described in SI Materials and Methods. Cells were transfected with plasmids using FuGENE HD transfection reagent (Roche Applied Sciences) according to the manufacturer’s instructions. Briefly, 5 × 105 cells were seeded 12–16 h before transfection, and 1.5 μg plasmid/4.5 μL transfection reagent was mixed in 150 μL opti-MEM (Invitrogen) and incubated for 25 min before addition to the cultures. For cotransfection, 1 μg plasmid/3 μL transfection reagent was used for each plasmid. All of the transiently transfected cells were incubated for 48 h before harvesting for analysis. For stable expression, Ngly1−/− MEF cells transfected with pEF/RTAΔN236Q/V5-myc/ER were maintained in medium supplemented with 0.8 mg/mL G418 (Nacalai Tesque Co.) for 7 d, and antibiotic resistant clones were isolated and RTAΔm expression was confirmed using Western blotting.
Cycloheximide Chase Assay.
Cells transiently transfected with pEF/RTAΔN236Q/V5-myc/ER were equally divided into six dishes 24 h after transfection (at 60% confluence), 50 μg/mL cycloheximide (Sigma) was added to the cultures 48 h after transfection, and the cells were collected at the indicated times. To inhibit proteasomal degradation, MG-132 (Peptide Institute Inc.) was added at final concentration of 5 μM upon addition of cycloheximide.
Cell Lysis, Western Blotting, Immunoprecipitation, and Mass Spectrometry.
Detailed methods for cell lysis, Western blotting, immunoprecipitation, and mass spectrometry are described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank the members of the Glycometabolome Team for the fruitful discussions related to this project, and Dr. Jianguo Gu (Tohoku Pharmaceutical University), Dr. Yukiko Yoshida (Tokyo Metropolitan Institute of Medical Science), and Dr. Nobuko Hosokawa (Kyoto University) for providing reagents. This work was partially supported by the RIKEN International Joint Graduate School Program, and Grants-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to Tadashi Suzuki, Y.H., and A.H.) and the Yamada Science Foundation (to Tadashi Suzuki).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1414593112/-/DCSupplemental.
References
- 1.Needham PG, Brodsky JL. How early studies on secreted and membrane protein quality control gave rise to the ER associated degradation (ERAD) pathway: The early history of ERAD. Biochim Biophys Acta. 2013;1833(11):2447–2457. doi: 10.1016/j.bbamcr.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aebi M, Bernasconi R, Clerc S, Molinari M. N-glycan structures: Recognition and processing in the ER. Trends Biochem Sci. 2010;35(2):74–82. doi: 10.1016/j.tibs.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Thibault G, Ng DT. The endoplasmic reticulum-associated degradation pathways of budding yeast. Cold Spring Harb Perspect Biol. 2012;4(12):a013193. doi: 10.1101/cshperspect.a013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki T, Harada Y. Non-lysosomal degradation pathway for N-linked glycans and dolichol-linked oligosaccharides. Biochem Biophys Res Commun. 2014;453(2):213–219. doi: 10.1016/j.bbrc.2014.05.075. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki T. The cytoplasmic peptide:N-glycanase (Ngly1)-basic science encounters a human genetic disorder. J Biochem. 2015;157(1):23–34. doi: 10.1093/jb/mvu068. [DOI] [PubMed] [Google Scholar]
- 6.Hirayama H, Hosomi A, Suzuki T. Physiological and molecular functions of the cytosolic peptide:N-glycanase. Semin Cell Dev Biol. 2014 doi: 10.1016/j.semcdb.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki T, Seko A, Kitajima K, Inoue Y, Inoue S. Identification of peptide:N-glycanase activity in mammalian-derived cultured cells. Biochem Biophys Res Commun. 1993;194(3):1124–1130. doi: 10.1006/bbrc.1993.1938. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki T, Seko A, Kitajima K, Inoue Y, Inoue S. Purification and enzymatic properties of peptide:N-glycanase from C3H mouse-derived L-929 fibroblast cells. Possible widespread occurrence of post-translational remodification of proteins by N-deglycosylation. J Biol Chem. 1994;269(26):17611–17618. [PubMed] [Google Scholar]
- 9.Suzuki T, Park H, Hollingsworth NM, Sternglanz R, Lennarz WJ. PNG1, a yeast gene encoding a highly conserved peptide:N-glycanase. J Cell Biol. 2000;149(5):1039–1052. doi: 10.1083/jcb.149.5.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim I, et al. The Png1-Rad23 complex regulates glycoprotein turnover. J Cell Biol. 2006;172(2):211–219. doi: 10.1083/jcb.200507149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanabe K, Lennarz WJ, Suzuki T. A cytoplasmic peptide: N-glycanase. Methods Enzymol. 2006;415:46–55. doi: 10.1016/S0076-6879(06)15004-1. [DOI] [PubMed] [Google Scholar]
- 12.Hosomi A, et al. Identification of an Htm1 (EDEM)-dependent, Mns1-independent Endoplasmic Reticulum-associated Degradation (ERAD) pathway in Saccharomyces cerevisiae: application of a novel assay for glycoprotein ERAD. J Biol Chem. 2010;285(32):24324–24334. doi: 10.1074/jbc.M109.095919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hosomi A, Suzuki T. Cytoplasmic peptide:N-glycanase cleaves N-glycans on a carboxypeptidase Y mutant during ERAD in Saccharomyces cerevisiae. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbagen.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Blom D, Hirsch C, Stern P, Tortorella D, Ploegh HL. A glycosylated type I membrane protein becomes cytosolic when peptide: N-glycanase is compromised. EMBO J. 2004;23(3):650–658. doi: 10.1038/sj.emboj.7600090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirsch C, Blom D, Ploegh HL. A role for N-glycanase in the cytosolic turnover of glycoproteins. EMBO J. 2003;22(5):1036–1046. doi: 10.1093/emboj/cdg107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Misaghi S, Pacold ME, Blom D, Ploegh HL, Korbel GA. Using a small molecule inhibitor of peptide: N-glycanase to probe its role in glycoprotein turnover. Chem Biol. 2004;11(12):1677–1687. doi: 10.1016/j.chembiol.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki T, Funakoshi Y. Free N-linked oligosaccharide chains: Formation and degradation. Glycoconj J. 2006;23(5-6):291–302. doi: 10.1007/s10719-006-6975-x. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki T, et al. Endo-beta-N-acetylglucosaminidase, an enzyme involved in processing of free oligosaccharides in the cytosol. Proc Natl Acad Sci USA. 2002;99(15):9691–9696. doi: 10.1073/pnas.152333599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato T, et al. Free oligosaccharides in the cytosol of Caenorhabditis elegans are generated through endoplasmic reticulum-golgi trafficking. J Biol Chem. 2007;282(30):22080–22088. doi: 10.1074/jbc.M700805200. [DOI] [PubMed] [Google Scholar]
- 20.Kim YC, et al. Identification and origin of N-linked β-D-N-acetylglucosamine monosaccharide modifications on Arabidopsis proteins. Plant Physiol. 2013;161(1):455–464. doi: 10.1104/pp.112.208900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobata A. Exo- and endoglycosidases revisited. Proc Jpn Acad Ser B Phys Biol Sci. 2013;89(3):97–117. doi: 10.2183/pjab.89.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki T. Introduction to “Glycometabolome”. Trends Glycosci Glycotechnol. 2009;21(120):219–227. [Google Scholar]
- 23.Suzuki T, Kwofie MA, Lennarz WJ. Ngly1, a mouse gene encoding a deglycosylating enzyme implicated in proteasomal degradation: Expression, genomic organization, and chromosomal mapping. Biochem Biophys Res Commun. 2003;304(2):326–332. doi: 10.1016/s0006-291x(03)00600-4. [DOI] [PubMed] [Google Scholar]
- 24.Need AC, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012;49(6):353–361. doi: 10.1136/jmedgenet-2012-100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Enns GM, et al. FORGE Canada Consortium Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet Med. 2014;16(10):751–758. doi: 10.1038/gim.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiertz EJ, et al. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84(5):769–779. doi: 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- 27.Hughes EA, Hammond C, Cresswell P. Misfolded major histocompatibility complex class I heavy chains are translocated into the cytoplasm and degraded by the proteasome. Proc Natl Acad Sci USA. 1997;94(5):1896–1901. doi: 10.1073/pnas.94.5.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu H, Kaung G, Kobayashi S, Kopito RR. Cytosolic degradation of T-cell receptor alpha chains by the proteasome. J Biol Chem. 1997;272(33):20800–20804. doi: 10.1074/jbc.272.33.20800. [DOI] [PubMed] [Google Scholar]
- 29.Halaban R, et al. Aberrant retention of tyrosinase in the endoplasmic reticulum mediates accelerated degradation of the enzyme and contributes to the dedifferentiated phenotype of amelanotic melanoma cells. Proc Natl Acad Sci USA. 1997;94(12):6210–6215. doi: 10.1073/pnas.94.12.6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bebök Z, Mazzochi C, King SA, Hong JS, Sorscher EJ. The mechanism underlying cystic fibrosis transmembrane conductance regulator transport from the endoplasmic reticulum to the proteasome includes Sec61beta and a cytosolic, deglycosylated intermediary. J Biol Chem. 1998;273(45):29873–29878. doi: 10.1074/jbc.273.45.29873. [DOI] [PubMed] [Google Scholar]
- 31.de Virgilio M, Weninger H, Ivessa NE. Ubiquitination is required for the retro-translocation of a short-lived luminal endoplasmic reticulum glycoprotein to the cytosol for degradation by the proteasome. J Biol Chem. 1998;273(16):9734–9743. doi: 10.1074/jbc.273.16.9734. [DOI] [PubMed] [Google Scholar]
- 32.Petaja-Repo UE, et al. Newly synthesized human delta opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J Biol Chem. 2001;276(6):4416–4423. doi: 10.1074/jbc.M007151200. [DOI] [PubMed] [Google Scholar]
- 33.Hirayama H, Suzuki T. Metabolism of free oligosaccharides is facilitated in the och1Δ mutant of Saccharomyces cerevisiae. Glycobiology. 2011;21(10):1341–1348. doi: 10.1093/glycob/cwr073. [DOI] [PubMed] [Google Scholar]
- 34.Hirayama H, Seino J, Kitajima T, Jigami Y, Suzuki T. Free oligosaccharides to monitor glycoprotein endoplasmic reticulum-associated degradation in Saccharomyces cerevisiae. J Biol Chem. 2010;285(16):12390–12404. doi: 10.1074/jbc.M109.082081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masahara-Negishi Y, Hosomi A, Della Mea M, Serafini-Fracassini D, Suzuki T. A plant peptide: N-glycanase orthologue facilitates glycoprotein ER-associated degradation in yeast. Biochim Biophys Acta. 2012;1820(10):1457–1462. doi: 10.1016/j.bbagen.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Grotzke JE, Lu Q, Cresswell P. Deglycosylation-dependent fluorescent proteins provide unique tools for the study of ER-associated degradation. Proc Natl Acad Sci USA. 2013;110(9):3393–3398. doi: 10.1073/pnas.1300328110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park S, et al. ERADication of EDEM1 occurs by selective autophagy and requires deglycosylation by cytoplasmic peptide N-glycanase. Histochem Cell Biol. 2014;142(2):153–169. doi: 10.1007/s00418-014-1204-3. [DOI] [PubMed] [Google Scholar]
- 38.Chalkley RJ, Thalhammer A, Schoepfer R, Burlingame AL. Identification of protein O-GlcNAcylation sites using electron transfer dissociation mass spectrometry on native peptides. Proc Natl Acad Sci USA. 2009;106(22):8894–8899. doi: 10.1073/pnas.0900288106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trinidad JC, et al. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics. 2012;11(8):215–229. doi: 10.1074/mcp.O112.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trinidad JC, Schoepfer R, Burlingame AL, Medzihradszky KF. N- and O-glycosylation in the murine synaptosome. Mol Cell Proteomics. 2013;12(12):3474–3488. doi: 10.1074/mcp.M113.030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Culyba EK, et al. Protein native-state stabilization by placing aromatic side chains in N-glycosylated reverse turns. Science. 2011;331(6017):571–575. doi: 10.1126/science.1198461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diepold A, Li G, Lennarz WJ, Nürnberger T, Brunner F. The Arabidopsis AtPNG1 gene encodes a peptide: N-glycanase. Plant J. 2007;52(1):94–104. doi: 10.1111/j.1365-313X.2007.03215.x. [DOI] [PubMed] [Google Scholar]
- 43.Funakoshi Y, et al. Evidence for an essential deglycosylation-independent activity of PNGase in Drosophila melanogaster. PLoS ONE. 2010;5(5):e10545. doi: 10.1371/journal.pone.0010545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gosain A, Lohia R, Shrivastava A, Saran S. Identification and characterization of peptide: N-glycanase from Dictyostelium discoideum. BMC Biochem. 2012;13:9. doi: 10.1186/1471-2091-13-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Habibi-Babadi N, Su A, de Carvalho CE, Colavita A. The N-glycanase png-1 acts to limit axon branching during organ formation in Caenorhabditis elegans. J Neurosci. 2010;30(5):1766–1776. doi: 10.1523/JNEUROSCI.4962-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seiler S, Plamann M. The genetic basis of cellular morphogenesis in the filamentous fungus Neurospora crassa. Mol Biol Cell. 2003;14(11):4352–4364. doi: 10.1091/mbc.E02-07-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.