

Significance
Maintaining physiological balance is vital in the primary response to infectious and other stress stimuli to avert damaging inflammation. Delineation of the cell regulatory processes that control inflammatory processes better enable the development of informed strategies to treat associated pathologies. Toward this end, we identify that the promyelocytic leukemia zinc finger (PLZF) transcription factor limits pathogen-induced inflammation. PLZF stabilizes a repressor complex that encompasses histone deacetylase activity, which modifies the state of chromatin. This activity maintains homeostasis by decreasing the scale of induction of select immune response genes. In the absence of PLZF, the chromatin structure is altered, enabling active transcriptional complexes to immediately assemble on gene promoters, resulting in inordinate production of inflammatory cytokines.
Keywords: inflammation, transcriptional regulation, chromatin structure
Abstract
Inflammation is critical for host defense, but without appropriate control, it can cause chronic disease or even provoke fatal responses. Here we identify a mechanism that limits the inflammatory response. Probing the responses of macrophages to the key sensory Toll-like receptors, we identify that the Broad-complex, Tramtrack and Bric-a-brac/poxvirus and zinc finger (BTB/POZ), transcriptional regulator promyelocytic leukemia zinc finger (PLZF) limits the expression of inflammatory gene products. In accord with this finding, PLZF-deficient animals express higher levels of potent inflammatory cytokines and mount exaggerated inflammatory responses to infectious stimuli. Temporal quantitation of inflammatory gene transcripts shows increased gene induction in the absence of PLZF. Genome-wide analysis of histone modifications distinguish that PLZF establishes basal activity states of early response genes to maintain immune homeostasis and limit damaging inflammation. We show that PLZF stabilizes a corepressor complex that encompasses histone deacetylase activity to control chromatin. Together with our previous demonstration that PLZF promotes the antiviral response, these results suggest a strategy that could realize one of the major goals of immune therapy to retain immune resistance to pathogens while curbing damaging inflammation.
The innate immune system is essential to protect against infectious pathogens, although the resulting response encodes potent antimicrobial and proinflammatory agents that are inherently harmful (1, 2). Therapeutic interventions to diminish damaging immune responses are desired in a variety of disease states. Thus far, however, attempts to inhibit the activity of select toxic cytokines has had limited success. Furthermore, efforts to inhibit proximal receptor and cytoplasmic signaling molecules have had confounding outcomes, including increased susceptibility to infection and, paradoxically, impaired resolution of inflammation (3, 4). Rather than blocking innate immune cell signaling, effective therapy must retain protective antimicrobial responses while limiting acute or chronic inflammation. Promisingly, analysis of transcripts induced during the innate immune response demonstrates varied transcriptional regulation of distinct functional gene subsets (5, 6). This specificity is afforded via distinct transcription complexes that alternately promote or repress select genes. Identifying the specific components of functionally distinct transcriptional complexes is essential to inform a rational therapeutic strategy against damaging innate immune responses.
Here we probe the transcriptional responses induced by Toll-like receptor (TLR) signaling. TLRs are key primary sensors of the innate immune response. Activation of TLRs triggers transcription factors that include the nuclear factor κB (NF-κB), IFN regulatory factor 3 (IRF3), activator protein 1 (AP1), and activating transcription factor 3 (ATF3), which associate with specific elements in the promoters of responsive genes. Coactivator and corepressor complexes modulate the activity of these transcription factors. Emerging evidence indicates that corepressor complexes containing histone deacetylase (HDAC) activity are important to repress inflammatory response genes (7). Related to this evidence, immune priming is correlated with histone marks, such as the acetylated lysine 27 (H3K27ac) and trimethylated lysine residue 4 (H3K4me3) that are a feature of active transcription. The occurrence of these histone marks with gene promoters corresponds with occupancy by transcription factors and with more rapid transcriptional induction (8, 9). Here we show that the transcriptional regulator promyelocytic leukemia zinc finger protein (PLZF) restrains TLR-induced inflammatory signaling programs by modulating the state of chromatin.
PLZF belongs to the Broad-complex, Tramtrack and Bric-a-brac/poxvirus and zinc finger (BTB/POZ) family of transcription factors that regulate a variety of biological processes, including development, differentiation, tumorigenesis and, as is emerging, the immune response (10–13). PLZF regulates transcription by either directly binding to a specific motif in gene promoters, or indirectly by assembling transcription complexes. Here, we identify a critical contribution of PLZF to the innate immune response, showing that it modulates the immediate transcriptional response to pathogens. Characterization of the basal and activated states of gene promoters shows that PLZF initially establishes appropriate basal activity states of a subset of early TLR response genes. In keeping with the demonstrated role of histone acetylation in transcription activity, we demonstrate that PLZF stabilizes a corepressor complex that includes HDAC3. This complex is shown to be an essential limiting factor for TLR signaling, because macrophages ablated for PLZF produce excessive levels of inflammatory cytokines and PLZF-null mice mount an exaggerated inflammatory response invoked by Salmonella infection. Hence, PLZF serves as a critical rheostat in the transition from basal to active transcriptional states, thereby limiting inflammation in response to TLR signals.
Results
PLZF Suppresses TLR-Induced Inflammation in Macrophages.
Because PLZF is emerging as an important regulator of innate immune cell development and function (10, 11, 14), we sought to determine the consequences of PLZF in the immediate response to pathogens. Therefore, we probed the response of bone marrow-derived macrophages (BMDM) from wild-type (WT) and PLZF-ablated (Zbtb16−/−) mice to a range of TLR ligands. Measuring the levels of the inflammatory cytokines interleukin (IL)-12p40 and tumor necrosis factor α (TNFα) in the cell culture supernatants by ELISA shows that Zbtb16−/− BMDM are hyperresponsive to the TLR challenge (Fig. 1 A and B). Assessment of a more extensive set of transcripts by a TLR signaling pathway-specific PCR array shows a general increase in the induction of early response genes in Zbtb16−/− BMDM (Fig. 1C). In keeping with the established importance of NF-κB in TLR-induced responses, transcripts that were deregulated in Zbtb16−/− macrophages appear to be NF-κB regulated (Fig. 1C). An assessment of cell signaling molecules showed there is no inherent defect in NF-κB activation in Zbtb16−/− BMDM (Fig. S1). To test the in vivo response, WT and Zbtb16−/− mice were challenged by i.p. injection of the TLR9 ligand, CpG-DNA. Measurement of the levels of a number of cytokines and chemokines by ELISA confirms that Zbtb16−/− mice have a heightened immune response (Fig. 1D). These data demonstrate that PLZF limits the TLR-dependent immune response.
Fig. 1.

PLZF limits the TLR-induced immune responses. (A and B) The levels of IL-12p40 and TNFα in cell supernatants from WT or Zbtb16−/− BMDMs untreated (Control) or treated for 6 h with Pam3Cys (100 ng/mL), polyI:C (50 μg/mL), LPS (100 ng/mL), and Gardiquimod (1 μM), which activates the indicated TLRs (n = 6). (C) Levels of the indicated transcripts measured by TLR signaling pathway-specific PCR array in WT or Zbtb16−/− BMDMs (n = 3) challenged with LPS (100 ng/mL) for 1 h. (D) Levels of the indicated cytokines, chemokines, and growth factors measured in the serum of WT or Zbtb16−/− mice (n = 3) treated with CpG DNA (500 μg) for 3 h. The graphs show the mean ± SEM from independent experiments (*P < 0.05, **P < 0.01 by Student’s t test; n = 8).
PLZF Limits Pathogen-induced Inflammatory Pathology.
To assess the consequence of PLZF in vivo, we infected WT and Zbtb16−/− mice with the Gram-negative bacterial pathogen Salmonella enterica serovar Typhimurium (S. typhimurium) (15). Mice were injected with live or heat-killed S. typhimurium then killed after 3 h to measure the immediate immune response, or 24–48 h following live S. typhimurium infection, to assess inflammatory pathology. Measurement of IL-12p40 by ELISA showed that Zbtb16−/− mice produced significantly more of the inflammatory cytokine early after infection, compared with WT mice (Fig. 2A). Examination of tissues from mice infected for longer periods with Salmonella demonstrated elevated liver damage, evident as microabscesses throughout the hepatic parenchyma, and increased recruitment of neutrophils and monocytes to the liver and spleen of PLZF-deficient, compared with WT, mice, as evident from increased myeloperoxidase (MPO) activity (Fig. 2 B and C). To identify the immunologic cell that affects the PLZF-dependent control of the in vivo immune response, we adoptively transferred WT or Zbtb16−/− BMDMs into mice depleted of their endogenous myeloid cells through treatment with dichloromethylene diphosphate (Fig. S2 A–C). Mice reconstituted with Zbtb16−/− BMDM produced more proinflammatory cytokines in response to lipopolysaccharide (LPS) or polyI:C challenge than mice reconstituted with WT macrophages (Fig. 2D and Fig. S2 A–C). There was no conspicuous defect in BMDMs isolated from Zbtb16−/− compared with WT mice, as judged by the expression of cell surface markers (CD45, CD11B, and F4/80) and phagocytic function (Fig. S2 D and E). Collectively, these data indicate that PLZF limits TLR-induced inflammatory pathology by restricting the induction of cytokines and chemokines in macrophages.
Fig. 2.

PLZF limits pathogen-induced inflammatory pathology. (A) The levels of IL-12p40 in serum taken from WT or Zbtb16−/− mice (n = 9 per group) 3 h after i.p. injection with 1 × 103 cfu of S. typhimurium. (B) Micrographs of hematoxylin and eosin-stained sections of the livers from WT and Zbtb16−/− mice collected 24–48 h after infection with S. typhimurium. Arrows indicate immune cell infiltrates. (C) The levels of neutrophil and macrophage accumulation in the spleen and liver from WT and Zbtb16−/− mice assessed by MPO activity. (D) The levels of TNFα, IL-6, and IL-12p40 measured by ELISA in serum from WT mice that were depleted of their endogenous macrophages and then transplanted with 1 × 107 of either WT or Zbtb16−/− BMDM 6 h before challenge for 2 h with LPS (20 mg/kg), or polyI:C (10 mg/kg). The graphs show the mean ± SEM from independent experiments (*P < 0.05, **P < 0.01 by Student’s t test; n = 8).
PLZF Modifies Chromatin.
The array experiment identified that the induction of IL-10 was reduced in Zbtb16−/− BMDM (Fig. 1C and Fig. S3A). Because IL-10 inhibits TLR-dependent gene induction (16), this inhibition appeared as a possible explanation for the PLZF effects. However, direct testing excluded any role for IL-10 in the PLZF-dependent repression of inflammation (Fig. S3 B–F). To investigate the mechanism underlying the PLZF-dependent repression of cytokine production, we measured temporal induction of cytokines by PCR (Tnfα, Ccl7, Il-6, and Il-12) and ELISA (IL-12p40, IL-6, and TNFα). These data demonstrate increased induction in the Zbtb16−/− compared with WT BMDMs (Fig. 3 A and B). To try to explain this result, we characterized histone modifications of gene promoters by chromatin immunoprecipitation sequencing (ChIP-seq) of cells at basal conditions or after TLR stimulation. Transcriptionally active gene promoters were enriched with antibodies against either the H3K4me3 or H3K27ac histone marks from BMDM before and after stimulation with LPS. The deduced sequence tags were assigned to a specific promoter if they occurred within 2 kb of the transcriptional start site. Both histone marks exhibited a similar distribution on the gene promoters. Representative genome browser images for the early TLR response gene Il-6 with sequence counts associated with the H3K4me3 mark in Zbtb16−/− and WT cells are illustrated (Fig. 3C). Promoter-associated tag counts from these early response genes are depicted as a heat map (Fig. 3D). Notably, Zbtb16−/− cells displayed a marked increase in H3K4me3- and H3K27ac-associated tags compared with the WT cells. Transcriptional analysis of selected genes confirmed that gene expression levels correlated with the corresponding H3K4me3 and H3K27ac allied promoter sequences (Fig. 3E). These data demonstrate that PLZF modifies the state of the chromatin.
Fig. 3.

PLZF limits the induction of early responses genes and modifies chromatin. (A and B) Induction of the indicated transcripts measured by Q-PCR (relative to 18S), or cytokines measured by ELISA in BMDMs from WT or Zbtb16−/− mice treated with LPS (100 ng/mL) for the indicated times (*P < 0.05, **P < 0.01 by Student’s t test of five replicates). (C) Genome browser images showing ChIP-sequence tag density enriched with the H3K4me3 histone marks at the Il-6 locus in BMDMs from WT or Zbtb16−/− mice untreated or treated for 30 min with LPS (100 ng/mL). Arrows indicate differentially recovered sequence tags. (D) A heat map representation of ChIP-sequence tag density differentially enriched with the H3K4me3 and H3K27ac histone marks in WT or Zbtb16−/− BMDMs. (E) Transcriptional analysis of selected genes differentially expressed in WT compared with Zbtb16−/− BMDMs treated with LPS (100 ng/mL) that had either enhanced or reduced association with the H3K4me3 histone mark (as indicated). Gene expression was normalized to housekeeping genes to determine the fold change (*P < 0.05, **P < 0.01 by Student's t test; ns, not significant; means ± SD, n = 3 individuals per genotype).
PLZF Regulates the Induction of Early Response Transcripts.
Gene profiling demonstrated that the genes regulated by PLZF in our ChIP-seq analysis were equivalent to those previously reported in BMDM treated with the active component of LPS, Kdo2-lipid (Table S1) (17). Through subtraction of the enriched sequences between WT and Zbtb16−/− cells, then use of a de novo sequence motif discovery method, we identified a greater enrichment of consensus binding sites for the NF-κB, hepatocyte nuclear factor (HNF), and ETS (E-twenty six) transcription factors in the promoters of Zbtb16−/− compared with WT cells in the resting state (Fig. 4A). After LPS treatment, the NF-κB binding site was the principal enriched motif in the Zbtb16−/− compared with the WT sequence tags (Fig. 4A). Hence, PLZF appears to control the NF-κB response, which has been established to be a key regulator of the TLR transcriptional response. Furthermore, many of the promoter sequences that are enriched in Zbtb16−/−, compared with WT cells treated with LPS, control genes that have been identified to be NF-κB responsive (18, 19). Gene Ontology Enrichment Analysis with network visualization of promoter sequences associated with both histone marks supports an immune-related function of the genes elevated in Zbtb16−/− compared with the WT cells (Fig. 4B and Table S1). The preponderance of inflammatory cytokines and chemokines identified by gene annotation that were elevated in the Zbtb16−/− BMDM reproduce the hyperresponsive phenotype identified in mice ablated for PLZF.
Fig. 4.

PLZF regulates the induction of early response transcripts. (A) De novo analysis of transcription factor binding motifs in the differentially enriched early TLR response gene promoters before (Upper) and after LPS treatment (Lower) in the promoters of Zbtb16−/− compared with the WT BMDMs. (B) A network generated by Gene Ontology (GO) Enrichment Analysis of the promoter sequences enriched in Zbtb16−/− compared with the WT BMDMs. Nodes (circles) represent enriched GO terms and lines between nodes represent genes shared between the GO terms. Select genes encoding inflammatory cytokines and chemokines that were identified as being regulated by PLZF in multiple experiments are indicated (in red) within the network.
PLZF Recruits Histone Deacetylase Activity.
The previously reported association between PLZF and HDACs (20, 21), and the key role of deacetylases in gene regulation via chromatin condensation, led us to postulate that the recruitment of HDACs accounted for the observed PLZF-dependent modification of chromatin. This association was initially assessed through overexpression of cyan fluorescent protein (CFP)-tagged PLZF and FLAG-tagged HDAC-1, -2, -3, and -7 in HEK293 cells. Immunoprecipitation of each HDAC, with an anti-FLAG antibody, demonstrated an association between PLZF and HDAC3 (Fig. 5A). We corroborated this protein association by recovery of PLZF and immune detection of HDAC3 by coimmunoprecipitation (co-IP) (using alternate epitope tags) and by colocalization (using fluorescent tags), through Western blot and confocal microscopy, respectively (Fig. 5B and Fig. S4 A and B). In accordance with the observed enrichment of NF-κB–regulated transcripts by PLZF (Figs. 1C and 4A), we also demonstrated an association between PLZF and the NF-κB p50 subunit by coimmunoprecipitation and colocalization (Fig. 5C and Fig. S4 B and C). The association between HDAC3 and PLZF, or NF-κB p50, was confirmed for endogenous levels of the proteins in WT and Zbtb16−/− macrophages by coimmunoprecipitation (Fig. 5D). Although an expected stimulus-dependent increase in the protein association was not evident by immune capture, this association was apparent in the formation of the trimeric protein complex in HEK293T cells transfected with PLZF, NF-κB p50, and HDAC3 by measuring Förster’s resonance energy transfer (FRET) between full-length Venus formed through an association between HDAC3 and NF-κB p50, each tagged with separate halves of a split Venus fluorophore and the donor CFP-PLZF (Fig. 5E).
Fig. 5.

PLZF functionally interacts with HDAC3 and NF-κB p50. (A–D) Immunoblots showing co-IP of PLZF with HDAC3 in HEK293T cells transfected with PLZF-CFP and FLAG-HDAC-1, HDAC-2, HDAC-3, or HDAC-7 (A); FLAG-PLZF and mCherry-HDAC3 in HEK293T cells (B); FLAG-PLZF and Myc-NF-κB p50 in HEK293T cells (C); Endogenous HDAC3 and PLZF, or NF-κB p50 in WT and Zbtb16−/− BMDM (D). Cells were stimulated for 0.5 h with TNFα (10 ng/mL) in B and C, or LPS (100 ng/mL) in D. A control anti-IgG antibody was used to test the specificity of the IP. The specific IP and detection (IB) antibodies are as indicated. (E) Micrographs of HEK293T cells showing an association among V1-HDAC3, V2-NF-κB p50, and PLZF-CFP in HEK293T cells stimulated for 0.5 h with TNFα (10 ng/mL). The association between the molecules is graphed at Right as the extent of FRET either in the entire nucleus (Nucleus) or within the nuclear speckles (Speckle) (n = 3). The schematics at Left depict the protein complex being detected. (Scale bars: 10 μm.) (F–H) Graphs quantitating sequence tags for the indicated gene promoters, from WT and Zbtb16−/− BMDMs treated for 0.5 h with LPS (100 ng/mL), enriched by ChIP with: Anti-H3K4me3, Anti-H3K27Ac, Anti-HDAC3, Anti-NF-κB p50, Anti-p65, and Anti-p300 antibodies (F); Anti-PLZF antibody (G); and Anti-NF-κB p50 followed by re-ChIP with an anti-PLZF antibody (H). An IgG antibody was used as a negative control. The graphs show the mean ± SEM from independent experiments (*P < 0.05, **P < 0.01 by Student’s t test; n = 3).
PLZF-dependent recruitment of HDAC3 and NF-κB p50 to gene promoters was confirmed by ChIP studies of the Il-6, Il-12, and Tnfα gene promoters in WT and Zbtb16−/− BMDMs (Fig. 5F). The increased induction of these transcripts seen in our preceding experiments correlated with increased recruitment of the transcription activators NF-κB p65 and p300 to the gene promoters in Zbtb16−/− compared with WT cells after treatment with LPS. This recruitment was specific to the RelA with no appreciable difference in the recruitment of NF-κB p50 to the promoter in WT and Zbtb16−/− cells. Importantly, there was a significant defect in the recruitment of HDAC3 to the gene promoter in the absence of PLZF, which coincides with the binding of NF-κB p65 and the histone acetyltransferase p300. Consonant with this defect, histones associated with the gene promoters had increased posttranslational modifications associated with active transcription (H3K27Ac and H3K4me3) in the absence of PLZF (Fig. 5F). ChIP of PLZF itself directly confirmed that the protein associated with the promoters of genes that were up-regulated (Il-6, Il-12, Tnfα, Cxcl12, and Cxcl11), and not those that were unchanged (Infβ and Csf3), in the Zbtb16−/− compared with the WT cells (Fig. 5G). Furthermore, ChIP of NF-κB p50, followed by re-ChIP of PLZF, validated the coassociation of these two transcription factors with the promoters of the Il-12 and Tnfα genes (Fig. 5H).
Collectively, these data support a model in which PLZF controls the state of chromatin by recruiting HDAC3 to the promoters of select genes that contain an NF-κB binding motif. Recruitment or stabilization of HDAC3 to gene promoters alters the state of the chromatin through histone modification and reduced binding of p65 and p300 with RNA polymerase II and other factors from the activating transcriptional complex (Fig. 6).
Fig. 6.
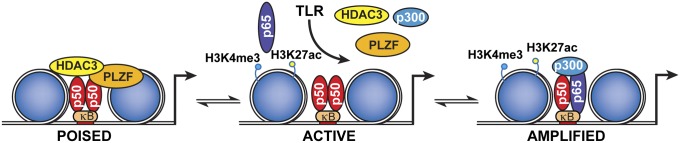
Proposed model of PLZF-dependent repression of inflammation. The schematic shows how PLZF limits the formation of the active transcriptional complex by stabilizing the transcriptionally inactive NF-κB p50–HDAC3 complex on the promoters of early response genes. This complex is not immutable, but rather there is an exchange of corepressor for coactivator complexes, so that both complexes only transiently occupy the promoters of regulated genes. In the absence of PLZF, histones have heightened acetylation and so the chromatin is more relaxed at homeostasis, evidenced as increased active histone marks (H3K4me3 and H3K27ac). As a result, there is a more rapid recruitment of active transcriptional complexes and increased induction of these gene transcripts, with the ensuing increase in inflammatory pathology.
Discussion
Inflammatory responses are crucial to survival and must be tightly regulated to ensure appropriate immune protection of the host. Key inducible factors do not function in isolation, but are modulated by a variety of transcriptional cofactors. Identification of these separate cofactors and their roles has the potential to establish strategies to more effectively control harmful immune responses. Toward this end, we identify PLZF as a transcriptional cofactor that limits damaging inflammation by controlling the state of chromatin. We propose that PLZF maintains homeostasis by stabilizing the transcriptionally inhibitory HDAC3 complex on gene promoters. After TLR stimulation, there is a progressive exchange of corepressor, encompassing PLZF, for coactivator complexes with accompanying modification of juxtaposed histones to enable phased amplification of the gene transcript. In the absence of PLZF, the chromatin is already active, enabling transcriptional complexes to immediately assemble on the gene promoter, resulting in inordinate production of inflammatory cytokines (Fig. 6).
Our findings for PLZF are consistent with recent reports of an analogous role for other members of this family of proteins in controlling inflammatory responses. The BTB and zinc-finger domain encoding B-cell lymphoma 6 (BCL6) protein (encoded by the Zbtb28 gene) was shown to regulate LPS-induced transcript (22). Accordingly, BCL6-deficient macrophages were hypersensitive to LPS. Similarly, deletion of the BTB domain from the Myc-interacting zinc finger protein 1 (MIZ1) (encoded by the Zbtb17 gene), induced a hyperinflammatory response to LPS (23). In this instance, phosphorylation of MIZ1 enabled the protein to recruit HDAC1 to establish a repressive transcriptional complex. The role that we have identified for PLZF in restricting the TLR response is distinct from known negative regulatory mechanisms (24, 25). Unlike MIZ1, which terminates the TLR response, we show that PLZF establishes the appropriate state of the chromatin to control gene induction and subsequently reinstate homeostasis.
In addition to the role we identify here in macrophages, PLZF has been reported to be critical for the development of a number of other immune cell lineages that are of importance in protection from pathogens (10–12, 14). The role of PLZF in the differentiation of these cells has not been established. Our data showing that PLZF regulates chromatin suggest a potential mechanism whereby epigenetic changes regulated through modification of the histones could mediate the development of these immune cell lineages.
It has recently been proposed that histone modifications, pertinently H3K4me3, establish a short-term memory that allows macrophages to more rapidly recall the transcriptional response to a pathogen (26–28). This proposition is in keeping with the effect that we demonstrate for PLZF on chromatin to delay the kinetics of transcriptional activation.
We demonstrated that PLZF forms a transcriptional complex with HDAC1 to induce the expression of a subset of IFN-regulated genes (13). Here, we demonstrate PLZF associates with HDAC3 to repress the inflammatory response. Formation of these different transcriptional complexes is regulated by alternate posttranscriptional modifications of PLZF (29). We established that phosphorylation of PLZF induces antiviral transcripts (13), whereas studies by others have demonstrated that repressor activity is contingent on acetylation of PLZF (30). At the initiation of an infection, the alternate cell signaling pathways that mediate these separate modifications of PLZF would be temporally separated, with the TLR-dependent regulation of PLZF repression being immediate, whereas the IFN-dependent regulation of PLZF transcriptional induction is delayed. The consequence of coordinated induction of each pathway is untested. Presuming that the levels of PLZF are limiting, sustained production of interferons might recruit PLZF to this pathway, thereby increasing inflammation. This activity might account for the cytokine storm induced by some viral infections. Conversely, increased commitment to the repressor function would be expected to diminish both the inflammatory and antiviral responses. This apparent tension in the immune response needs to be resolved in order to retain immune resistance to pathogens while curbing damaging inflammation.
Materials and Methods
Mice and Reagents.
All mice used in experiments were treated in accordance with practices approved by the Monash University, Monash Medical Centre animal ethics committee (Melbourne, Australia) as obligated by the Australian Code of Practice for the care and use of animals for scientific purposes, in compliance with standards mandated by the Australian National Health and Medical Research Council and National Institutes of Health. Mice were bred in high barrier-specific pathogen-free conditions on a C57BL/6 genetic background. PLZF (Zbtb16−/−) mice were killed for bone marrow between 7 and 10 wk of age. BMDMs from mice were generated and cultured in l-cell–derived colony-stimulating factor 1, as described (13, 31). Pam3CSK4, polyI:C, LPS, Gardiquimod, and CpG oligodeoxynucleotides were purchased from Invivogen.
Quantitative Real-Time PCR.
Quantitative PCR (Q-PCR) was done in triplicate on an Applied Biosystems 7700 Prism real-time PCR machine with TaqMan probes for Il-6, Il-12, Ccl2, Tnfα, Ifnβ, Cxcl10, Ifit1, and Oasl1 (Applied Biosystems). Expression of mRNA was normalized to the expression of 18S ribosomal RNA by the change in cycling threshold (ΔCT) method and calculated based on 2−ΔCT. Results are expressed as relative gene expression for each target gene. All Q-PCR primers used are listed in Table S2.
TLR Signaling Pathway-Specific PCR Array.
An RT2 Profiler PCR array system was used to quantify gene expression by using a TLR signaling-focused array as per the manufacturer's instructions (SuperArray Bioscience). Briefly, WT and Zbtb16−/− macrophages were treated with LPS (100 ng/mL) for 60 or 120 min, then total RNA was extracted from 2 × 106 cells by using the RNeasy Mini Kit (Qiagen) and was DNase treated. Reverse transcription was carried out by using the RT2 PCR array first-strand kit (SuperArray). Quality control of all samples was carried out on a SuperArray QC array. The threshold cycle (Ct) difference (mean of three independent experiments) between the experimental and control groups was calculated and normalized to housekeeping genes, and data were analyzed by the ΔΔCt method using data analysis templates from SuperArray.
ChIP Assay.
ChIP assays were done according to the manufacturer’s instructions (Millipore), with modifications. Cells (1 × 107) were treated with LPS at different time points, and then fixed with formaldehyde. Detailed procedures are described in SI Materials and Methods. Primers used for ChIP are listed in Table S3.
ChIP-seq Sequence Analysis.
ChIP assays were performed according to the protocol of Dahl and Collas (32), with modifications, as described in SI Materials and Methods. ChIP-seq analysis was performed by using HOMER (33) and samples were visualized by creating custom tracks in the University of California, Santa Cruz genome browser (34). Detailed procedures are described in SI Materials and Methods.
ChIP-seq Enrichment and Gene Ontology Analysis.
This analysis was performed by using overlapping gene sets of the differentially enriched genes identified in the comparisons between Zbtb16−/− (untreated and treated with LPS) and their WT counterparts. This gene set included a subset of 44 immediate/early response genes (17). For this analysis, combined sequencing tags for H3K4me3 and H3K27ac markers and ±2000 interval from the transcription start site of the gene for peak detection were used.
Immunofluorescent and FRET Assay.
Cells were transfected with either PLZF-CFP alone (control) or equimolar ratios of PLZF-CFP and mCherry-HDAC3, or mCherry-NF-κB p50, or NF-κB p50 and HDAC3, each tagged with separate halves of the Venus fluorophore (V1 and V2) at a maximum of 100 ng of DNA per coverslip. Cells were imaged on a Lambert Instruments FLIM microscope with a CFP excitation filter (>438 nm) and the Acousto-Optic Tunable filter at ∼479–498 nm. A minimum of 10 images was collected per sample with between two and seven cells per image. FRET efficiency was calculated by the LI-FLIM software 1.2.9 by using CFP-PLZF alone as the donor-only lifetime and FITC 4.1 ns as a reference.
Immunoprecipitation and Western Blotting Analysis.
For immunoprecipitation, cells were lysed with triple detergent lysis buffer (and incubated with each antibody). Antibody complexes were isolated by using protein A/G–agarose beads (Pharmacia Biotech), analyzed by SDS/PAGE, and the target epitope was resolved and detected by Western blotting using the specific antibodies as indicated. Targeted proteins were detected and quantified on a Li-Cor Odyssey (LI-COR Biosciences) infrared imaging system.
Establishment of Endotoxin Shock and Bacterial Infection Models.
The endotoxin shock mouse model was generated by i.p. injection of LPS, as described (35). S. enterica serovar Typhimurium SL1344 was grown overnight in Luria–Bertani broth medium. Bacteria were subsequently washed twice in sterile PBS, diluted to 1 × 104 cfu/mL, and administered by i.p. injection with 100 µL of 1 × 103 cfu bacterial suspension per mouse. The number of bacteria contained within the inoculum was determined by performing viable counts.
Macrophage Reconstitution.
To deplete endogenous macrophages, mice were treated with 50 mg of clodronate-loaded liposomes (Sigma) by i.p. injection. Two days later, 1 × 107 BMDMs from either WT or Zbtb16−/− mice were transplanted by injection into the tail vein. After reconstitution with macrophages for 6 h, mice were challenged with LPS or polyI:C, as described (36).
Histology.
Livers and spleens were removed and fixed in 4% (vol/vol) paraformaldehyde overnight at room temperature, then were embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Sections were examined under a microscope (Leica Instruments).
Myeloperoxidase Activity Assay.
MPO activity was evaluated as an index of neutrophil accumulation in the tissues by using the MPO Activity Assay Kit from Invitrogen, following the manufacturer’s protocol.
Statistical Analysis.
All statistical analysis was performed with GraphPad Prism software. Differences in survival rates were assessed by the log-rank test, using the unpaired, two-tailed t test to analyze the data and generate P values. P values of less than 0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank T. Wilson for technical support for the ChIP-seq analysis, B. Jenkins for contributing the Il-6−/− mice, R. Evans (Salk Institute for Biological Studies) for the HDAC constructs, and F. Cribbin for assistance with preparation of the manuscript. This work was supported by National Basic Research Program of China Grant 2012CB911204, National Natural Science Foundation of China Grants 81273247 and 81472655, National Health and Medical Research Council of Australia Grants 606425 and 1066665, and the Victorian Government’s Operational Infrastructure Support Program.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequences reported in this paper have been deposited in the Sequence Read Archive database (accession no. SRP051792).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1409728112/-/DCSupplemental.
References
- 1.Akira S, Takeda K, Kaisho T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2(8):675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 2.Suntharalingam G, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 3.Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science. 2007;317(5838):675–678. doi: 10.1126/science.1142953. [DOI] [PubMed] [Google Scholar]
- 4.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9(10):692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 5.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 6.Chevrier N, et al. Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell. 2011;147(4):853–867. doi: 10.1016/j.cell.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138(1):129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganal SC, et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity. 2012;37(1):171–186. doi: 10.1016/j.immuni.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 9.Qiao Y, et al. Synergistic activation of inflammatory cytokine genes by interferon-γ-induced chromatin remodeling and toll-like receptor signaling. Immunity. 2013;39(3):454–469. doi: 10.1016/j.immuni.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovalovsky D, et al. The BTB-zinc finger transcriptional regulator PLZF controls the development of invariant natural killer T cell effector functions. Nat Immunol. 2008;9(9):1055–1064. doi: 10.1038/ni.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savage AK, et al. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity. 2008;29(3):391–403. doi: 10.1016/j.immuni.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raberger J, et al. The transcriptional regulator PLZF induces the development of CD44 high memory phenotype T cells. Proc Natl Acad Sci USA. 2008;105(46):17919–17924. doi: 10.1073/pnas.0805733105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu D, et al. Promyelocytic leukemia zinc finger protein regulates interferon-mediated innate immunity. Immunity. 2009;30(6):802–816. doi: 10.1016/j.immuni.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature. 2014;508(7496):397–401. doi: 10.1038/nature13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vazquez-Torres A, et al. Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: Importance of the Kupffer cell network. J Immunol. 2004;172(10):6202–6208. doi: 10.4049/jimmunol.172.10.6202. [DOI] [PubMed] [Google Scholar]
- 16.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 17.Escoubet-Lozach L, et al. Mechanisms establishing TLR4-responsive activation states of inflammatory response genes. PLoS Genet. 2011;7(12):e1002401. doi: 10.1371/journal.pgen.1002401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sen R, Smale ST. Selectivity of the NF-kappaB response. Cold Spring Harb Perspect Biol. 2010;2(4):a000257. doi: 10.1101/cshperspect.a000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grignani F, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391(6669):815–818. doi: 10.1038/35901. [DOI] [PubMed] [Google Scholar]
- 21.Hong SH, David G, Wong CW, Dejean A, Privalsky ML. SMRT corepressor interacts with PLZF and with the PML-retinoic acid receptor alpha (RARalpha) and PLZF-RARalpha oncoproteins associated with acute promyelocytic leukemia. Proc Natl Acad Sci USA. 1997;94(17):9028–9033. doi: 10.1073/pnas.94.17.9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barish GD, et al. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010;24(24):2760–2765. doi: 10.1101/gad.1998010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Do-Umehara HC, et al. Suppression of inflammation and acute lung injury by Miz1 via repression of C/EBP-δ. Nat Immunol. 2013;14(5):461–469. doi: 10.1038/ni.2566. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Natoli G, Saccani S, Bosisio D, Marazzi I. Interactions of NF-kappaB with chromatin: The art of being at the right place at the right time. Nat Immunol. 2005;6(5):439–445. doi: 10.1038/ni1196. [DOI] [PubMed] [Google Scholar]
- 25.Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012;33(9):449–458. doi: 10.1016/j.it.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Byun JS, et al. Dynamic bookmarking of primary response genes by p300 and RNA polymerase II complexes. Proc Natl Acad Sci USA. 2009;106(46):19286–19291. doi: 10.1073/pnas.0905469106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quintin J, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12(2):223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monticelli S, Natoli G. Short-term memory of danger signals and environmental stimuli in immune cells. Nat Immunol. 2013;14(8):777–784. doi: 10.1038/ni.2636. [DOI] [PubMed] [Google Scholar]
- 29.Ozato K. PLZF outreach: A finger in interferon’s pie. Immunity. 2009;30(6):757–758. doi: 10.1016/j.immuni.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guidez F, et al. Histone acetyltransferase activity of p300 is required for transcriptional repression by the promyelocytic leukemia zinc finger protein. Mol Cell Biol. 2005;25(13):5552–5566. doi: 10.1128/MCB.25.13.5552-5566.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baccarini M, Bistoni F, Lohmann-Matthes ML. In vitro natural cell-mediated cytotoxicity against Candida albicans: Macrophage precursors as effector cells. J Immunol. 1985;134(4):2658–2665. [PubMed] [Google Scholar]
- 32.Dahl JA, Collas P. A rapid micro chromatin immunoprecipitation assay (microChIP) Nat Protoc. 2008;3(6):1032–1045. doi: 10.1038/nprot.2008.68. [DOI] [PubMed] [Google Scholar]
- 33.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ulloa L, et al. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc Natl Acad Sci USA. 2002;99(19):12351–12356. doi: 10.1073/pnas.192222999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han C, et al. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol. 2010;11(8):734–742. doi: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.