

Significance
In forward genetics, a mutagen is used to randomly induce germline mutations that cause variant phenotypes. Forward genetics permits discovery of genes necessary for biological phenomena, but identifying the mutations that cause variant phenotypes is time-consuming and in the past usually occurred long after the phenotype was first recognized. Here we introduce a method and software tool, Linkage Analyzer, for identifying causative mutations present in the germline of mutant mice concurrent with recognition of variant phenotypes. It requires knowledge of genotype at all mutation sites in members of a pedigree prior to phenotypic assessment. Using this method and software, forward genetic studies in mice are limited only by the rates of mutant production and screening.
Keywords: N-ethyl-N-nitrosourea, genetic mapping, forward genetics, mutagenesis, massively parallel sequencing
Abstract
With the wide availability of massively parallel sequencing technologies, genetic mapping has become the rate limiting step in mammalian forward genetics. Here we introduce a method for real-time identification of N-ethyl-N-nitrosourea-induced mutations that cause phenotypes in mice. All mutations are identified by whole exome G1 progenitor sequencing and their zygosity is established in G2/G3 mice before phenotypic assessment. Quantitative and qualitative traits, including lethal effects, in single or multiple combined pedigrees are then analyzed with Linkage Analyzer, a software program that detects significant linkage between individual mutations and aberrant phenotypic scores and presents processed data as Manhattan plots. As multiple alleles of genes are acquired through mutagenesis, pooled “superpedigrees” are created to analyze the effects. Our method is distinguished from conventional forward genetic methods because it permits (1) unbiased declaration of mappable phenotypes, including those that are incompletely penetrant (2), automated identification of causative mutations concurrent with phenotypic screening, without the need to outcross mutant mice to another strain and backcross them, and (3) exclusion of genes not involved in phenotypes of interest. We validated our approach and Linkage Analyzer for the identification of 47 mutations in 45 previously known genes causative for adaptive immune phenotypes; our analysis also implicated 474 genes not previously associated with immune function. The method described here permits forward genetic analysis in mice, limited only by the rates of mutant production and screening.
Phenotypic variation in mice can be induced with N-ethyl-N-nitrosourea (ENU), which creates single base pair substitutions in germ line DNA. However, the positional cloning of ENU-induced mutations causative for phenotypes of interest has historically been a time-consuming process, beginning with generation of an outcrossed recombinant mapping population of phenotypically mutant and WT mice, genotyping individual mice at genetic markers across the genome to create a linkage map, and finally targeted sequencing to identify the causative mutation within the critical region. The advent of massively parallel sequencing techniques has given rise to more rapid “mapping-by-sequencing” methods in which genome-wide marker genotyping and DNA sequencing are combined into a single step applied to either individual or pooled groups of organisms (1). For ENU-mutagenized mice, early experiments used massively parallel sequencing for mutation identification within a critical region defined by traditional or bulk segregation mapping using recombinant mapping populations produced by outcrossing the mutant to another inbred laboratory strain and backcrossing or intercrossing a second time (2–4). Later reports demonstrated mapping with the identified sequence variants themselves as markers, which eliminated the need for outcrossing and its potential for altering the mutant phenotype (5, 6). However, whole genome/exome sequencing currently remains very costly to apply as the sole method for genotyping large collections of G3 mice.
Despite the acceleration of conventional genetic mapping and mutation identification made possible by massively parallel sequencing, determining a causative mutation after initial detection of a phenotype is still slowed by the need to retrospectively identify mutations in the original pedigree and to obtain sufficient animals to establish a mutation’s segregation pattern, which may be problematic in the case of incompletely penetrant phenotypes. Moreover, because only causative (rather than noncausative) mutations are identified it has not been possible to progressively exonerate genes from involvement in biological processes.
In ongoing forward genetic studies aimed at discovering genes essential for immune function in mice, we developed an alternative approach and computational software that permit declaration of causative and noncausative mutations concurrent with the development of data in phenotypic screens. For quantitative traits, phenovariance is identified statistically, facilitating the detection of incompletely penetrant phenotypes and precluding biased interpretations of phenotypic data by the human observer. Complex phenotypes may also be resolved. We avoid the traditional pathway of breeding phenovariant mice to confirm transmissibility, establishment of a stock, and the outcross and backcross (or intercross) of an F1 hybrid generation to permit the assignment of a map location in F2 mice. Within a sample of 15,055 G3 mice derived from 610 pedigrees, we detected causative mutations in 45 genes with known immunological function. We also identified mutations within 474 genes not previously known to have immunological function, many of which have been validated as causative based on the detection of multiple allelic variants linked to phenotype.
Results and Discussion
We optimized breeding procedures to achieve two goals: (i) phenotypic screening of 30–50 G3 mice from each pedigree within the same experiment and (ii) determination of genotype at all mutation sites in every G3 mouse before phenotypic assessment.
G3 mice carrying homozygous and heterozygous mutations induced by ENU in germ cells of G0 male C57BL/6J mice were generated using one of two breeding plans that differed in whether the female mated to the mutagenized G0 male carried ENU-induced mutations from her father (Fig. 1). If so, each G3 descendant carries on average 67 mutations (∼6 homozygous and 61 heterozygous) including X-linked mutations, whereas G3 descendants of a mutagenized G0 male mated to a WT female carry on average 45 mutations (∼4 homozygous and 41 heterozygous) but no X-linked mutations. Because of the lower mutation load, a greater number of G3 mice is ultimately produced from G0 × C57BL/6J breeding than from G0 × G0′ breeding; because a large number of G3 mice is desired, G0 × C57BL/6J mating is preferred. G1 males were bred to G2 females over a period of 12 wk to produce ∼50 G3 mice that would be 7–16 wk of age when the youngest mice were ready for screening. A G3 mouse produced in this way has a 12.5% chance of homozygosity for a mutation with a neutral effect on viability and 50 G3 mice from a single pedigree provide ample statistical power to detect concordance between traits of moderate strength and homozygosity at a particular locus. For example, screening 48 G3 mice will provide 91.9% power to detect an effect size of 1.5 under the recessive model (assuming 12.5% homozygosity occurs) and control type I error at 5%. Screening the mice in the same experiment on the same day was desirable to minimize phenotypic differences due to variations in experimental procedures, although data could be normalized to control C57BL/6J data when screening was performed on multiple days.
Fig. 1.
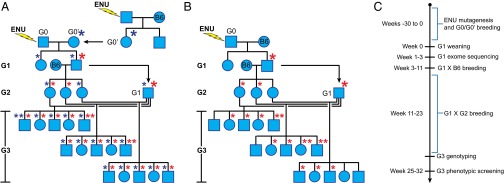
Inbreeding protocols for generating 30–50 G3 mice with ENU-induced mutations. Mutagenized G0 males are bred to G0′ females carrying germ-line mutations derived from other mutagenized males (A) or to WT C57BL/6J (B6) females (B). G1 males are crossed to B6 females to produce G2 mice. G3 mice result from backcross of G2 females to their G1 father. G3 mice are subjected to screening. Asterisks represent mutations derived from the G0 male (red) and G0′ female (blue); larger asterisks indicate initial germ-line transmission of the mutation. (C) Timeline for G3 mouse production, genotyping, and screening.
Real-time linkage assignment requires knowledge of genotype at all mutation sites in all G3 mice, and if possible G2 mice, by the time phenotypic data are acquired. Because the majority of causative ENU-induced mutations exist in protein-coding exons rather than in cis-acting gene regulatory elements (7, 8), this was accomplished by exome sequencing of the G1 male progenitor of each pedigree to identify all coding and splice site mutations that could possibly be present in the G3 mice. The G2 and G3 mice were then genotyped at each mutation site before phenotypic screening. Three genotypes were possible for each mutation site: REF, homozygous for C57BL/6J reference allele; HET, heterozygous for reference allele and variant allele; or VAR, homozygous for variant allele. Genotypes were reliably called by deep sequencing pooled, barcoded target DNA amplified from G1, G2, and G3 mice (Fig. 2). The results of exome sequencing of 1,252 G1 mice derived from G0 × C57BL/6J crosses and 667 G1 mice derived from G0 × G0′ crosses demonstrated an ENU mutation rate of 1.06 and 1.55/Mbp, respectively, consistent with previous reports (5, 6). Genotype data were stored in the Mutagenetix database (mutagenetix.utsouthwestern.edu) for later analysis together with phenotype data. The G3 mice then underwent phenotypic screening (Fig. 3).
Fig. 2.

Histogram of genotyping calls for 191 mutations sites in 132 mice from three pedigrees. The percentage of mutant allele reads out of total reads reflects the genotype; for example, 100% mutant allele reads/total reads represents a VAR genotype, i.e., homozygous mutant. Genotyping was performed by deep sequencing of genomic DNA after targeted amplification and yielded essentially REF, HET, and VAR genotypes exclusively; 7,636 data points are represented. Each data point represents one mutation site sequenced in one mouse.
Fig. 3.

Phenotypic screening pipeline. The flow of mice through the pipeline has been optimized for efficient use of animals based on the requirements of individual screens, with noninvasive screening preceding invasive screens. Groups of ∼500 G3 mice (15–20 pedigrees) enter the pipeline each week. A total of 9.5 wk are required for a group of mice to pass through all screening procedures. TLR, Toll-like receptor; NLR, NOD-like receptor; dsDNA, double stranded DNA; MCMV, mouse cytomegalovirus.
Analyses of genotype and phenotype data were performed using Linkage Analyzer, a program that tests the probability of single locus linkage to phenotypes using recessive, semidominant (additive), and dominant transmission models and assesses the probability of preweaning lethal effects due to single locus mutations. It detects phenovariance when it is statistically linked to genotype as determined by a linear regression model. For each mutation, the null hypothesis of nonlinkage is tested assuming a normal or a binomial distribution of phenotype scores for quantitative and qualitative phenotypes, respectively. The P value of association between genotype and phenotype is calculated using a likelihood ratio test from a generalized linear model or generalized linear mixed effect model (Materials and Methods). Manhattan plots for each inheritance mode are generated, along with displays of the phenotypic performance of every variant allele in homozygous and heterozygous state. Linkage Analyzer operates at a scalable speed depending on the capabilities of the cluster on which it is run. As presently configured in our laboratory it processes data at a rate that exceeds our capacity to produce mutations and develop screening data and delivers linkage assessments in real time. When phenotypic data are uploaded, the genetic cause of any phenovariance that may exist in the dataset is usually known within a few minutes. The production and phenotypic analysis of G3 mutant mice have thus become the rate-limiting steps in forward genetics.
We use PolyPhen-2 (9) and a splice site prediction program (Materials and Methods) to assess the predicted effect of all mutations in a pedigree. PolyPhen-2 classifies mutations as probably damaging (score, ∼0.95–1.0), possibly damaging (score, ∼0.45–0.95), or probably benign (score, ≤∼0.45); nonsense and critical splice site mutations (corresponding to first or last two nucleotides of an intron) are classified probably null, as are certain mutations identified by the splice site prediction program. Such information aids in distinguishing authentic genotype-phenotype associations from others that may be less likely. In addition, we sort genotype-phenotype associations by P value (for both raw and normalized datasets) and by the number of mice with VAR genotype tested to prioritize mutations for further study. The effect of varying the cutoff values for assigning linkage among all mutated genes and among a set of 21 genes with known immunological function is shown in Tables S1 and S2. We refer to a requirement that both raw and normalized datasets meet the same P value cutoff as the Raw+Norm restriction, and its effect on the number of mutations implicated is also shown. A range of parameters with varying stringency should be explored when initially searching a dataset for genotype-phenotype associations, with lower stringency parameters to identify all plausible linkages and higher stringency parameters to discern authentic associations.
Mapping a Qualitative Trait: The Teeny Phenotype.
We tested automated mapping in the analysis of the visible (qualitative) phenotype Teeny, characterized by decreased body size and weight compared with unaffected littermates. Within pedigree R0491, a total of 24 G3 mice were examined. Nine displayed small body sizes and were designated affected (Fig. 4A). Fifteen appeared normal and were designated unaffected. Automated mapping by recessive, additive, and dominant models of inheritance (Fig. 4B) implicated a putative null allele of kelch repeat and BTB (POZ) domain containing 2 (Kbtbd2) as the causative mutation, displaying predominantly recessive inheritance (P = 9.056 × 10−6) with a detectable semidominant effect (P = 1.036 × 10−5). Notably, only eight of the nine affected mice were homozygotes for the Kbtbd2 mutation; one was heterozygous.
Fig. 4.

Mapping teeny, a phenotype characterized by growth retardation. (A) Homozygous teeny and WT mice. (B and C) Manhattan plots showing P values calculated using the indicated transmission models. The −log10 P values (y axis) are plotted vs. the chromosomal positions of 64 mutations (x axis) identified in the G1 founder of the pedigree. Horizontal red and yellow lines represent thresholds of P = 0.05 with or without Bonferroni correction, respectively. (B) Binary, researcher-specified phenotype data (affected vs. unaffected) were used for linkage analyses (n = 9 affected, 15 unaffected). (C) Quantitative phenotype data (body weight) were used for linkage analysis. Corresponding phenotypic data plotted vs. genotype at the Kbtbd2 mutation site are shown (Lower). Each data point represents one mouse. Mean (μ) and SD (σ) are indicated. (D) Protein domains of mouse Kbtbd2, a member of the BTB (Broad-complex, Tramtrack and Bric à brac)-BACK (BTB and C-terminal kelch)-Kelch family of proteins. The teeny mutation is an arginine to premature stop codon substitution at position 121 of the protein. NTE, N-terminal extension.
The Teeny phenotype was also mapped as a quantitative trait, reduced body weight, using a smaller number of mice. A total of 21 animals were weighed, and weights were scaled with respect to age and sex. Although only two homozygotes were represented in the sample, the Kbtbd2 mutation showed strong linkage (P = 3.233 × 10−9), cosegregating with a mutation in Clcn1, using a recessive model of inheritance (Fig. 4C).
Kbtbd2 is a putative ubiquitin ligase with a BTB domain (Fig. 4D), for which no function has previously been reported. Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 KO confirmed the assessment of causality based on linkage (Table 1).
Table 1.
Weights of male Kbtbd2 CRISPR KO mice
| Genotype | N | Weight (g) | P value* |
| +/+ | 2 | 20.65 ± 2.65 | |
| Kbtbd2 CRISPR KO | 5 | 11.28 ± 1.25 | 0.0031 |
Mice were 6 wk old.
P value determined by Student t test.
Mapping a Quantitative Trait: T Cell-Dependent Antibody Response to Immunization.
Many immunological phenotypes are incompletely penetrant or show relatively high variance. If definitive discrimination between affected and nonaffected populations cannot be made, phenotypes are best mapped as quantitative traits. As an example, we used Linkage Analyzer to identify mutations that alter T cell-dependent antibody responses in mice immunized with ovalbumin (OVA) or β-galactosidase (βgal). Assessments of allele effects were made computationally based on statistical associations between the magnitude of a quantitative trait and the predetermined zygosity of specific variant alleles.
Phenotypic screening involved measurement by ELISA of antigen-specific IgG in the blood 14.5 d after immunization with alum plus OVA (OVA/Alum) or with βgal encoded by a recombinant Semliki Forest Virus vector (rSFV-βgal) (10). Of 7,436 genes screened (11,010 variant alleles present in 363 pedigrees encompassing 12,007 G3 mice), 24 genes (24 alleles) were implicated with the following specifications for linkage: ≥3 homozygous mice for each implicated mutation site, ≥16 mice in the pedigree, a linkage P value cutoff of 0.002 with Bonferroni correction, implication using both raw and normalized phenotype data, and a single linkage peak representing the implicated mutation at least three logarithms greater than the next highest peak in the Manhattan plot. Forty additional genes were implicated by the first three criteria but failed to meet the requirements for implication by both raw and normalized datasets or by a single linkage peak at least three logarithms greater than the next highest peak. The 24 implicated genes were located in 14 pedigrees containing a total of 482 G3 mice, and among them were 9 genes previously associated with T- or B-cell development or T cell-dependent antibody responses (Table 2) and 10 genes closely linked to them. There were also five novel genes not previously known to affect antibody responses. Manhattan plots for the transmission models giving strongest linkage between mutations in the nine known genes and phenotype are shown in Fig. 5 and Fig. S1.
Table 2.
Known genes implicated in screens for altered T cell-dependent antibody responses to immunization
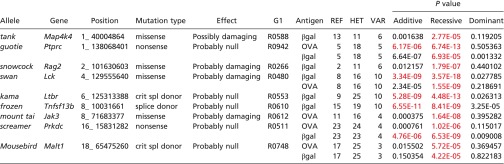 |
Criteria for linkage: VAR ≥ 3; pedigree size ≥ 16 mice; P ≤ 0.002 w/Bonferroni correction; “Raw+Norm” restriction applied; single linkage peak 3 logarithms > next highest peak. P values ≤ 0.002 w/Bonferroni correction are in red. crit spl donor, critical splice donor.
Fig. 5.

Mapping mutations in three known genes required for T cell-dependent antibody responses. (A–C, Left) Manhattan plots showing P values calculated using a recessive transmission model. The −log10 P values (y axis) are plotted vs. the chromosomal positions of all mutations (x axis) identified in the G1 founder of each pedigree. Causative mutations are shown in red. Horizontal red and yellow lines represent thresholds of P = 0.05 with or without Bonferroni correction, respectively. (Right) Quantitative phenotypic data are plotted vs. genotype at the causative mutation sites. Each data point represents one mouse. Mean (μ) and SD (σ) are indicated.
Conversely, for 653 putative null alleles of 631 genes examined in the homozygous state three or more times in pedigrees containing ≥16 mice, we failed to detect altered antigen-specific IgG production with a P value of 0.10 or less (with Bonferroni correction; Dataset S1). These 653 putative null alleles of 631 genes all failed to reject the null hypothesis of dispensability in the OVA/Alum and/or rSFV-βgal assays. Notably, we find no published suggestion that any of these genes plays an essential role in the T cell-dependent antibody response.
By tracking the amount of mutational damage done by mutagenesis and brought to homozygosity through inbreeding within a specified sample of G3 mice, a bracketed measurement of genome saturation can be made. The number of genes for which homozygosity for putative null alleles (the gold standard for loss of function) has been analyzed can be taken as the lower limit of saturation, whereas the number of genes with homozygosity for putative null alleles or alleles judged probably damaging by the program PolyPhen-2, which may or may not impair protein function (8), can be taken as the upper limit of saturation. Minimum and maximum estimates of saturation can be offered even with a relatively small dataset. Within the mice screened for defects of T cell-dependent IgG responses to OVA or βgal from pedigrees of at least 16 total mice, 652 putative null alleles of 632 genes were represented in homozygous form at least three times, whereas 4,177 putative null or probably damaging alleles of 3,439 genes were represented in homozygous form at least three times. The minimum genome saturation achieved was therefore 2.7%, whereas the maximum saturation achieved was 14.9%. The inferred number of target genes detectable in this screen might range between 94 and 518 (based on 14 implicated genes, 9 known and 5 unknown, identified using the following specifications for linkage: ≥3 homozygous mice for each implicated mutation site, ≥16 mice in the pedigree, a linkage P value cutoff of 0.002 with Bonferroni correction, implication using both raw and normalized phenotype data, and a single linkage peak representing the implicated mutation at least three logarithms greater than the next highest peak in the Manhattan plot).
Identification of Causative Mutations in 275 Pedigrees Screened for 45 Phenotypes.
Using Linkage Analyzer, we have tested a total of 35,764 mutations in 14,142 genes, derived from 594 pedigrees and distributed within 14,586 G3 mice, for their ability to cause phenovariance in 45 screens of immunological function (SI Text). Some of the screens measured interdependent phenotypes, for example, flow cytometric measurements of the frequency of B cells or T cells and the B-cell:T-cell ratio. Most but not all screens were applied to all mice. The 35,764 validated mutations were queried for individual phenotypic effects ∼2 × 106 times, and ∼1 × 106 individual determinations of genotype were made to permit mapping. Mutant alleles were represented in homozygous form at least once per pedigree for 84.1% of mutated genes; the percentage decreased to 59.4% for those with at least three instances of homozygosity.
When linkage was assessed for only probably null or probably damaging mutations in all 45 screens together, with requirements for VAR ≥ 0 within pedigrees of any size, P ≤ 0.01 with Bonferroni correction, application of the Raw+Norm restriction, and requirement for a single linkage peak representing the implicated mutation at least two logarithms greater than the next highest peak in the Manhattan plot, a total of 541 alleles of 519 genes (present in 276 pedigrees encompassing 6,245 G3 mice) were implicated out of 14,222 alleles of 8,620 genes screened (present in 610 pedigrees encompassing 15,055 G3 mice). An additional 1,982 genes failed to meet the requirements for implication by both raw and normalized datasets or by a single linkage peak at least two logarithms greater than the next highest peak. Forty-seven alleles of 45 genes with previously known function(s) in the immune phenotypes surveyed were implicated as causative for at least one of the phenotypes (Dataset S2). The involvement of 474 genes in one or more immune phenotypes was previously unknown. Based on the number of “implicated” noncausative mutations in linkage with the 45 known implicated genes, we calculated an average of 1.62 mutations per linkage peak. A similar rate of implication by linkage (rather than direct causation) might be expected among the 494 alleles of 474 unknown implicated genes. Determining which of two or more cosegregating mutations is causative for a phenotype depends on knowledge of the effect of a second mutant allele or on transgenic rescue of the mutant phenotype with the WT allele.
Detection of Lethal Alleles.
Linkage Analyzer detects associations with lethality based on underrepresentation of homozygous mutations in G3 mice. When linkage to lethality was assessed with requirements for VAR ≥ 0, pedigree size ≥ 16 mice, and P < 0.05 (Bonferroni correction applied), a total of 306 alleles of 299 genes, although viable in the heterozygous state, were found to bias against survival in the homozygous state before the age of weaning (4 wk). Many of these assessments might reflect linkage to a causative lethal mutation. The low level of monogenic lethality is likely attributable to the low power to detect statistically significant lethality in pedigrees containing small numbers of G3 mice. If the analysis is confined to alleles for which ≥20 instances of heterozygosity were observed in a pedigree (4,681 alleles of 3,828 genes), 253 alleles of 248 genes are implicated. Assuming once more that there are 1.62 mutations per linkage peak, one of which is causative, only 3.3% of ENU-induced exomic mutations with a potential for causing a change in coding sense are homozygous lethal (an average of 2.1 such mutations would exist per pedigree produced without a G0′ female). This estimate is consistent with the inference of Kile et al. (11), who observed monogenic lethality emanating from a 34.2-Mb region containing 758 genes on chromosome 11 in 7.5% of all pedigrees studied or about two to six lethal mutations per pedigree if we extrapolate to the whole genome.
Mapping Traits in Superpedigrees.
Approximately 3.1% of ENU-induced mutations in our colony are shared between two or more pedigrees, inherited from a common ancestral G0 male. Moreover, multiple alleles have already been identified for 9,296 of the 14,583 genes with validated mutations, a frequency of 63.7%. Because the genotypes at all mutation sites in all G3 mice are known, combining pedigrees with identical or nonidentical allelic mutations to make superpedigrees is possible. A superpedigree is then analyzed for genotype-phenotype associations as if it were a single pedigree. This increases the power to detect linkage, especially for weak or low penetrance phenotypes, and can help resolve a causative mutation where causative and noncausative mutations are closely linked. Relative to single pedigree analysis, combining pedigrees can greatly increase the strength of a genotype-phenotype association or eliminate it from consideration. As data accumulate from many pedigrees over time, the power to implicate or exonerate genes from participation in defined biological processes increases. Superpedigrees are automatically generated and analyzed by Linkage Analyzer whenever allelic mutations and phenotypic data are added to the database; lethal effects of mutations are also monitored in superpedigrees when possible.
Low IgD expression on peripheral blood B cells was detected in two pedigrees (R0522 and R0525) arising from a single G0 male. The phenotypes were designated lentil and chives in each of the two pedigrees, respectively. The recessive lentil phenotype was mapped to three colocalizing mutations [in Gpr176, Pla2g4b, and transformation related protein 53 binding protein 1 (Trp53bp1)] with equivalent P values (P = 1.715 × 10−6), whereas the chives phenotype was linked to a recessive mutation in Zfp708 (P = 7.071 × 10−7), although equivalent weak linkage with Olfr1056, Olfr1118, Olfr1200, and Trp53bp1 (P = 0.001139) was also detected (Fig. 6 A and B). A third pedigree (R0543) also contained the Trp53bp1 mutation, but only in the heterozygous state (four REF and five HET genotypes). By combining the three pedigrees, the significance of linkage to Trp53bp1 was greatly increased (P = 8.23 × 10−12), convincingly implicating the Trp53bp1 mutation as causative (Fig. 6C), as later confirmed by CRISPR knockout analysis.
Fig. 6.

A missense mutation of Trp53bp1 is linked to reduced expression of IgD by PBMC. Manhattan plots of (A) lentil (R0522) and (B) chives (R0525) pedigrees individually, along with corresponding phenotypic data plotted vs. genotype at the Trp53bp1 mutation site. (C) Manhattan plot of the superpedigree (R0522, R0525, and R0543) and corresponding phenotypic data plotted vs. genotype at the Trp53bp1 mutation site. Each data point represents one mouse. Mean (μ) and SD (σ) are indicated. MFI, mean fluorescence intensity.
Complex Traits.
We occasionally observed ENU-induced phenovariance that has a complex origin. For example, mutations of SRY (sex determining region Y)-box 10 (Sox10) and tyrosinase (Tyr) in the same pedigree produced mice with white-spotted cream colored coats, the cream color resulting from the Tyr mutation and the white spots from the Sox10 mutation. We also identified immunological phenotypes resulting from a combination of mutations in Itgb2 (joker) and Unc13d (jinx), which caused an NK cell maturation defect combined with mouse cytomegalovirus susceptibility (12). Hence, additive effects produced by a relatively small collection of ENU-induced mutations can cause distinct phenotypes within single pedigrees, and similar effects might be anticipated where other biological processes are concerned. If a variant phenotype depends on two unlinked mutations acting together in any combination of zygosities, Linkage Analyzer will declare this interpretation. However, because on average only 1 of 64 mice will be homozygous for two unlinked nonlethal mutations, large numbers of G3 mice are needed to implicate double homozygosity in the production of a complex trait.
Materials and Methods
Mice and Breeding Pipeline.
All animals were housed at the University of Texas Southwestern Medical Center, and experiments were performed in accordance with institutionally approved protocols. Male and female C57BL/6J mice were obtained from The Jackson Laboratory. Male C57BL/6J mice were mutagenized with ENU as previously described (13). Breeding plans are shown in Fig. 1, and the production pipeline is described in SI Materials and Methods.
Whole Exome Sequencing of G1 Males.
A total of 58,755,622 bp, which include ∼50 bp of intronic sequence upstream and downstream of each exon, were targeted for whole exome sequencing using oligonucleotide probes from Life Technologies’ TargetSeq Custom Enrichment Kit and modified to run on an Illumina HiSeq 2500 platform. Paired-end 2 × 100-bp sequencing was performed using an Illumina HiSeq 2500 instrument to detect heterozygous autosomal and hemizygous X-linked mutations. Reads were demultiplexed using CASAVA according to their index sequence and lane numbers. Reads were mapped to the University of California Santa Cruz mm10 genome reference sequence for C57BL/6J and paired using Burrows-Wheeler Aligner (BWA) v 0.6.2. Duplicate reads were removed by SAMtools and indel regions left aligned by Genome Analysis Toolkit (GATK). Coverage was calculated over targeted regions using BEDTools. Variants relative to the C57BL/6J reference sequence (GRCm38) were called and annotated by a combination of SAMtools, SnpEff, SnpSift and ANNOVAR, and then filtered to eliminate SNPs listed in dbSNP (build 137) and common variants observed in a rolling total of 40 previously sequenced mice with unshared G0 sires. Synonymous mutations and mutations not predicted to affect splicing or coding sense were also eliminated. Remaining mutations with a quality score ≥ 20 were listed in BED format and targeted in AmpliSeq panel design. All mutations assumed to be true [quality score ≥ 80 (corresponds to a true positive rate of 99% for 13,899 mutations validated in preliminary analysis of HiSeq accuracy)] were provisionally uploaded to the Incidental Mutations list on Mutagenetix. The Incidental Mutations list was revised after validation by Ion AmpliSeq custom panel sequencing to display all true positive mutations, regardless of quality, and eliminate all false mutations. All mutations contained in this list were analyzed by Simple Modular Architecture Research Tool (SMART) and PolyPhen-2 (HumDiv trained model) or a splice site prediction program. References for the software packages used in the pipeline are listed in SI Materials and Methods.
An average coverage depth of 85× was achieved per targeted nucleotide coordinate, and <4% of all nucleotides were covered fewer than 10×, suggesting that fewer than 4% of all mutations induced by ENU within the region of the exome were overlooked.
Splice Site Prediction Program.
Prediction of the effect of ENU-induced mutations on splicing of a transcript is based on the maximum entropy model developed by Yeo and Burge (14), in which scores are assigned to 9-mer splice donor sites and 23-mer splice acceptor sites. Higher scoring sequences have a greater probability of being used in splicing. All ENU-induced mutations were evaluated for their effect on the score of native sequences, and a gene splicing model was then predicted and stored in the Mutagenetix database. Mutations predicted to disrupt native splice sites leading to exon skipping or use of a cryptic splice site are classified as probably null. Intronic mutations not predicted to affect splicing are classified as probably benign.
Amplicon Resequencing of Targeted Loci.
Data from whole exome sequencing of each G1 mouse were processed, and loci from mutations that caused coding changes, splice site variations, etc., were added to a custom AmpliSeq panel from Life Technologies. All loci were amplified in single PCR via custom AmpliSeq panel primer mixes. Each pedigree included the G1, G0′, female G2s, and all G3s, plus the addition of a WT control. The amplicons were made into Ion Torrent barcoded libraries and run on the Ion PGM (Life Technologies) in 316 or 318 chips via 200-bp sequencing. Alignment was performed by TMAP software within the Torrent Suite Software package to the UCSC mm10 genome reference sequence for C57BL/6J. Variants were called using the Torrent Variant Caller plugin available in Torrent Suite software, and an output file was generated containing the total number of reads for REF and VAR alleles for each barcoded sample.
Automated Genotype Assignment at All Loci in G0′, G2, and G3 Mice.
Unambiguous genotypic assignments at each variant site in each mouse were made computationally and uploaded into Mutagenetix for later use.
The REF/VAR ratio was coded as x1/x2 on each locus of every mouse, where x1 refers to the count of reads supporting REF and x2 supporting VAR. If x1 + x2 < 5, the dosage is marked as N/A and masked from the following analysis. Otherwise, the dosage is calculated as x2/(x1 + x2). For the additive model, this raw dosage value is used. For the recessive and dominant models, a further transformation of the dosages is used:
In the recessive transformation, the use of this function brought the raw dosage between (0, 0.7) to (0–0.00669) and the raw dosage between (0.8, 1) to (0.99331–1). The raw dosage that falls into the gray area of (0.7, 0.8) will have a large span from almost 0 to almost 1 after transformation to reflect the uncertainties. The raw/transformed dosage scores within (0, 1) for each locus are tested for association with phenotype. A typical distribution of genotyping calls is shown in Fig. S2.
Phenotypic Screening.
After weaning, all G3 mice from each pedigree were screened for phenovariance according to 109 parameters, most of which were quantitative measures of immune performance (Fig. 3). Screens were performed in a precise order on a regimented schedule. Age-matched WT animals were screened together with G3 mice, and normalization was performed relative to the screening results of the WT controls on a day by day basis. When possible, whole pedigrees were screened together on a single day. If visible or behavioral phenotypes were observed within the pedigree (a relatively common occurrence), these phenotypes were recorded using a binomial scoring system (affected vs. unaffected). Phenotypic data were uploaded to the Mutagenetix database and combined with genotype data for automated mapping. Protocols for the antibody response screens and blood flow cytometry screens, for which data are presented in this report, are described in SI Materials and Methods.
Automated Mapping Using Linkage Analyzer.
Overview.
For each phenotype identified by screening, automated computation of single locus linkage was performed for every mutation in the pedigree using recessive, semidominant (additive), and dominant models of transmission using the program Linkage Analyzer, an R-based program. Linkage Analyzer is freely available for download and online data analysis of selected pedigrees described herein via the Mutagenetix website (https://mutagenetix.utsouthwestern.edu/linkage_analysis/linkage_analysis.cfm).
In each case, the presence or absence of a qualitative phenotype, or the magnitude of a quantitative phenotype, was correlated with genotype (REF, homozygous for reference allele; HET, heterozygous for reference allele and variant allele; or VAR, homozygous for variant allele) at each mutation site in all mice in the pedigree. Continuous variables were analyzed using a linear regression model to assess linkage between specific mutations and quantitative traits. Binomial calculations were performed to assess linkage between specific mutations and qualitative traits. The output of automated mapping is a Manhattan plot of the P value of genotype-phenotype association using recessive, semidominant, and dominant models of transmission for every mutation in the pedigree.
In addition, the likelihood that each mutation influences viability was tested. In this latter assessment, the composition of the mutations carried by each G2 mother was important, insofar as noncarrier status for a particular mutation will prevent homozygosity in any G3 offspring derived from this mother. Computational checks of maternity and paternity were also carried out, insofar as a mouse bearing none of the target mutations in either homozygous or heterozygous form is not likely derived from the pedigree being tested; a mouse homozygous for mutation(s) not carried by the putative G2 mother cannot be derived from that mother.
Mapping monogenic traits.
Depending on whether the phenotype scores are continuous or binary, they are assumed to be generated from the Gaussian or binomial distribution, respectively. It is possible that different G2 mothers, by virtue of maternal effect mutations, influence the phenotypes of their offspring. Therefore, G2 mother identity is optionally treated as a potential confounder in the statistical analysis to control this effect. When maternal effects are considered, a generalized linear mixed effect model (15) is used, where maternal effect information is treated as the random variable. When maternal effects are not considered, a generalized linear model is used without a G2 mother covariate. In the recessive, additive, and dominant settings, respectively, genotypes of (REF, HET, VAR) are coded numerically as (0,0,1), (0,1,2), and (0,1,1). For each gene, a likelihood ratio test from a generalized linear model or a generalized linear mixed effect model is used to assess the association of the numerical genotypes with binary or continuous phenotypes controlling for the effect of G3 mouse sex. P value is corrected by Bonferroni procedure and adjusted based on whether a one-tailed or two-tailed test is desired (in practice, we use the two-tailed test and adjust the P value according to the sign of coefficient and the desired direction if one-tailed test is needed).
Mapping complex traits (double locus linkage).
The procedure for detecting interactions between two loci follows the framework of testing for one locus. In the generalized linear (or mixed effect) model, the main effects of two loci and the interaction term between two loci are included in the model. We assumed four disease models (recessive, additive, dominant, and inhibitory models) for the phenotype/two-locus associations. Let the first locus be A and the second locus be B. The coding of the nine possible combinations of G3 genotypes is given in Table S3.
Detecting lethal effects (single and synthetic lethals).
Lethal test for a single locus.
To test lethality for a single mutation, we calculate the probability of observing equal or rarer G3 mice with VAR genotypes using Fisher’s exact test. For any G3 mouse, it can be an offspring of a G2 mouse with HET genotype with probability 1/4, or an offspring of a G2 mouse with unknown genotype with probability 1/8. We denote G3 mice with VAR genotypes as a random variable Nvar = NfromG2Het + NfromG2unknown, where NfromG2Het follows binomial distribution Binomial (NG2Het, 1/4) and NfromG2unknown similarly follows binomial (NG2unknown, 1/8). The exact P value can be calculated as
Synthetic lethality test.
The synthetic lethality test is carried out to identify possible detrimental effect on survival of G3 mice rendered by combinatory effect of two mutations. Again, the G2 mothers could have REF and HET genotypes, but some G2 mothers’ genotypes are unknown (coded as Un). Given the genotypes of the G2 mother at the two mutation sites (A and B), the probability of giving birth to G3 offspring of (HET, VAR), (VAR, HET), and (VAR, VAR) genotypes is given in Table S4.
Given the number of G3 offspring and the genotype pairs of each G2 mother, we deduced a similar formula to the single locus lethal test. Then Fisher’s exact P value can be calculated as the summation of the probability of observing equal or less counts of G3 offspring of (HET, VAR), (VAR, HET), and (VAR, VAR) than observed counts.
Dichotomization of continuous phenotype scores into binary phenotype scores.
For dichotomizing continuous phenotype scores into binary phenotype scores, we used the model based clustering method (Mclust) (16). Mclust chooses the optimal model according to Bayesian Information Criterion (BIC) for EM initialized by hierarchical clustering for Gaussian mixture models. If the BIC of a two-component mixture model with equal variance is larger than that of a one-component model, the phenotype scores can be optionally transformed to a binary variable.
Mapping monogenic traits by combining multiple pedigrees (superpedigree analysis).
Superpedigrees incorporate genotypic and phenotype data from two or more pedigrees containing identical or nonidentical allelic mutations increasing the power to detect causal relationships between mutations and phenotypes. Both continuous and binary traits can be analyzed with optional adjustments for sex, age, and maternal effects. A correction for the phenotypic performance of each allele within its component pedigree is made to exclude nongenetic pedigree effects that might differ between pedigrees analyzed at different times. For example, if REF, HET, and VAR CD8+ T-cell counts are uniformly high in one pedigree and uniformly low in another pedigree, both of which have allelic mutations of a certain gene, the mean phenotypic performance of each allele will be normalized to correct for interpedigree assay differences. Pedigrees derived from different G1 males have mostly unshared mutations. For such unshared mutations, the G3 genotypes at loci mutated in one pedigree are assumed to be REF in the other pedigree and vice versa. For quantitative or qualitative traits, we applied a linear regression model or a generalized linear mixed model, respectively. We obtained P values using the likelihood ratio test and performed a Bonferroni correction according to the total counts of mutated alleles.
Hardware.
Linkage Analyzer was run on a PSSC Powerwulf Bio Titanium Cluster with 1 head node, 8 compute nodes, 204 total Intel Xeon E5-2600 v2 Cloud Series CPU cores, and 192 total 2.4-GHz cores on 8 compute nodes and 12 total 2.1-GHz cores on 1 Head Node; 288 GB of total system memory was distributed with 1.33 GB per compute processor core and 2.66 GB per head node processor core. There were 40 TB of raw storage space with 2 TB in the head node configured as RAID1 and 19 TB configured as RAID6 storage space; 1 TB scratch/data space per compute node 10 GbE Network Backplane; CentOS 6.5 64-bit Linux Operating System.
Data from Illumina whole exome sequencing were processed using a Dell PowerEdge R620 Cluster (iCompute Cluster) with 64 Total Intel Xeon CPU cores, 256 GB total system memory, and 40 TB raw storage space. Data from the Ion Torrent sequencers were processed using a Dell T7600 n-series server with 12 Total Intel Xeon CPU cores and 48 GB total system memory.
The Mutagenetix Database Server runs on a Dell PowerEdge R720 with Intel Xeon E5-2630 2.30 GHz, 12 total Intel Xeon CPU cores, and 64 GB total system memory.
Verification by CRISPR Knockout or Knockin Mutations.
CRISPR target sites for genes were chosen using web resource CRISPR Design (crispr.mit.edu). Oligo DNA pairs corresponding to each CRISPR target were synthesized, annealed, and cloned into plasmid pX330 (Addgene) (17). Neuro-2A cells transfected with CRISPR plasmids were subjected to surveyor assay to determine CRISPR activity (18). For in vitro transcription of CRISPR small guide RNA (sgRNA) and cas9 mRNA, T7 promoter was first added to template by PCR. PCR products were purified using QiaQuick Purification Kit (Qiagen) and served as the templates for in vitro transcription with T7 Quick High Yield RNA Synthesis Kit (New England Biolabs) for sgRNA or using mMESSAGE mMACHINE T7 ULTRA kit (Life Technologies) for cas9 mRNA. Both Cas9 mRNA and sgRNA were purified using MEGAclear kit (Life Technologies) and eluted in RNase-free water.
Microinjection of Zygotes for CRISPR Targeting of Genes.
Female C57BL/6J mice were superovulated by injecting them with 6.5 U of pregnant mare's serum gonadotropin (PMSG; Millipore, 367222) and then 6.5 U of human chorionic gonadotropin (hCG; Sigma-Aldrich, C1063) 48 h later. The superovulated females were subsequently mated with C57BL/6JJcl male mice (The Jackson Laboratory) overnight. The following day, fertilized eggs were collected from the oviducts of the female mice, and in vitro transcribed Cas9 mRNA (50 ng/μL) and sgRNA (20–50 ng/μL) were injected into the pronucleus or cytoplasm of the fertilized eggs. The injected embryos were cultured in M16 medium (Sigma-Aldrich, M7292) at 37 °C and 95% air/5% CO2. For the production of mutant mice, two-cell stage embryos were transferred into the ampulla of the oviduct (10–20 embryos per oviduct) of pseudopregnant Hsd:ICR (CD-1) females (Harlan Laboratories).
Supplementary Material
Acknowledgments
We thank Peter Jurek for expert assistance in preparing the figures and the administrative staff of the Center for the Genetics of Host Defense (Linda Watkins, Betsy Layton, Gail Wright, Laurie Hughes, and Wanda Simpson) for supporting the laboratory effort described in this paper. J.S. is a Howard Hughes Medical Institute Medical Research Fellow. This work was also supported by generous donations from the Lyda Hill Foundation and the Kent and JoAnn Foster Family Foundation and by National Institutes of Health Grants P01 AI070167, U19 AI100627, and R37 GM067759.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1423216112/-/DCSupplemental.
References
- 1.Schneeberger K. Using next-generation sequencing to isolate mutant genes from forward genetic screens. Nat Rev Genet. 2014;15(10):662–676. doi: 10.1038/nrg3745. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Z, et al. Massively parallel sequencing identifies the gene Megf8 with ENU-induced mutation causing heterotaxy. Proc Natl Acad Sci USA. 2009;106(9):3219–3224. doi: 10.1073/pnas.0813400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold CN, et al. Rapid identification of a disease allele in mouse through whole genome sequencing and bulk segregation analysis. Genetics. 2011;187(3):633–641. doi: 10.1534/genetics.110.124586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yabas M, et al. ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat Immunol. 2011;12(5):441–449. doi: 10.1038/ni.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrews TD, et al. Massively parallel sequencing of the mouse exome to accurately identify rare, induced mutations: An immediate source for thousands of new mouse models. Open Biol. 2012;2(5):120061. doi: 10.1098/rsob.120061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bull KR, et al. Unlocking the bottleneck in forward genetics using whole-genome sequencing and identity by descent to isolate causative mutations. PLoS Genet. 2013;9(1):e1003219. doi: 10.1371/journal.pgen.1003219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fairfield H, et al. Mutation discovery in mice by whole exome sequencing. Genome Biol. 2011;12(9):R86. doi: 10.1186/gb-2011-12-9-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnold CN, et al. ENU-induced phenovariance in mice: Inferences from 587 mutations. BMC Res Notes. 2012;5:577. doi: 10.1186/1756-0500-5-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hidmark AS, et al. Humoral responses against coimmunized protein antigen but not against alphavirus-encoded antigens require alpha/beta interferon signaling. J Virol. 2006;80(14):7100–7110. doi: 10.1128/JVI.02579-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kile BT, et al. Functional genetic analysis of mouse chromosome 11. Nature. 2003;425(6953):81–86. doi: 10.1038/nature01865. [DOI] [PubMed] [Google Scholar]
- 12.Crozat K, et al. Impact of β2 integrin deficiency on mouse natural killer cell development and function. Blood. 2011;117(10):2874–2882. doi: 10.1182/blood-2010-10-315457. [DOI] [PubMed] [Google Scholar]
- 13.Georgel P, Du X, Hoebe K, Beutler B. ENU mutagenesis in mice. Methods Mol Biol. 2008;415:1–16. doi: 10.1007/978-1-59745-570-1_1. [DOI] [PubMed] [Google Scholar]
- 14.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11(2-3):377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 15. Bates D, Maechler M, Bolker B, Walker S (2014) lme4: Linear mixed-effects models using Eigen and S4. R package version 1.1-7. Available at CRAN.R-project.org/package=lme4. Accessed January 7, 2015.
- 16.Fraley C, Raftery AE. MCLUST: Software for model-based cluster analysis. Journal of Classification. 1999;16(2):297–306. [Google Scholar]
- 17.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guschin DY, et al. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.