

Abstract
The MAGMa software for automatic annotation of mass spectrometry based fragmentation data was applied to 16 MS/MS datasets of the CASMI 2013 contest. Eight solutions were submitted in category 1 (molecular formula assignments) and twelve in category 2 (molecular structure assignment). The MS/MS peaks of each challenge were matched with in silico generated substructures of candidate molecules from PubChem, resulting in penalty scores that were used for candidate ranking. In 6 of the 12 submitted solutions in category 2, the correct chemical structure obtained the best score, whereas 3 molecules were ranked outside the top 5. All top ranked molecular formulas submitted in category 1 were correct. In addition, we present MAGMa results generated retrospectively for the remaining challenges. Successful application of the MAGMa algorithm required inclusion of the relevant candidate molecules, application of the appropriate mass tolerance and a sufficient degree of in silico fragmentation of the candidate molecules. Furthermore, the effect of the exhaustiveness of the candidate lists and limitations of substructure based scoring are discussed.
Keywords: mass spectrometry, small molecule identification, metabolomics, MS/MS fragmentation, substructures
INTRODUCTION
The identification of unknown molecules is a major bottleneck in untargeted LC-MS based metabolite profiling.1,2) In the absence of data from reference compounds or NMR experiments, fragmentation data from tandem mass spectrometry (MS/MS) may be used to derive chemical information about the unknown molecules. The interpretation of the product ion spectra by experts is labor intensive, and several academic and commercial projects are dedicated to the development of software tools to facilitate this task.3) For example, computer algorithms are used to derive the most likely elemental formula of a mass peak, based on accurate mass and/or isotope ratios,4–6) or to provide candidate chemical structures on the basis of MS fragmentation patterns. One strategy to achieve the latter is to search for similar spectra in mass spectral databases.7) However, in the absence of informative reference spectra, computational methods are needed to annotate fragment ion spectra solely on the basis of the chemical structures of potential candidate molecules. A common approach is to select the best matching chemical structures from comprehensive sets of candidate molecules.8,9) Such a procedure must assess how well the observed fragment peaks can be explained by a given candidate structure, e.g. based on substructures,10–13) or rule-based fragments.9) Different scoring functions based on matching the in silico fragments with observed peaks have been applied to prioritize lists of candidate structures, including different regio-isomers.9,10,13,14)
Recently, we have introduced an extended substructure-based algorithm (MAGMa) capable of handling spectral trees generated by multistage accurate MSn, resulting in hierarchical and internally consistent trees of substructures representing the subsequent fragmentation steps.15) The method has been used to perform a complete annotation of an LC-MSn metabolite profile of green tea, illustrating its utility at the scale of a metabolomics experiment.16) Although the MAGMa algorithm was developed to be able to handle MSn multistage fragmentation data with n>2, its performance on MS/MS data was demonstrated to be comparable to other methods.15)
In 2012, the CASMI (Critical Assessment of Small Molecule Identification) contest was initiated, which challenged scientists to interpret a number of blinded MS/MS spectra and to provide ranked lists of solutions, both for the correct molecular formula and the correct chemical structure.17) This allowed identification experts and algorithm developers to objectively compare the reliability of their methods. The second edition of the CASMI contest, organized in 2013, comprised of accurate MS and MS/MS spectra for 16 compounds. The competition was aimed at finding the correct chemical structure (category 2), as well as the correct molecular formulas for 12 of these 16 challenges (category 1). Ranked lists of multiple solutions could be submitted. Here, we describe the application of the MAGMa algorithm to solve these challenges. We submitted solutions to 12 of the 16 challenges in category 2, as well as the corresponding unique molecular formulas for 8 challenges in category 1. In a retrospective analysis we also present the MAGMa results on the remaining challenges and discuss the influence of some of the MAGMa processing parameters in relation to specific challenges.
METHODS
Input file preparation
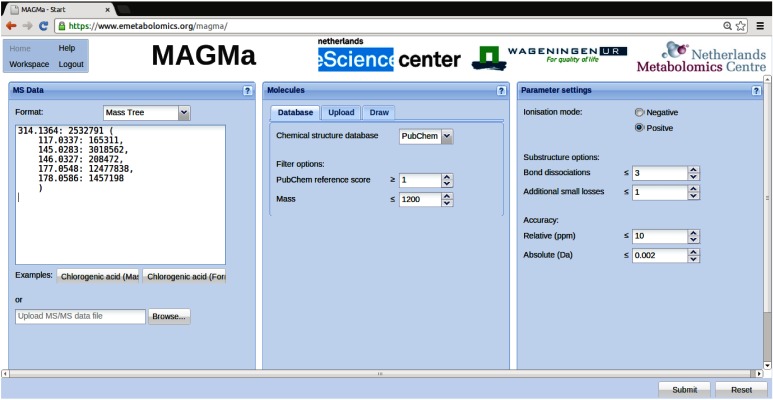
The peak lists provided by the CASMI contest were first converted to MAGMa input files. The MAGMa input format for multistage MS fragmentation data is an extension of the more commonly used peak lists, such as those provided for the CASMI challenges, enabling the description of a hierarchy of fragment peaks. After a given precursor peak, the next level MS fragment peaks can be listed between parenthesis. Parenthesis can be used recursively, but for the MS/MS data supplied by CASMI only two hierarchical levels were involved: the precursor mono-isotopic peak taken from the MS file and all fragment peaks taken from the MS/MS file. As an example, the MAGMa input for challenge 1 is shown in Fig. 1.
Fig. 1. The completed MAGMa input page for challenge 1 of CASMI 2013. Peaks are defined by m/z : intensity, and separated by commas. Parentheses are used to indicate the next level of fragment peaks observed for the preceding (precursor-) ion. Line breaks and spaces can be inserted freely, without influencing the processing. Candidate molecules were retrieved from PubChem and m/z values were matched with theoretical masses using a tolerance of 10 ppm and 0.002 Da (i.e. the larger of the two depending on the peak mass).
MAGMa processing
The input data was processed with MAGMa in command line mode or via the web interface (www.emetabolomics.org/magma). The allowed mass deviation was set to 10 ppm, with an absolute minimum of 0.002 Da, except for challenges 4, 5, and 6, for which the mass tolerance for retrieving candidate molecules was set to 4 ppm, in accordance with the provided technical details. MAGMa retrieved candidate molecules from a local database consisting of all PubChem compounds on 2 December 2013, up to a mass of 1200 Da and composed of the elements C, H, N, O, P, and S only, assuming [M+H]+ or [M−H]− precursor ions for positive and negative mode data, respectively. For challenges 7 and 8, candidates with the provided elemental composition were retrieved directly from the online PubChem service in SDF format and used as input for the MAGMa calculations. Also for challenge 9, where we recognized from the isotope ratios in the MS file that the molecule contained chlorine atoms, we obtained candidates from an online query on mono-isotopic mass. Since the CASMI deadline, we have added a local MAGMa database containing halogenated PubChem compounds. This extension allowed us to apply the default MAGMa processing retrospectively on the data for the halogenated compounds of challenges 9, 15, and 16.
MAGMa systematically fragments all candidate molecules one by one, as described previously.15) Substructures were generated by applying up to 3 bond dissociations. This set of substructures was expanded with single losses of hydroxyl groups (representing neutral loss of water) or primary amine groups (representing neutral loss of ammonia). A substructure penalty score is calculated as the sum of the penalty values for all dissociated bonds (1, 2, and 3 for single, double and triple or aromatic bonds involving at least one heteroatom, respectively, and 2, 4, and 6 for single, double and triple or aromatic carbon–carbon bonds), with a maximum of 10. m/z values of the MS/MS peaks are matched with the best scoring substructures. A total penalty score for a given candidate is calculated as an intensity weighted average of the substructure penalties for all peaks in the MS/MS spectrum, including a (intensity weighted) penalty of 10 for non-matched peaks.
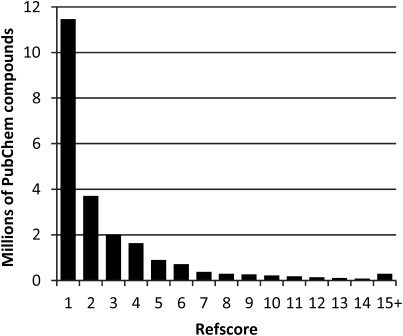
In addition to the MS/MS based candidate score, MAGMa provides a second parameter for each candidate, which we refer to as “refscore” and can be used as a general indicator of its relevance. PubChem does not systematically provide information about the (e.g. biological) relevance or occurrence of compounds. However, MAGMa extracts a simple statistic for each PubChem compound, equal to the total number of compound-related “substances” in PubChem, which represents the presence of the (stereoisomers of) a given compound in different mixtures and salt forms in a large number of depositor databases. Figure 2 presents the distribution of refscore values up to 15, which shows that the largest fraction of compounds is known from only one source and in only one chemical form, corresponding to a refscore of 1, and that an even larger fraction has a low refscore, e.g. below 5. We have used the refscore to reduce the large numbers of candidates obtained for challenges 1, 2, and 14, by applying a threshold value of 5.
Fig. 2. Distribution of refscore values of non-halogenated PubChem compounds in the local MAGMa database.
Candidate ranking
The fragment based penalty score provided by MAGMa is lowest for the most likely candidates and highest for the poorly matching candidates, which is opposite to the requirements for CASMI. For the CASMI entries we therefore provided a reversed ranking value, which was primarily based on the candidate score obtained from the substructure annotation. Equally scoring candidates were further ordered on the basis of their PubChem refscore, consistent with the “refined ranking” described previously.16) Candidates equal in both scores were assigned the same value corresponding to their average ranking position.
RESULTS
Solutions for twelve of the sixteen challenges in category 2 were submitted to the CASMI competition (Table 1, column 3). In six of the MAGMa solutions, the correct candidate was ranked in first position. In three other cases the correct candidate was in the top 5, while for the remaining three challenges lower rankings were obtained. For challenge 3, the ranking of the correct molecule among the first 17 candidates was the best solution among all participants.
Table 1. Overview of the MAGMa results on the CASMI 2013 challenges, category 2.
| # | Compound | Entry candidates | Rank | All PubChem candidates | Rank | <1000 candidates | Refsc. filter | Rankd | <500 candidates | Refsc. filter | Rank | <200 candidates | Refsc. filter | Rank |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Feruloyl tyramine | 1084a | 1 | 4694 | 1 | 858 | >6 | 1 | 422 | >10 | 1 | 181 | >14 | 1 |
| 2 | Feruloyl putrescine | 631a | 3 | 11548 | 116 | 856 | >4 | 7 | 486 | >6 | 2 | 185 | >11 | 1 |
| 3 | Acetyl-Gln-Leu-amide | 370 | 17 | 370 | 17 | 370 | >0 | 17 | 370 | >0 | 17 | 85 | >1 | — |
| 4 | Dihydrochalcone | 825 | 78 | 825 | 78 | 825 | >0 | 78 | 464 | >1 | 60 | 188 | >4 | 35 |
| 5 | Isoprothiolane | 350 | 2 | 350 | 2 | 350 | >0 | 2 | 350 | >0 | 2 | 162 | >1 | 2 |
| 6 | Phosphatidyl-6-acetyl-glucose | 2 | 1 | 2 | 1 | 2 | >0 | 1 | 2 | >0 | 1 | 2 | >0 | 1 |
| 7 | Cinnamtannin A3 | 17 | 1 | 17 | 1 | 17 | >0 | 1 | 17 | >0 | 1 | 17 | >0 | 1 |
| 8 | Prodelphinidin C2 | 1 | 1 | 1 | 1 | 1 | >0 | 1 | 1 | >0 | 1 | 1 | >0 | 1 |
| 9 | Chlorpyrifos | 113 | 1 | 178b | 1 | 178 | >0 | 1 | 178 | >0 | 1 | 178 | >0 | 1 |
| 10 | VAL-HIS-LEU-THR-PRO-VAL-GLU-LYS | 20 | 1 | 20 | 1 | 20 | >0 | 1 | 20 | >0 | 1 | 20 | >0 | 1 |
| 11 | Demethoxycurcumin | 4719 | 51 | 906 | >5 | 6 | 422 | >9 | 3 | 172 | >12 | 2 | ||
| 11 | Demethoxycurcumin (tautomer 1) | 4719 | 25 | 906 | >5 | 4 | 422 | >9 | 1 | 172 | >12 | 1 | ||
| 12 | Baicalein | 813 | 271 | 813 | >0 | 271 | 445 | >1 | 192 | 194 | >4 | 95 | ||
| 13 | EST; Aloxistatin | 207 | 42 | 207 | 42 | 207 | >0 | 42 | 207 | >0 | 42 | 104 | >1 | 22 |
| 14 | Tetrahydroalstonine | 1583a | 5 | 7321 | 20 | 736 | >6 | 4 | 449 | >8 | 1 | 195 | >11 | 1 |
| 15 | 2-(Perfluorooctyl)ethanol | 720 | 2 | 720 | >0 | 2 | 382 | >2 | 1 | 182 | >12 | 1 | ||
| 16 | Ofloxacin | 8172 | 699 | 998 | >7 | 78 | 447 | >11 | 33 | 189 | >18 | 12 | ||
| 16 | Ofloxacinc | 8172 | 126 | 998 | >7 | 18 | 447 | >11 | 6 | 189 | >18 | 3 |
a Number of candidates were reduced by filtering on refscore >5. b New list of candidates retrieved after implementing local database of halogenated PubChem compound. c Results obtained with increased number of substructures by breaking up to 5 bonds. d The MAGMa results obtained with the selected (<1000) candidate lists can be accessed online via http://www.emetabolomics.org/results_CASMI2013.html.
MAGMa results for the remaining challenges 11, 12, 15, and 16
For challenges 11, 12, 15, and 16 no solutions were submitted. Here we describe the initial results as well as a number of retrospective calculations for further analysis. For challenge 11, three fragment peaks at m/z=119.0538, m/z=143.0534, and m/z=158.0396 could initially not be explained by substructures even from the best scoring candidates, assuming a maximum mass error of 0.002 Da. This lead us to the assumption that the correct chemical structure was not present in the candidate list. In retrospect, two of the three tautomers of the correct candidate were present and it is more likely that the assumed maximum error of 10 ppm and 0.002 Da was too restrictive for a proper interpretation of the MS/MS spectrum. After the solutions were announced, we reprocessed the data using a minimum mass tolerance of 0.005 Da, resulting in acceptable substructure matches for the above mentioned peaks and in reasonable ranking positions of the two tautomers of demethoxycurcumin present (Table 1). Also for challenge 12, the correct candidate was in fact present in the initial PubChem candidate set, although at a low ranking position, as presented in Table 1. In this case the poor ranking was due to chemical rearrangements not properly represented by substructures as discussed later.
For challenges 15 and 16 the correct molecules were not present in the initial MAGMa candidate list, because the MAGMa database of PubChem compounds did not contain fluorine containing molecules. After the CASMI deadline we have added a second MAGMa compound database with halogenated compounds from PubChem, from which additional candidates can be retrieved optionally. The results with this extended candidate sets are now included in Table 1. The input data was constructed by generating composite spectra of the MS/MS spectra obtained at the different collision energies for both challenges, i.e. peaks obtained at both energies are merged into a single spectrum, taking the highest intensity for ions present at both energies.10,12) Nominal m/z values were replaced by accurate m/z values when available from the MS spectra and otherwise ignored. For challenge 16, several peaks from the higher energy spectrum (with m/z=122.0408, m/z=178.03072, m/z=179.03857, m/z=193.04172, m/z=194.02574, m/z=199.05114, m/z=205.04175, m/z=219.05744, and m/z=221.07309) could not be explained by substructures generated with the standard number of bond dissociations (up to 3 bond dissociations and one further loss of an OH or NH2 groups). Because of the fused ring system, we suspected that a number of the observed fragments may involve more than 3 bond cleavages. Indeed, repeating the MAGMa analysis with up to 5 bond dissociations (without OH or NH2 losses) resulted in substructure annotation of all fragment peaks, and a corresponding improvement of the ranking (Table 1, bottom row). When the same extensive fragmentation was applied to the other challenges, the rankings did not change significantly (results not shown), except for challenge 13, where the ranking of aloxistatin deteriorated slightly from 42 to 58, and for challenge 15, where 2-(perfluorooctyl)ethanol was ranked on top instead of the second position of the full list of 720 candidates.
Reducing the number of candidate structures
Automatic methods allow the evaluation of large numbers of molecules for a given MS/MS spectrum. MAGMa, like other automatic methods, can retrieve thousands of candidate molecules from PubChem. The evaluation of such comprehensive lists of candidate molecules is not possible in a manual approach, which is often guided by literature and experience with known compound classes. Manual analysis will therefore, at least initially, be biased towards well-known molecules, whereas automatically evaluated candidate molecules from PubChem may include large numbers of synthetic molecules with limited relevance to the sample of interest. To mimic the bias of manual analysis, MAGMa reports the refscore, as explained in the Methods section, which can be considered as an indicator of the general relevance of a compound, with high values indicating the more well-known and widely studied molecules.16) In the initial solution submitted to the CASMI contest, we have used a threshold value of 5 to filter the large numbers of candidates initially obtained for challenges 1, 2, and 14 (Table 1).
Table 1 (columns 4–7) also presents a more systematic analysis of the effect of filtering the candidate sets on the basis of the refscore. Without filtering, the total number of matching candidates in PubChem varies significantly between the different challenges, ranging from 1 to over 104 candidates. For each challenge we stepwise raised the threshold on the refscore until the candidates sets were reduced to <1000, <500, and <200 molecules. Figure 3 shows the filter option in the MAGMa results page obtained for challenge 1. Table 1 presents the actual numbers of candidates obtained for each challenge following this procedure, as well as the MAGMa rankings of the correct molecules within these candidate sets. All but one of the filtered candidate lists still contained the correct compound, indicating that all compounds in the CASMI 2013 challenge are relatively well-known molecules. Only for challenge 3, the correct compound, N2-acetyl glutaminyl leucinamide, was lost when the candidate list was reduced to less than 200 molecules. The MAGMa results obtained for all challenges, with selected (<1000) candidate lists, can be accessed online via http://www.emetabolomics.org/results_CASMI2013.html.
Fig. 3. The MAGMa results page for challenge 1. The optional filter on the refscore column is highlighted.

Chemical rearrangements
From Table 1 it can be concluded that the most difficult cases for MAGMa were challenges 4, 12, and 13. In challenge 4, the MS/MS spectrum of dihydrochalcone includes a peak due to loss of water. This water loss must consist of the carbonyl oxygen and two hydrogen atoms from the aliphatic backbone, which is not covered by substructures generated by MAGMa and results in the maximum penalty score. Consequently, a large number of molecules containing hydroxyl, epoxide or another cyclic ether, from which MAGMa does allow loss of water, obtained a better score. For baicalein (challenge 12), closer inspection of the data indicated that major MS/MS fragments involve loss of CO and CH2O2. These neutral losses are associated with chemical rearrangements of the heterocyclic part of the flavon structure.18,19) Such chemical rearrangements are not accounted for by MAGMa, and as a result, the substructures matched with these m/z values obtained high penalty scores. Finally, aloxistatin obtained a poor score in MAGMa because the most intense peak at m/z=246.1331 could not be matched with a substructure. This ion most likely represents an unusual rearrangement product. Entries for this challenge came only from three participants applying automatic methods on large candidate sets and none of them identified aloxistatin within the top 10. The results for challenges 4, 12, and 13 highlight a limitation of substructure-based methods by not accounting for chemical rearrangements.
To assess whether rule-based fragmentation could overcome this limitation for the above challenges we submitted the three molecules, dihydrochalcone, baicalein, and aloxistatin to one of the commercial rule-based software packages, Mass Frontier 7.0, using a fragmentation library search to predict fragments for baicalein in negative ion-mode. The fragments predicted by Mass Frontier included the loss of water from dihydrochalcone as well as the rearrangement of the heterocyclic part of baicalein corresponding to loss of carbon monoxide, illustrating the potential advantage of rule-based fragmentation. However, two other fragments of baicalein, with m/z=169.0675 (C3O4 loss) and m/z=223.0396 (CH2O2 loss), as well as the m/z=246.13 fragment of aloxistatin were not predicted by Mass Frontier 7.0, emphasizing that they are difficult to annotate in an automatic way, by both substructure and rule-based methods.3)
Category 1 entries
Based on the MAGMa entries in category 2, we also submitted lists of unique molecular formulas to the category 1 contest. Each molecular formula was assigned the highest score (reversed ranking value) at which it was present in the category 2 entry list. Isotope ratios given in the MS data files were not used for the category 1 submissions. Based on the additional MAGMa analyses described above, Table 2 also lists the results for the remaining challenges in which we did not participate. For all challenges, except challenge 12, MAGMa identified the correct molecular formula, even if the top ranking chemical structure was not correct. This is in line with earlier findings based on other applications of the MAGMa algorithm.15,16) It indicates that accurate mass fragmentation data often provides sufficient information to pick the right molecular formula from a large set of molecules without using additional information, such as isotope peak ratios of the precursor ion, and even if the top-ranked chemical structure is not correct.
Table 2. Overview of the MAGMa results on CASMI 2013 challenge data, category 1.
| # | Compound | Entry formulas | Rank | All PubChem formulas | Rank |
|---|---|---|---|---|---|
| 1 | Feruloyl tyramine | 5a | 1 | 9 | 1 |
| 2 | Feruloyl putrescine | 2a | 1 | 4 | 1 |
| 3 | N2-Acetyl glutaminyl leucinamide | 5 | 1 | 5 | 1 |
| 4 | Dihydrochalcone | 2 | 1 | 2 | 1 |
| 5 | Isoprothiolane | 4 | 1 | 4 | 1 |
| 6 | Phosphatidyl-6-acetyl-glucose | 2 | 1 | 2 | 1 |
| 9 | Chlorpyrifos | 15 | 1 | 22b | 1 |
| 10 | VAL-HIS-LEU-THR-PRO-VAL-GLU-LYS | 7 | 1 | 7 | 1 |
| 11 | Demethoxycurcumin | 14 | 1 | ||
| 12 | Baicalein | 13 | 2 | ||
| 15 | 2-(Perfluorooctyl)ethanol | 95 | 1 | ||
| 16 | Ofloxacin | 30 | 1 |
a Number of candidates were reduced by filtering on refscore >5. b New list of candidates retrieved after implementing local database of halogenated PubChem compound.
DISCUSSION
Although MAGMa performed well on most CASMI challenges, a number of lessons can be learned about the advantages and limitations of its application. For example, we did not participate in four challenges of the contest and the retrospective analyses provided hints to avoid such misses in future exercises.
The first obvious enhancement of MAGMa is the inclusion of halogenated compounds in the local databases of candidate molecules, which was required to obtain good solutions for challenges 15 and 16. This holds for challenge 9 as well, but in this case the need for halogenated candidate compounds had been recognized on the basis of the isotope peaks typical of chlorine and therefore relevant candidates had been retrieved from the online PubChem database already. We have now extended the MAGMa webinterface with an option to supplement the candidate sets with halogen containing compounds, by which satisfactory entries for challenges 15 and 16 could be generated. Secondly, the ofloxacin dataset of challenge 16 provided a clear example of a molecule where the default level of in silico fragmentation in MAGMa was insufficient. Retrospectively, up to 5 bonds had to be broken to generate the proper substructures for some of the MS/MS peaks. A third lesson learned was not to apply a too narrow mass tolerance, which caused us to incorrectly conclude from the initial results for challenge 11 that no correct molecule was present in the candidate list. These observations illustrate that although automated methods can facilitate certain aspects of the identification process, experts are still needed to properly apply these methods by integrating different factors and types of information.
After having recognized and “tackled” these basic aspects of the MAGMa processing, the rankings obtained for some of the challenges remain unsatisfactory. Automatic methods allow the consideration of large numbers of candidate molecules, e.g. retrieved from comprehensive chemical databases such as Pubchem. On the positive site, this may yield relevant results for mass data where manual methods that consider only small numbers of candidates may result in incomplete annotation. This is illustrated by the CASMI results for challenge 3, where the correct molecule was only present in two automatic entries with larger numbers of candidates. However, another consequence of processing large candidate sets is that also molecules with limited (biological or clinical) relevance may be included. These may include homologues of the correct molecules that may not be distinguishable on the basis of MS/MS data. To deliberately bias the results towards known compounds, the MAGMa interface allows filtering of the PubChem candidates (before or after in silico fragmentation) based on “refscore,” a simple statistic derived from the PubChem substances table. The filtering threshold on refscore could be stepwise increased to restrict candidate sets to less than 1000, less than 500, and less than 200 molecules. The rankings of the correct molecules improved, in absolute terms, with decreasing list sizes, indicating that this filter may help to focus the analysis towards the most relevant candidates. Although, this approach is not based on the actual experimental data, it may help expert users to do what they would do anyway, i.e., consider well-known molecules first. In the solutions we submitted for challenges 2 and 14, this filtering step improved the result in comparison to the non-filtered lists. Similarly, it improved the rankings in the analysis of challenges 11, 15, and 16. It should be noted, however, that while this filter can be useful for the annotation of known compounds, as in the case of most challenges in this contest, it should be applied carefully in practice when samples also contain less common compounds. This is exemplified by challenge 3 for which the synthetic product N2-acetyl glutaminyl leucinamide had the lowest possible refscore of 1 and is lost when a filter on refscore is applied.
In addition to the above considerations, the results show that there is room for improving the substructure based scoring. Major peaks in the spectra of baicalein and aloxistatin result from chemical rearrangements that cannot be adequately described by substructures. Either no or very unfavorable substructures are matched with these peaks resulting in maximum penalties contributing to the overall candidate scores. In these cases, more favorable scores are obtained for alternative molecules for which these fragment peaks can be matched with simpler substructures. It indicates that there is still a need for improved computational algorithms that combine the capability to handle chemical rearrangements3) with the speed to evaluate large numbers of candidate molecules.
Finally, the fact that the selection of candidate molecules, prior to MS/MS based scoring, has a significant effect on the rankings might be considered in the design and evaluation of the contest. A format in which candidate selection and subsequent automatic scoring are evaluated independently may provide more detailed insight into the differences between the automated methods. One possibility could be to define a separate category in which participants are asked to apply their computational (MS/MS based) scoring algorithm to the same set of candidate molecules. This would enable a more direct comparison of the performance of different in silico fragmentation and scoring algorithms and thereby help to identify bottlenecks and provide directions for further developments.
Acknowledgments
This research was supported by the Netherlands eScience Center, grant number 660.011.302.
L. Ridder, et al., Automatic Compound Annotation from Mass Spectrometry Data Using MAGMa, Mass Spectrom (Tokyo) 2014; 3(3): S0033; DOI: 10.5702/massspectrometry.S0033
References
- 1) S. Neumann, S. Böcker. Computational mass spectrometry for metabolomics: Identification of metabolites and small molecules. Anal. Bioanal. Chem. 398: 2779–2788, 2010. [DOI] [PubMed] [Google Scholar]
- 2) J. J. J. van der Hooft, R. C. H. de Vos, L. Ridder, J. Vervoort, R. J. Bino. Structural elucidation of low abundant metabolites in complex sample matrices. Metabolomics 9: 1009–1018, 2013. [Google Scholar]
- 3) F. Hufsky, K. Scheubert, S. Böcker. Computational mass spectrometry for small-molecule fragmentation. Trends Analyt. Chem. 53: 41–48, 2014. [Google Scholar]
- 4) T. Kind, O. Fiehn. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinformatics 7: 234, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5) T. Kind, O. Fiehn. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics 8: 105, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6) S. Böcker, M. C. Letzel, Z. Lipták, A. Pervukhin. SIRIUS: Decomposing isotope patterns for metabolite identification. Bioinformatics 25: 218–224, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7) H. Oberacher, M. Pavlic, K. Libiseller, B. Schubert, M. Sulyok, R. Schuhmacher, E. Csaszar, H. C. Köfeler. On the inter-instrument and the inter-laboratory transferability of a tandem mass spectral reference library: 2. Optimization and characterization of the search algorithm. J. Mass Spectrom. 44: 494–502, 2009. [DOI] [PubMed] [Google Scholar]
- 8) C. Hildebrandt, S. Wolf, S. Neumann. Database supported candidate search for metabolite identification. J. Integr. Bioinform. 8: 157, 2011. [DOI] [PubMed] [Google Scholar]
- 9) D. W. Hill, T. M. Kertesz, D. Fontaine, R. Friedman, D. F. Grant. Mass spectral metabonomics beyond elemental formula: Chemical database querying by matching experimental with computational fragmentation spectra. Anal. Chem. 80: 5574–5582, 2008. [DOI] [PubMed] [Google Scholar]
- 10) B. Bonn, C. Leandersson, F. Fontaine, I. Zamora. Enhanced metabolite identification with MSE and a semi-automated software for structural elucidation. Rapid Commun. Mass Spectrom. 24: 3127–3138, 2010. [DOI] [PubMed] [Google Scholar]
- 11) M. Heinonen, A. Rantanen, T. Mielikäinen, J. Kokkonen, J. Kiuru, R. A. Ketola, J. Rousu. FiD: A software for ab initio structural identification of product ions from tandem mass spectrometric data. Rapid Commun. Mass Spectrom. 22: 3043–3052, 2008. [DOI] [PubMed] [Google Scholar]
- 12) A. W. Hill, R. J. Mortishire-Smith. Automated assignment of high-resolution collisionally activated dissociation mass spectra using a systematic bond disconnection approach. Rapid Commun. Mass Spectrom. 19: 3111–3118, 2005. [Google Scholar]
- 13) S. Wolf, S. Schmidt, M. Muller-Hannemann, S. Neumann. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinformatics 11: 148, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14) L. Leclercq, R. J. Mortishire-Smith, M. Huisman, F. Cuyckens, M. J. Hartshorn, A. Hill. IsoScore: Automated localization of biotransformations by mass spectrometry using product ion scoring of virtual regioisomers. Rapid Commun. Mass Spectrom. 23: 39–50, 2009. [DOI] [PubMed] [Google Scholar]
- 15) L. Ridder, J. J. J. van der Hooft, S. Verhoeven, R. C. H. de Vos, R. van Schaik, J. Vervoort. Substructure-based annotation of high-resolution multistage MSn spectral trees. Rapid Commun. Mass Spectrom. 26: 2461–2471, 2012. [DOI] [PubMed] [Google Scholar]
- 16) L. Ridder, J. J. J. van der Hooft, S. Verhoeven, R. C. H. de Vos, R. J. Bino, J. Vervoort. Automatic chemical structure annotation of an LC-MSn based metabolic profile from green tea. Anal. Chem. 85: 6033–6040, 2013. [DOI] [PubMed] [Google Scholar]
- 17) E. Schymanski, S. Neumann. The critical assessment of small molecule identification (CASMI): Challenges and solutions. Metabolites 3: 517–538, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18) N. Fabre, I. Rustan, E. de Hoffmann, J. Quetin-Leclercq. Determination of flavone, flavonol, and flavanone aglycones by negative ion liquid chromatography electrospray ion trap mass spectrometry. J. Am. Soc. Mass Spectrom. 12: 707–715, 2001. [DOI] [PubMed] [Google Scholar]
- 19) J. J. J. van der Hooft, J. Vervoort, R. J. Bino, J. Beekwilder, R. C. H. de Vos. Polyphenol identification based on systematic and robust high-resolution accurate mass spectrometry fragmentation. Anal. Chem. 83: 409–416, 2011. [DOI] [PubMed] [Google Scholar]